
Abstract
Background
Treatment of neuromyelitis optica spectrum disorder (NMOSD) has so far been based on retrospective case series. The results of six randomized clinical trials including five different monoclonal antibodies targeting four molecules and three distinct pathophysiological pathways have recently been published.
Methods
Literature search on clinical trials and case studies in NMOSD up to July 10. 2020.
Results
We review mechanism of action, efficacy and side effects, and consequences for reproductive health from traditional immunosuppressants and monoclonal antibodies including rituximab, inebilizumab, eculizumab, tocilizumab and satralizumab.
Conclusion
In NMOSD patients with antibodies against aquaporin 4, monoclonal antibodies that deplete B cells (rituximab and inebilizumab) or interfere with interleukin 6 signaling (tocilizumab and satralizumab) or complement activation (eculizumab) have superior efficacy compared to placebo. Tocilizumab and rituximab were also superior to azathioprine in head-to-head studies. Rituximab, tocilizumab and to some extent eculizumab have well-known safety profiles for other inflammatory diseases, and rituximab and azathioprine may be safe during pregnancy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neuromyelitis optica (NMO) was previously characterized by bilateral optic neuritis and transverse myelitis. After the discovery of antibodies against aquaporin 4 (AQP4-IgG), it was acknowledged that clinical presentation can be more diverse, and the term NMO spectrum disorder (NMOSD) was introduced in 2007 [1]. In 2015, the International Panel for NMO Diagnosis decided to only use this unifying term [2]. In patients with AQP4-IgG, the diagnosis only requires one of the six core clinical criteria including optic neuritis and acute myelitis. In patients without AQP-4 IgG, it requires two core clinical characteristics disseminated in space, and at least one of these must be myelitis, optic neuritis, or area postrema syndrome supported by MRI [2]. Some AQP4-IgG-negative NMOSD patients may have antibodies against myelin oligodendrocyte glycoprotein (MOG-IgG). MOG-IgG-associated disease (MOGAD) is increasingly recognized as a distinct entity [3, 4], characterized by optic neuritis, transverse myelitis and/or brain stem syndrome in combination with positive MOG-IgG [5]. NMOSD with AQP4-IgG is rarely monophasic, attacks are often severe, and up to 25% have other autoimmune disease [6].
Several treatments that are effective in multiple sclerosis (MS), including interferon beta, fingolimod, alemtuzumab and natalizumab are associated with severe outcome including catastrophic exacerbations in patients with NMOSD [7,8,9].
Treatment of NMOSD has been based on case series and consensus reports [10]. Very recently, the results of six randomized clinical trials with five monoclonal antibodies (Table 1) targeting four different molecules and three pathways (Fig. 1) have been published [11,12,13,14,15,16]. We here review existing and emerging NMOSD treatments about to be implemented in clinical practice. Experimental treatments were recently reviewed elsewhere [17]. AQP4-IgG is expressed on the placenta, and expecting mothers with NMOSD have high risk of miscarriage, pre-eclampsia and eclampsia, particularly if untreated or unstable [18,19,20]. The relapse risk during pregnancy and particularly postpartum is also high [21,22,23,24]. Compatibility with pregnancy is, therefore, important in treatment decisions and will be highlighted in this paper.

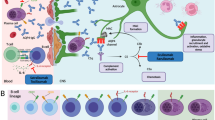
Pathogenesis and drug targets in NMOSD. Aquaporin 4 (AQP4)-specific B cells mature and differentiate in the periphery to memory cells or AQP4-IgG secreting cells (1). T cells may interact with B cells or dendritic cells, and upon stimuli including IL-6 T cells differentiate to pro-inflammatory Th17 cells that can cross the blood brain barrier (2). Inflammatory conditions allow both antibodies and complement factors to traverse the blood brain barrier and bind to AQP4 on astrocytic end feet (3). This activates complement through C1q ligation leading to formation of C5a and C5b. C5b is part of the membrane attack complexes (MAC) (4). C5a recruits pro-inflammatory leukocytes, including eosinophils, neutrophils, natural killer (NK) cells, and macrophages (5), capable of antibody dependent cellular cytotoxicity or inflammatory degranulation through Fc receptor activation (6). Astrocytes targeted by these mechanisms undergo destruction, and oligodendrocytes and neurons lose their supportive functions (7). The IL-6R blockers satralizumab and tocilizumab (A) suppress differentiation of AQP-IgG antibody secreting cells (ASC), as well as generation of pro-inflammatory Th17 T cells and M1 macrophages in favour of regulatory T cells and M2 macrophages. Rituximab (B1) kills cells expressing CD20 (mainly naïve and memory B cells, but also some T cells), while inebilizumab (B2) depletes a wider proportion of the B cell repertoire expressing CD19, including some antibody secreting cells. Both rituximab and inebilizumab deplete antigen presenting memory B cells. Eculizumab (C) blocks the complement cascade by binding complement component 5 (C5), halting generation of MAC through C5b and recruitment of pro-inflammatory cells through C5a. Printed with permission from © Kari C. Toverud
Traditional immunosuppressive treatment
Traditional immunosuppressive treatment, including azathioprine or mycophenolate mofetil in combination with low-dose corticosteroids has been a cornerstone in treatment of NMOSD, and is still widely used [25]. The use of low-dose corticosteroids for maintenance treatment is supported by retrospective evidence [26], but has not been studied systematically.
Azathioprine is a prodrug of 6-mercaptopurine, which inhibits purine synthesis and lymphocyte proliferation [27]. Positive results in NMOSD were first reported in 1998 in a prospective study of seven patients followed for at least 18 months [28]. Mean Expanded Disability Status Scale (EDSS) score improved from 9.0 to 3.0, and no relapses occurred.
Most evidences on azathioprine in NMOSD come from retrospective case series. Of 99 patients treated until 2009 at the Mayo Clinic, 38 discontinued treatment due to adverse events or disease activity [29]. In the remaining 61 patients, annualized relapse rate (ARR) fell from 1.5 to 0.2, and 26 became relapse-free. The failure rate for azathioprine was later reported to be higher than for rituximab and mycophenolate mofetil [30]. In 36 patients from Sao Paulo, mean ARR fell from 5.0 to 1.0 after treatment [31]. Of 100 Brazilian patients followed for median seven years, 69 had no disability progression but 11 discontinued treatment due to side effects [32]. Similar results were reported for 28 Iranian patients [33]. Of 103 AQP4-IgG-positive patients from UK, 63 remained relapse-free for a median follow-up of 18 months. Azathioprine was, however, discontinued in 47 patients, mostly because of side effects [34].
Recently, azathioprine was shown inferior to rituximab and tocilizumab on relapses and disability progression in head-to-head studies [11, 16], supporting previous case series and one open clinical trial indicating that azathioprine may be inferior to rituximab and mycophenolate mofetil [30, 35,36,37,38].
Azathioprine is transferred through placenta, but because the fetus lacks the enzyme generating active metabolites, concentrations of these are lower than in the mother [39, 40]. A recent expert recommendation emphasized that the number of exposed pregnancies exceeds 2000, and that azathioprine might be relatively safe during pregnancy [22]. Two large retrospective studies did, however, suggest increased risk of preterm birth and low birthweight [41, 42], and possibly also cardiac septal defects [42]. Another large study did not confirm this [43]. Bone marrow suppression in the infant may occur [44], and regular laboratory testing and ultrasound screening are necessary to evaluate fetal growth, organ development, and need of dose reduction [22]. The active drug is largely absent in breast milk (the maximum exposure of the drug to the infant is < 1% of the maternal dose) and blood of the infant [45, 46]. Breastfeeding is possible, although asymptomatic neutropenia has been reported [47]. The American College of Rheumatology strongly recommends continuing azathioprine during pregnancy and conditionally recommends during breastfeeding (118).
Mycophenolate mofetil is a reversible inhibitor of inosine monophosphate dehydrogenase, which is involved in guanosine nucleotide synthesis needed for lymphocyte proliferation [48]. In 2006, it was reported that a girl with NMO had been treated successfully with mycophenolate for two years [49]. The drug has later been increasingly used in NMOSD, and the first retrospective study was published in 2009 [50]. In 24 patients followed at the Mayo Clinic for a median of 28 months, median ARR fell from 1.3 to 0.09, and disability stabilized or decreased in 22 patients. Among 28 patients treated at the Mayo Clinic and the Johns Hopkins Hospital, failure rate (36%) was similar to that of rituximab and better than for azathioprine [30]. Corroborating these results, 36 of 62 Chinese patients remained relapse-free for a median of 1.5 years [51]. Case series and a meta-analysis indicate that the efficacy of mycophenolate mofetil is comparable to rituximab, and mycophenolate mofetil was also most tolerable in meta-analysis [36, 37].
Methotrexate is a folate derivative that inhibits nucleotide synthesis. In a retrospective study of 14 AQP4-IgG seropositive patients followed for a median of 21.5 months, median ARR decreased from 1.4 to 0.2, and 43% of the patients became relapse-free [52]. Disability stabilised or improved in 79%, and no patients stopped treatment due to adverse effects. In another nine patients, mean ARR dropped from 3.1 to 1.1 after treatment.[53].
Mitoxantrone intercalates into DNA causing crosslinks and strand breaks. In 2006, it was reported that five patients followed prospectively for two years had two relapses [54]. In 20 patients treated up to a cumulative dose of 120 mg/m2 and followed for a mean of 41 months, relapse rate decreased by 75% and disability improved or stabilized in all patients [55].
Cyclophosphamide is an alkylating agent that cross-links guanine bases in DNA. In four AQP4-IgG-positive patients, EDSS improved from 8.0 to 5.74 after treatment [56]. In another retrospective cohort of 41 patients treated for a median of 13.6 months, median ARR dropped from 0.7 to 0.0 [57]. Reports are, however, conflicting. Thus, six of seven patients treated with pulse doses of cyclophosphamide at the Federal University of São Paulo continued to have relapses from which one died [58].
Cyclosporine A is a natural immunosuppressant isolated from the fungus Beauveria nivea. In nine AQP4-IgG-positive patients NMOSD treated up to 51 months, ARR decreased from 2.7 to 0.4 [59].
Methotrexate, mitoxantrone, cyclophosphamide and mycophenolate mofetil are teratogenic [60,61,62], and should if possible be avoided in fertile women with NMOSD.
Autologous hematological stem cell transplantation (HSCT)
HSCT has not been extensively used in NMOSD, and results may depend on the conditioning regime. Thus, of 16 patients with refractory disease receiving different conditioning regimens comprising either intermediate-intensity myeloablative conditioning with carmustine, etoposide, cytarabine and melphalan (BEAM) plus anti-thymocyte globulin (ATG, n = 9) or low-intensity non-myeloablative regimen with thiotepa-cyclophosphamide (n = 3) cyclophosphamide and ATG (n = 4), only three patients remained relapse-free and 13 had relapse or progression in disability after a median of 47 months [63]. More encouraging results have been reported for non-myeloablative cyclophosphamide, ATG and rituximab conditioning [64]. In a prospective study, 80% of 12 patients treated with this non-myeloablative regime remained relapse-free without any other immunomodulatory treatment after a median of 57 months, and most patients also became AQP-IgG seronegative [65].
Interleukin 6 (IL-6) pathway inhibitors
The pro-inflammatory cytokine IL-6 was identified as a B cell stimulator in 1986. IL-6 is produced by several cell types including monocytes, macrophages and lymphocytes, and regulates the expression of proteins involved in inflammation, immune responses, and cell differentiation and homeostasis [66]. The IL-6 receptor (IL-6R) is expressed on the membrane of hepatocytes and leukocytes (mIL-6R), and as soluble forms (sIL-6R) in the circulation [66]. Both mIL-6R and sIL-6R transduce IL-6 signaling through glycoprotein 130 (gp130) which is ubiquitously expressed on hematopoietic and non-hematopoietic cells [67]. Classic signaling through mIL-6R induces anti-inflammatory responses, such as differentiation from M1 to M2 macrophages, while binding of sIL-6R/IL-6 to gp130 (trans-signaling) induces pro-inflammatory responses [66, 68]. In a third signaling pathway (trans-presentation), dendritic cells present mIL-6R/IL-6 to naïve T cells during differentiation into Th17 cells [69]. AQP4-specific Th17 cells are frequent in the blood of patients with NMOSD [70], and Th17 cells play an important role in the pathogenesis [71] (Fig. 1).
IL-6 levels are elevated in blood and cerebrospinal fluid of NMOSD patients and correlate with AQP4-IgG levels and disease severity [72, 73]. IL-6 facilitates disruption of the blood–brain barrier and enhances lesion severity [74, 75], and promotes AQP4-IgG production in vitro and ex vivo [76].
Tocilizumab was the first humanized monoclonal antibody against IL-6R. It is licenced for treatment of rheumatoid arthritis, giant cell arteritis and cytokine release syndrome, but has not been considered by EMA or FDA for NMOSD. Case reports suggest beneficial effects in NMOSD on relapses, disability progression, pain, and fatigue [77,78,79,80]. In 2014, a pilot study with seven NMOSD patients treated with intravenous tocilizumab reported a fall in mean ARR from 2.9 to 0.4 [81]. In another eight highly active patients followed for 10–51 months, median ARR dropped from 4.0 to 0.4 and median EDSS score from 7.3 to 5.5 [82]. Recently, similar results were reported in 12 patients treated with subcutaneous tocilizumab [83].
Tocilizumab has been used for 10 years and by more than one million rheumatoid arthritis (RA) patients, also in combination with methotrexate. The safety profile is, therefore, well established [84]. Tocilizumab induces a modest increase in lipoproteins and risk of neutropenia and bacterial infections, most markedly in combination with methotrexate [85]. This does not seem to increase with treatment duration [84, 86]. In the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis, the risk of serious infections tended to be higher for tocilizumab than for rituximab [87], whereas an opposite trend appeared in Danish and Swedish RA registries [88]. So far, neutropenia and serious infections seem to be less frequent in NMOSD than in RA [77].
TANGO was an open-label, multicentre, randomised, phase-2 trial comparing intravenous tocilizumab (8 mg/kg every 4 weeks) with azathioprine (2–3 mg/kg per day) in 118 Chinese patients followed for up to 90 weeks [16] (Table 1). Almost all patients used immunosuppressive therapy at baseline. Patients randomized to tocilizumab had to stop these within 12 weeks. Patients in the azathioprine arm used azathioprine as monotherapy from week 24. Those who had used azathioprine for less than 24 weeks before randomization received supplementary immunosuppressants until 24 weeks of azathioprine treatment. Time to relapse (primary outcome) was longer in the tocilizumab than in the azathioprine group (78.9 vs 56.7 weeks; p = 0.0026). Eight (14%) patients on tocilizumab and 28 (47%) on azathioprine had an attack during the study, corresponding to a risk reduction of 76% (p < 0.0001). Among the AQP4-IgG-negative patients, two of nine (22%) on tocilizumab and three of six (50%) on azathioprine relapsed during the study. AQP4-IgG levels dropped by 50% in the tocilizumab group and remained unchanged in the azathioprine group. Tocilizumab reduced the relative risk of 24 weeks confirmed disability progression by 78% compared to azathioprine (exploratory analysis).
Overall adverse events were equally frequent, but some adverse events were more common in the azathioprine group compared to tocilizumab: elevation of alanine transferase (31% vs 46%), upper respiratory tract infections (29% vs 39%), and urinary tract infections (29% vs 36%). There were 10-grade 3–5 adverse events in the tocilizumab group and 23 in the azathioprine group. Two patients stopped tocilizumab and three patients stopped azathioprine because of adverse events. One patient on tocilizumab died from myelitis and respiratory failure, and one patient on azathioprine died from listeriosis.
The concentration of tocilizumab in cord blood serum of an infant was recently reported to be 80–90% of maternal concentrations [89]. Prospective (n = 180) and retrospective (n = 108) data on pregnancies exposed to tocilizumab (beyond the first trimester in altogether 17 patients) indicated slightly increased risks of malformation without distinct pattern, spontaneous abortion, and preterm birth [90]. One-third of the patients with adverse outcomes were, however, also treated with methotrexate/leflunomide. No evidence of increased risks was found in a Japanese cohort of 61 pregnancies including 30 patients who were treated during the first trimester [91]. NMOSD experts recently recommended that tocilizumab can be used during pregnancy in patients with very severe NMOSD, and that breastfeeding could be considered under close monitoring [22]. The American College of Rheumatology concluded that treatment until conception and breastfeeding during treatment are supported by conditional evidence [92]. Drugs and Lactation Database recommend particular caution in mothers of preterm infants [93].
Satralizumab is modified from tocilizumab through amino acid sequence alterations in the CDR domains, variable regions, and constant regions. The mutations in the CDR and variable regions reduce binding affinity with IL-6R at low pH (Fig. 2). Satralizumab, therefore, dissociates from IL-6R in the acidic environment found in early endosomes, and is excreted rather than being digested [94], allowing extended dosage intervals (Fig. 2). Satralizumab is approved for AQP4 NMOSD by FDA, and decision by EMA is pending.

Endosomal processing of tocilizumab and satralizumab. The IL-6 receptor (IL6R) is present on a vast array of cells in the immune system, and also exists in soluble form in circulation. Membrane bound IL-6R (mIL-6R) or the soluble variant (sIL-6R) interacts with glycoprotein 130 (gp130) upon ligation with IL-6. Gp130 acts as a signal transducer into the cell that regulates expression of proteins involved in inflammation and cell homeostasis. Upon binding to mIL-6R, the receptor is brought intracellularly into endosomal compartments, where pH drops. Satralizumab, unlike the closely related tocilizumab, was specifically engineered with alterations in both variable regions to dissociate from IL-6R at low pH, and changes in the constant regions to simultaneously maintain affinity for neonatal Fc receptors (FcRn). FcRn are present in the endosomes, and allow satralizumab to recirculate to the cell surface and re-bind another s/mIL-6Rs thus enabling extended dosage protocol. Printed with permission from © Kari C. Toverud
Subcutaneous satralizumab 120 mg at week 0, 2 and every four weeks thereafter, has been tested as monotherapy and as add-on therapy in two double-blind placebo-controlled phase III trials (SAkuraStar and SAkuraSky), comprising altogether 188 patients aged 13–74 years [13, 14] (Table 1). In both trials, all reported relapses were evaluated by a clinical endpoint committee (adjudicated relapses).
In SAkuraSky, patients continued using azathioprine or mycophenolate mofetil plus oral glucocorticoids, but not rituximab [14]. At baseline, 24 of 42 patients on satralizumab and 21 of 41 patients on placebo used either azathioprine or mycophenolate mofetil. Adjudicated relapses (primary outcome) occurred in eight patients (20%) on satralizumab and in 18 (43%) on placebo (relative risk reduction 62%; p = 0.02). At 96 weeks, 78% of patients on satralizumab and 59% on placebo were relapse-free. The study was negative for AQP4-IgG-seronegative patients, as 5 of 14 patients on satralizumab and 6 of 14 patients on placebo relapsed. The first key secondary endpoint, reduction in pain, was negative. The number of adverse events and severe adverse events did not differ, and there were no deaths. Three patients on satralizumab and 10 patients on placebo discontinued the study during the double-blind period.
SAkuraStar was a phase-3, placebo-controlled parallel-group study of satralizumab as monotherapy [13]. Nineteen of the 63 patients (30%) receiving satralizumab and 16 of the 32 (50%) receiving placebo relapsed (HR 0.45, p = 0·018). After 96 weeks, 72% of patients on satralizumab and 51% on placebo were relapse-free. Again, the effect of satralizumab on ARR was only observed among AQP4-IgG-positive patients, as 10 of 22 (46%) patients on satralizumab in the AQP4-IgG-seronegative subgroup relapsed versus three of nine (33%) on placebo. As in SakuraSky, pain was not significantly reduced by satralizumab. Whereas the frequency of serious adverse events was quite similar (19% and 16%), severe adverse events were more common on satralizumab than on placebo (27% vs 6%). These were mostly considered unrelated to the study drug and did not lead to discontinuation, unless in one case of severe pneumonia in the satralizumab group. No deaths occurred in either group. One patient in each group withdrew from the study due to adverse events.
Collectively, these three trials provided evidence for the efficacy and safety of intravenous and subcutaneous anti-IL6R treatment mainly in AQP4-IgG-seropositive NMOSD, both as monotherapy and add-on treatment.
Pregnancy outcomes with satralizumab are unknown, but considerations may be similar to tocilizumab.
Complement blocking therapy
Complement deposits were early recognized in NMOSD lesions [95], and complement markedly enhances the pathogenicity of AQ4-IgG in vivo and ex vivo [74, 96,97,98]. Eculizumab is a monoclonal IgG2 antibody targeting C5, and inhibits cleaving and prevents release of pro-inflammatory C5a and the involvement of C5b in the membrane attack complex [99] (Fig. 3). Eculizumab could, thus, downregulate adaptive and innate immune responses either through C5a in the periphery, or through C5b on astrocytes in the CNS [100] (Figs. 1, 3). Eculizumab has been approved by EMA and FDA for AQP4-IgG positive NMOSD, and is also licenced for paroxysmal nocturnal hemoglobinuria and myasthenia gravis.

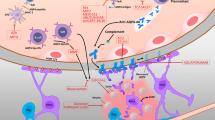
Mechanism of eculizumab. Eculizumab binds complement component 5 (C5) and prevents cleavage into C5a and C5b by C5 convertase. C5a is a potent attractant for leukocytes. C5b can form a complex with C6, and form the basis for formation of the membrane attack complex (MAC), which includes additional complement components. Printed with permission from © Kari C. Toverud
In an open-label phase II trial with 14 AQP4-IgG-positive NMOSD patients, 12 patients were relapse-free and none progressed during twelve months on eculizumab, whereas five relapsed within five months after withdrawal [101]. One patient had meningococcal sepsis and sterile meningitis during the treatment.
PREVENT was a phase III, randomized, double-blind, placebo-controlled trial comprising 143 AQP4-IgG-positive NMOSD patients randomized 2:1 to eculizumab (900 mg weekly for 4 weeks and 1200 mg every 2 weeks thereafter) or placebo [15] (Table 1). Most (76%) patients continued their previous immunosuppressive therapy. Forty-six patients had previously used rituximab, which was stopped within three months before inclusion. Given the uncertainty of when the final relapse would occur, the sponsor terminated the trial after 23 of the predefined 24 adjudicated relapses. Three of 96 patients on eculizumab and 20 of 47 on placebo had adjudicated relapse (primary outcome), corresponding to a hazard ratio 0.06 (p < 0.001). None of the 34 patients on eculizumab monotherapy experienced adjudicated relapses, versus 7 of 13 on placebo. Physician-determined (non-adjudicated) relapse (original primary end point) occurred in 14 patients on eculizumab and 29 on placebo (p < 0.001). There was no difference in disability progression as measured by EDSS (− 0.18 on eculizumab group and 0.12 on placebo). Patients receiving eculizumab had higher rates of upper respiratory tract infections and headache. There were no pneumococcus infections, but one patient on eculizumab and azathioprine died from pulmonary empyema with cultures yielding Peptostreptococcus micros and Streptococcus intermedius, which are part of the normal microbiota and common causes of opportunistic infections [102]. More patients on eculizumab (17%) than on placebo (6%) discontinued the study.
Data about eculizumab during pregnancy are available in paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, and HELLP syndrome (hemolysis, elevated liver enzymes and low platelet levels). In these diseases, higher dose of eculizumab is required during pregnancy to block complement activity [103]. Transfer to newborns is low [103], and newborns of mothers treated with eculizumab have normal complement function [104]. The concentrations in breast milk are also low, and eculizumab is suggested to be safe during pregnancy and lactation [105]. Data are, however, limited [93], and therapeutic concentration with transient low complement levels in a newborn was recently reported [106].
B cell depletion
B cell involvement in NMOSD may include production of autoantibodies, T cell activation and cytokine production [107] (Fig. 1).
Rituximab targets CD20 and depletes B cell lineage cells from late pro-B cells through early plamablasts, and also some T cells [108]. During the last 15 years, several retrospective case series have reported promising results in NMOSD. Rituximab has been included in treatment guidelines [109], but has not been reviewed or approved by EMA or FDA for this indication. In the first report from 2005, six of eight patients became relapse-free and seven experienced substantial disability improvement [110]. In two later retrospective studies on 25 and 23 patients, median ARR dropped from 1.7 and 1.9 to 0.0 and disability improved or stabilized in most patients [111, 112]. In the 10-year material from Johns Hopkins Hospital and the Mayo Clinic, rituximab reduced ARR up to 88.2%, and two in three patients achieved complete remission [30]. In 30 patients followed for five years in Korea, ARR fell from 2.4 to 0.3, 18 patients became relapse-free, and disability either improved or stabilized in 28 [113]. In another retrospective study of 32 patients treated first-line with rituximab, ARR was reduced by 97% [114]. Significant reduction in relapse rate was also reported in 16 children followed for a mean of 6.1 years [115].
In a 1-year open controlled trial comprising 86 patients, rituximab was significantly more effective than azathioprine [38]. ARR (primary outcome) decreased from 1.0 to 0.5 in the azathioprine group and from 1.3 to 0.2 in the rituximab group. Nineteen patients (54%) in the azathioprine group and 26 patients (79%) in the rituximab group became relapse-free. Patients receiving rituximab also improved significantly more in mean EDSS score (0.98 vs 0.44). Results were not specified for AQP4-IgG- seropositive and seronegative patients. It is, therefore, not known whether the high proportion of AQP4-IgG-seronegative patients (60.6% in the rituximab arm and 42.9% in the azathioprine arm for per protocol analysis) may have influenced the results. Any misclassification of MS as seronegative NMOSD could possibly have favoured rituximab.
In a randomized double-blind RCT (RIN-1), 19 patients were randomized to rituximab 375 mg/m2 weekly for four weeks and then 1000 mg every 2 week at week 24 and 48, and 19 patients were randomized to placebo [11] (Table 1). All patients were previously AQP4-IgG-seropositive, but 11 were seronegative at baseline. Mean ARR two years before inclusion was 1.4 for patients randomized to rituximab and 0.9 for patients randomized to placebo. Three patients on rituximab who discontinued treatment during the 72 weeks of follow-up (one withdrew consent, one used contraindicated drug, one had adverse event) were included in the primary analysis. Seven relapses occurred in the placebo group and none in the rituximab group (p = 0.0058). Change in EDSS score did not differ between groups. Eight of the 11 AQP4-IgG-seronegative patients at baseline became positive during the study. AQP4-IgG titers decreased in six patients on rituximab but in none of the patients on placebo, and increased > 10% in one patient on rituximab and in five patients on placebo.
The optimal dosage of rituximab in NMOSD is not determined. Studies using dosages ranging from 100 mg [116] to 1000–2000 mg [117] every 6 month have reported similar results. Both in adults and children, disease activity seems to correlate with depletion of B cells, but less clearly with AQP4-IgG levels [115, 118, 119]. In contrast to MOGAD, relapses mainly occur after repopulation of memory B cells in blood in AQP4-IgG NMOSD, supporting the effect of rituximab in AQP4-IgG-positive NMOSD [120]. Re-dosing based on measurements of CD27+ memory B cells may possibly allow disease control at a lower dose than fixed-dose intervals [113, 121].
Inebilizumab is a humanized, monoclonal antibody targeting the B cell surface antigen CD19. In contrast to CD20, CD19 is also expressed by pro-B cells, plasmablasts, and plasma cells, but not by any T cells. Inebilizumab has been studied in preclinical models [122] and was reported safe in phase-1 clinical studies of systemic sclerosis [123] and multiple sclerosis [124]. Inebiluzumab was approved by FDA for AQP4-IgG NMOSD June 2020, and has been granted orphan designation by EMA.
N-MOmentum was a double-blind phase-2/3 study comprising 231 patients randomized (3:1) to inebilizumab or placebo as monotherapy [12] (Table 1). The patients received intravenous inebilizumab (total dose 600 mg) or placebo on days 1 and 15, with no further doses. Two-thirds of the patients had used other immunosuppressants previously. Cyclophosphamide. Methotrexate, cyclosporine and mitoxantrone had to be stopped at least three months prior to randomization, and no other immunosupressants were allowed during the study. To minimize the risk of an attack following withdrawal of previous treatment and initiation of inebilizumab, all participants received oral prednisone 20 mg per day or equivalent from day one tapered to day 21. The double-blinded period lasted up to 197 days, or to a new NMOSD attack or termination of enrolment. All patients were thereafter offered open-label therapy.
Because of a clear demonstration of efficacy, enrolment was stopped before reaching the target of 252 patients and 67 adjudicated attacks. A total of 169 of 175 patients on inebilizumab and 54 of 56 patients on placebo completed the study period. Inebilizumab significantly increased time to onset of a new attack compared with placebo (p < 0.0001). In intention-to-treat analyses, 21 of 174 patients on inebilizumab experienced an attack versus 22 of 56 on placebo (hazard ratio 0.27, p < 0.0001). Among the 17 AQP4-IgG-seronegative patients, attacks were recorded in three of 13 on inebilizumab and none of four on placebo. Fewer patients had EDSS worsening on inebilizumab compared to placebo (p = 0.0049). No differences in change in low-contrast visual acuity binocular score from baseline were detected, but significantly fewer inebilizumab-treated patients experienced optic neuritis compared to placebo (10 patients in each group, corresponding to HR 0.288). Patients on inebilizumab had fewer new MRI lesions (p = 0.0034) and hospitalizations (p = 0.010) compared to placebo.
Adverse events, serious adverse events, infusion reactions and infections were equally frequent among patients on inebilizumab and on placebo, and no malignancies were reported. No death occurred during the placebo-controlled phase, but two patients died during the open-label phase, one (originally receiving placebo) because of respiratory insufficiency due to the ongoing disease process. Another patient (originally receiving inebilizumab) developed new neurological symptoms including seizures nine days after receiving 300 mg inebilizumab in the open-label period. MRI showed new large lesions in white and grey matter, considered not representative for progressive multifocal leukoencephalopathy (PML). PCR for John Cunningham virus in cerebrospinal fluid were negative in two laboratories but positive in another. No definitive diagnosis was made, and it could not be excluded that the death was treatment-related.
Monoclonal antibodies are not transported to the fetus until the second trimester [125]. Given their prolonged effect on B cells and half-life around three weeks [126], rituximab and inebiluzumab administered before conception may protect the mother against relapses without exposing the fetus to potential harmful effects. Data from 153 pregnancies in mothers treated with rituximab for hematological malignancies and autoimmune diseases, including 21 treated during pregnancy, indicated increased risk of miscarriage and premature birth [127]. Many of these mothers had, however, also used other immunosuppressants. Limited data in MS and NMOSD do not suggest increased risk of adverse pregnancy outcomes beyond reversible hematological abnormalities [128, 129], also in those very few treated during pregnancy [129, 130]. Both the American College of Rheumatology and NMOSD experts conditionally recommend rituximab until conception, and if necessary also during pregnancy [22, 92]. B cells should be measured in the infant, and vaccination planned accordingly.
Rituximab concentrations in breast milk are low, and rituximab is likely not absorbed from the gastrointestinal tract [131, 132]. The American College of Rheumatology recently concluded that recommending breastfeeding during treatment with rituximab is supported by strong evidence [92]. Drugs and Lactation Database recommend caution, particularly in preterm infants [133].
Pregnancy outcomes with inebilizumab are unknown, but considerations may be similar to anti-CD20 therapy.
Discussion
Differences in study design (active comparator versus placebo, monotherapy versus add-on therapy, open versus blinded) preclude conclusions on the relative efficacy of the monoclonal antibodies in NMOSD. Patients in the RIN-1 study of rituximab, N-MOmentum of inebiluzumab and SakuraSTAR of satralizumab had to stop ongoing immunosuppressive treatment [11,12,13]. The use of prednisolone was also restricted in these studies and in the TANGO study of tocilizumab (Table 1). It is, therefore, conceivable that control patients in these studies had poorer outcome than they would have had in routine practice [134]. On the other hand, co-treatment with immunosupressants that has not been proven effective in randomized trials may possibly have confounded the results of the PREVENT study of eculizumab and the SAkuraSky study of satralizumab [100]. It is, therefore, reassuring that satralizumab was superior to placebo also as monotherapy [13], and that rituximab and tocilizumab were more effective than azathioprine [16, 38]. Data on de novo treatment are scarce in NMOSD, particularly for the novel drugs, as all or more than two-thirds of the patients in the recent clinical trials had used other immunosuppressants at enrolment.
The robust treatment responses to eculizumab and to satralizumab monotherapy compared to placebo prove that IL-6 signaling and complement activation are relevant therapeutic targets in NMOSD patients with AQP4-IgG, who have the highest risk of relapse [135]. The effect of these novel treatment options is, however, less clear for patients without AQP4-IgG. Eculizumab was only tested in AQP4-IgG-positive patients [15], and the effect of satralizumab was not convincingly shown in seronegative patients [13, 14].
In MS patients, inebilizumab induced a 10.5% decrease in total immunoglobulin levels after 24 weeks and 15.0% after 18 months [126], exceeding that recorded for rituximab [136].
It could be speculated that the broader depletion of the B cell linage by inebilizumab, including antibody secreting cells, could be an advantage compared to rituximab [134]. Although theoretically appealing in an autoantibody-mediated disease, this remains to be proven. More pronounced drop in immunoglobulins may also lead to more infections.
Anti-drug antibodies may reduce therapeutic effects and can also cause adverse reactions through formation of immune complexes. This is particularly relevant for B cell therapies, as rituximab is a chimeric, whereas inebilizumab is a humanized antibody. Whereas no data exist in NMOSD, about one-third of MS patients develop antibodies against rituximab [137]. The clinical effect of such antibodies is not firmly established. They are, however, associated with poorer B cell depletion [137], which likely is unfavorable in NMOSD [118, 119], and may rarely also cause serum sickness [138]. The immunogenicity of inebilizumab seems much lower. Anti-inebilizumab antibodies were measurable in only 3% of NMOSD patients on inebilizumab, which was not more frequent than in patients on placebo [12]. Anti-drug antibodies are rare in rheumatoid arthritis patients treated with tocilizumab [139]. It is not known whether modifications have increased the immunogenicity of satralizumab.
Complement blocking increases the risk of meningococcal and encapsulated bacterial infection [140,141,142]. Therefore, patients in PREVENT and also the REGAIN study of eculizumab in myasthenia gravis were vaccinated against Neisseria meningitidis, and meningitis was not reported in either study [143]. Long-term treatment with rituximab and possibly also tocilizumab and satralizumab are also associated with infection risk [144, 145], and pneumococcal vaccination is often recommended before starting rituximab. Vaccination may, however, increase relapse risk in untreated NMOSD patients [146]. On the other hand, eculizumab, rituximab and most likely also inebilizumab reduce vaccine responses [142, 147]. Whether or not to start treatment before vaccination must be based on individual evaluation of infection and relapse risk.
Conclusion
The therapeutic armamentarium for NMOSD has expanded, and now includes evidence-based oral, subcutaneous and intravenous medications given daily, biweekly, monthly and every 6 months. The efficacy of monoclonal antibodies in NMOSD-IgG-positive patients is better documented, including class I evidence for eculizumab, inebilizumab and satralizumab, and likely also greater than traditional immunosuppressive therapy with azathioprine. Based on current evidence, we suggest that most NMOSD patients, particularly those with AQP4-IgG, should start with one of the monoclonal antibodies as a first-line treatment. Currently, we do not know which of these offers the best efficacy. Thus, treatment decisions will depend on factors like availability, price, co-morbidity and future pregnancy planning. For women who might become pregnant, rituximab has the best documented and possibly also most favorable safety profile, but azathioprine and some other monoclonal antibodies may also be compatible with pregnancy in selected patients.
Given the rarity of NMOSD, head-to-head studies of the monoclonal antibodies will not likely be conducted. Registry-based follow-up and real-life life studies will hopefully clarify optimal sequencing and combination, and particularly for B cell depleting drugs also long-term dosing regimens.
References
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6(9):805–815
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85(2):177–189
Mader S, Kumpfel T, Meinl E (2020) Novel insights into pathophysiology and therapeutic possibilities reveal further differences between AQP4-IgG- and MOG-IgG-associated diseases. Curr Opin Neurol 33(3):362–371
Takai Y, Misu T, Kaneko K, Chihara N, Narikawa K, Tsuchida S et al (2020) Myelin oligodendrocyte glycoprotein antibody-associated disease: an immunopathological study. Brain 143(5):1431–1446
Lopez-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP et al (2018) Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders. JAMA Neurol 75(11):1355–1363
Jarius S, Ruprecht K, Wildemann B, Kuempfel T, Ringelstein M, Geis C et al (2012) Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflamm 2012(9):14. https://doi.org/10.1186/1742-2094-9-14
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ (2012) Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler 18(10):1480–1483
Azzopardi L, Cox AL, McCarthy CL, Jones JL, Coles AJ (2016) Alemtuzumab use in neuromyelitis optica spectrum disorders: a brief case series. J Neurol 263(1):25–29
Kitley J, Evangelou N, Kuker W, Jacob A, Leite MI, Palace J (2014) Catastrophic brain relapse in seronegative NMO after a single dose of natalizumab. J Neurol Sci 339(1–2):223–225
Duchow A, Paul F, Bellmann-Strobl J (2020) Current and emerging biologics for the treatment of neuromyelitis optica spectrum disorders. Expert Opin Biol Ther 13:1–12
Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H et al (2020) Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 19(4):298–306
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM et al (2019) Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 394(10206):1352–1363
Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S et al (2020) Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol 19(5):402–412
Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B et al (2019) Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med 381(22):2114–2124
Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J et al (2019) Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 381(7):614–625
Zhang C, Zhang M, Qiu W, Ma H, Zhang X, Zhu Z et al (2020) Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol 19(5):391–401
Tradtrantip L, Asavapanumas N, Verkman AS (2020) Emerging therapeutic targets for neuromyelitis optica spectrum disorder. Expert Opin Ther Targets 24(3):219–229
Kim W, Kim SH, Nakashima I, Takai Y, Fujihara K, Leite MI et al (2012) Influence of pregnancy on neuromyelitis optica spectrum disorder. Neurology 78(16):1264–1267
Nour MM, Nakashima I, Coutinho E, Woodhall M, Sousa F, Revis J et al (2016) Pregnancy outcomes in aquaporin-4-positive neuromyelitis optica spectrum disorder. Neurology 86(1):79–87
Delgado-Garcia G, Chavez Z, Rivas-Alonso V, Corona T, Flores-Rivera J (2018) Obstetric outcomes in a Mexican cohort of patients with AQP4-antibody-seropositive neuromyelitis optica. Mult Scler Relat Disord 25:268–270
Klawiter EC, Bove R, Elsone L, Alvarez E, Borisow N, Cortez M et al (2017) High risk of postpartum relapses in neuromyelitis optica spectrum disorder. Neurology 89(22):2238–2244
Mao-Draayer Y, Thiel S, Mills EA, Chitnis T, Fabian M, Katz SI et al (2020) Neuromyelitis optica spectrum disorders and pregnancy: therapeutic considerations. Nat Rev Neurol 16(3):154–170
Shosha E, Pittock SJ, Flanagan E, Weinshenker BG (2017) Neuromyelitis optica spectrum disorders and pregnancy: interactions and management. Mult Scler 23(14):1808–1817
Borisow N, Hellwig K, Paul F (2018) Neuromyelitis optica spectrum disorders and pregnancy: relapse-preventive measures and personalized treatment strategies. EPMA J 9(3):249–256
Fragoso YD, Sousa NAC, Alves-Leon SV, Dias RM, Pimentel MLV, Gomes S et al (2019) Clinical characteristics of 153 Brazilian patients with neuromyelitis optica spectrum disorder (NMOSD). Mult Scler Relat Disord 27:392–396
Watanabe S, Misu T, Miyazawa I, Nakashima I, Shiga Y, Fujihara K et al (2007) Low-dose corticosteroids reduce relapses in neuromyelitis optica: a retrospective analysis. Mult Scler 13(8):968–974
Tiede I, Fritz G, Strand S, Poppe D, Dvorsky R, Strand D et al (2003) CD28-dependent Rac1 activation is the molecular target of azathioprine in primary human CD4+ T lymphocytes. J Clin Investig 111(8):1133–1145
Mandler RN, Ahmed W, Dencoff JE (1998) Devic's neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprine. Neurology 51(4):1219–1220
Costanzi C, Matiello M, Lucchinetti CF, Weinshenker BG, Pittock SJ, Mandrekar J et al (2011) Azathioprine: tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 77(7):659–666
Mealy MA, Wingerchuk DM, Palace J, Greenberg BM, Levy M (2014) Comparison of relapse and treatment failure rates among patients with neuromyelitis optica: multicenter study of treatment efficacy. JAMA Neurol 71(3):324–330
Bichuetti DB, Lobato de Oliveira EM, Oliveira DM, de Amorin SN, Gabbai AA (2010) Neuromyelitis optica treatment: analysis of 36 patients. Arch Neurol 67(9):1131–1136
Bichuetti DB, Perin MMM, Souza NA, Oliveira EML (2019) Treating neuromyelitis optica with azathioprine: 20-year clinical practice. Mult Scler 25(8):1150–1161
Sahraian MA, Moinfar Z, Khorramnia S, Ebrahim MM (2010) Relapsing neuromyelitis optica: demographic and clinical features in Iranian patients. Eur J Neurol 17(6):794–799
Elsone L, Kitley J, Luppe S, Lythgoe D, Mutch K, Jacob S et al (2014) Long-term efficacy, tolerability and retention rate of azathioprine in 103 aquaporin-4 antibody-positive neuromyelitis optica spectrum disorder patients: a multicentre retrospective observational study from the UK. Mult Scler 20(11):1533–1540
Torres J, Pruitt A, Balcer L, Galetta S, Markowitz C, Dahodwala N (2015) Analysis of the treatment of neuromyelitis optica. J Neurol Sci 351(1–2):31–35
Jeong IH, Park B, Kim SH, Hyun JW, Joo J, Kim HJ (2016) Comparative analysis of treatment outcomes in patients with neuromyelitis optica spectrum disorder using multifaceted endpoints. Mult Scler 22(3):329–339
Huang W, Wang L, Zhang B, Zhou L, Zhang T, Quan C (2019) Effectiveness and tolerability of immunosuppressants and monoclonal antibodies in preventive treatment of neuromyelitis optica spectrum disorders: a systematic review and network meta-analysis. Mult Scler Relat Disord 35:246–252
Nikoo Z, Badihian S, Shaygannejad V, Asgari N, Ashtari F (2017) Comparison of the efficacy of azathioprine and rituximab in neuromyelitis optica spectrum disorder: a randomized clinical trial. J Neurol 264(9):2003–2009
Saarikoski S, Seppala M (1973) Immunosuppression during pregnancy: transmission of azathioprine and its metabolites from the mother to the fetus. Am J Obstet Gynecol 115(8):1100–1106
de Boer NK, Jarbandhan SV, Mulder CJ, van Elburg RM, van Bodegraven AA (2006) Azathioprine use during pregnancy: unexpected intrauterine exposure to metabolites. Am J Gastroenterol 101(6):1390–1392
Goldstein LH, Dolinsky G, Greenberg R, Schaefer C, Cohen-Kerem R, Diav-Citrin O et al (2007) Pregnancy outcome of women exposed to azathioprine during pregnancy. Birth Defects Res A Clin Mol Teratol 79(10):696–701
Cleary BJ, Kallen B (2009) Early pregnancy azathioprine use and pregnancy outcomes. Birth Defects Res A Clin Mol Teratol 85(7):647–654
Francella A, Dyan A, Bodian C, Rubin P, Chapman M, Present DH (2003) The safety of 6-mercaptopurine for childbearing patients with inflammatory bowel disease: a retrospective cohort study. Gastroenterology 124(1):9–17
Jharap B, de Boer NK, Stokkers P, Hommes DW, Oldenburg B, Dijkstra G et al (2014) Intrauterine exposure and pharmacology of conventional thiopurine therapy in pregnant patients with inflammatory bowel disease. Gut 63(3):451–457
Christensen LA, Dahlerup JF, Nielsen MJ, Fallingborg JF, Schmiegelow K (2008) Azathioprine treatment during lactation. Aliment Pharmacol Ther 28(10):1209–1213
Sau A, Clarke S, Bass J, Kaiser A, Marinaki A, Nelson-Piercy C (2007) Azathioprine and breastfeeding: is it safe? BJOG 114(4):498–501
National Institute of health. Azathioprine. Drugs and lactation Database (lactMed). https://www.ncbi.nlm.nih.gov/books/NBK501050/. Accessed 20 Apr 2020
Allison AC, Eugui EM (1993) The design and development of an immunosuppressive drug, mycophenolate mofetil. Springer Semin Immunopathol 14(4):353–380
Falcini F, Trapani S, Ricci L, Resti M, Simonini G, de Martino M (2006) Sustained improvement of a girl affected with Devic's disease over 2 years of mycophenolate mofetil treatment. Rheumatology (Oxford) 45(7):913–915
Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E et al (2016) Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol 66(9):1128–1133
Chen H, Zhang Y, Shi Z, Feng H, Yao S, Xie J et al (2016) The efficacy and tolerability of mycophenolate mofetil in treating neuromyelitis optica and neuromyelitis optica spectrum disorder in western China. Clin Neuropharmacol 39(2):81–87
Kitley J, Elsone L, George J, Waters P, Woodhall M, Vincent A et al (2014) Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry 84(8):918–921
Ramanathan RS, Malhotra K, Scott T (2014) Treatment of neuromyelitis optica/neuromyelitis optica spectrum disorders with methotrexate. BMC Neurol 15(14):51
Weinstock-Guttman B, Ramanathan M, Lincoff N, Napoli SQ, Sharma J, Feichter J et al (2006) Study of mitoxantrone for the treatment of recurrent neuromyelitis optica (Devic disease). Arch Neurol 63(7):957–963
Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ (2011) Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol 68(4):473–479
Yaguchi H, Sakushima K, Takahashi I, Nishimura H, Yashima-Yamada M, Nakamura M et al (2013) Efficacy of intravenous cyclophosphamide therapy for neuromyelitis optica spectrum disorder. Intern Med 52(9):969–972
Xu Y, Wang Q, Ren HT, Qiao L, Zhang Y, Fei YY et al (2016) Comparison of efficacy and tolerability of azathioprine, mycophenolate mofetil, and cyclophosphamide among patients with neuromyelitis optica spectrum disorder: a prospective cohort study. J Neurol Sci 370:224–228
Bichuetti DB, Oliveira EM, Boulos FC, Gabbai AA (2012) Lack of response to pulse cyclophosphamide in neuromyelitis optica: evaluation of 7 patients. Arch Neurol 69(7):938–939
Kageyama T, Komori M, Miyamoto K, Ozaki A, Suenaga T, Takahashi R et al (2013) Combination of cyclosporine A with corticosteroids is effective for the treatment of neuromyelitis optica. J Neurol 260(2):627–634
Hyoun SC, Obican SG, Scialli AR (2012) Teratogen update: methotrexate. Birth Defects Res A Clin Mol Teratol 94(4):187–207
Asgari N, Henriksen TB, Petersen T, Lillevang ST, Weinshenker BG (2014) Pregnancy outcomes in a woman with neuromyelitis optica. Neurology 83(17):1576–1577
Coscia LA, Armenti DP, King RW, Sifontis NM, Constantinescu S, Moritz MJ (2015) Update on the teratogenicity of maternal mycophenolate mofetil. J Pediatr Genet 4(2):42–55
Greco R, Bondanza A, Oliveira MC, Badoglio M, Burman J, Piehl F et al (2015) Autologous hematopoietic stem cell transplantation in neuromyelitis optica: a registry study of the EBMT Autoimmune Diseases Working Party. Mult Scler 21(2):189–197
Aouad P, Li J, Arthur C, Burt R, Fernando S, Parratt J (2015) Resolution of aquaporin-4 antibodies in a woman with neuromyelitis optica treated with human autologous stem cell transplant. J Clin Neurosci 22(7):1215–1217
Burt RK, Balabanov R, Han X, Burns C, Gastala J, Jovanovic B et al (2019) Autologous nonmyeloablative hematopoietic stem cell transplantation for neuromyelitis optica. Neurology 93(18):e1732–e1741
Uciechowski P, Dempke WCM (2020) Interleukin-6: a masterplayer in the cytokine network. Oncology 98(3):131–137
Silver JS, Hunter CA (2010) gp130 at the nexus of inflammation, autoimmunity, and cancer. J Leukoc Biol 88(6):1145–1156
Garbers C, Heink S, Korn T, Rose-John S (2018) Interleukin-6, designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov 17(6):395–412
Heink S, Yogev N, Garbers C, Herwerth M, Aly L, Gasperi C et al (2017) Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat Immunol 18(1):74–85
Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, CCree BA et al (2012) Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 72(1):53–64
Lin J, Li X, Xia J (2016) Th17 cells in neuromyelitis optica spectrum disorder: a review. Int J Neurosci 126(12):1051–1060
Uzawa A, Mori M, Ito M, Uchida T, Hayakawa S, Masuda S et al (2009) Markedly increased CSF interleukin-6 levels in neuromyelitis optica, but not in multiple sclerosis. J Neurol 256(12):2082–2084
Lotan I, McGowan R, Levy M (2020) Anti-IL-6 therapies for neuromyelitis optica spectrum disorders: a systematic review of safety and efficacy. Curr Neuropharmacol. https://doi.org/10.2174/1570159X18666200429010825
Zhang H, Bennett JL, Verkman AS (2011) Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol 70(6):943–954
Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E et al (2017) Effects of neuromyelitis optica-IgG at the blood–brain barrier in vitro. Neurol Neuroimmunol Neuroinflamm 4(1):e311
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T et al (2011) Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA 108(9):3701–3706
Rosso M, Saxena S, Chitnis T (2020) Targeting IL-6 receptor in the treatment of neuromyelitis optica spectrum: a review of emerging treatment options. Expert Rev Neurother 20(5):509–516
Araki M (2019) Blockade of IL-6 signaling in neuromyelitis optica. Neurochem Int 130:104315
Ayzenberg I, Kleiter I, Schroder A, Hellwig K, Chan A, Yamamura T et al (2013) Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol 70(3):394–397
Kieseier BC, Stuve O, Dehmel T, Goebels N, Leussink VI, Mausberg AK et al (2013) Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol 70(3):390–393
Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M et al (2014) Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology 82(15):1302–1306
Ringelstein M, Ayzenberg I, Harmel J, Lauenstein AS, Lensch E, Stogbauer F et al (2015) Long-term therapy with interleukin 6 receptor blockade in highly active neuromyelitis optica spectrum disorder. JAMA Neurol 72(7):756–763
Lotan I, Charlson RW, Ryerson LZ, Levy M, Kister I (2019) Effectiveness of subcutaneous tocilizumab in neuromyelitis optica spectrum disorders. Mult Scler Relat Disord. https://doi.org/10.1016/j.msard.2019.101920
Choy EH, De BF, Takeuchi T, Hashizume M, John MR, Kishimoto T (2020) Translating IL-6 biology into effective treatments. Nat Rev Rheumatol 16(6):335–345
Campbell L, Chen C, Bhagat SS, Parker RA, Ostor AJ (2011) Risk of adverse events including serious infections in rheumatoid arthritis patients treated with tocilizumab: a systematic literature review and meta-analysis of randomized controlled trials. Rheumatology (Oxford) 50(3):552–562
Jones G, Panova E (2018) New insights and long-term safety of tocilizumab in rheumatoid arthritis. Ther Adv Musculoskelet Dis 10(10):195–199
Rutherford AI, Subesinghe S, Hyrich KL, Galloway JB (2018) Serious infection across biologic-treated patients with rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Ann Rheum Dis 77(6):905–910
Gron KL, Arkema EV, Glintborg B, Mehnert F, Ostergaard M, Dreyer L et al (2019) Risk of serious infections in patients with rheumatoid arthritis treated in routine care with abatacept, rituximab and tocilizumab in Denmark and Sweden. Ann Rheum Dis 78(3):320–327
Tada Y, Sakai M, Nakao Y, Maruyama A, Ono N, Koarada S (2019) Placental transfer of tocilizumab in a patient with rheumatoid arthritis. Rheumatology (Oxford) 58(9):1694–1695
Hoeltzenbein M, Beck E, Rajwanshi R, Gotestam SC, Berber E, Schaefer C et al (2016) Tocilizumab use in pregnancy: analysis of a global safety database including data from clinical trials and post-marketing data. Semin Arthritis Rheum 46(2):238–245
Nakajima K, Watanabe O, Mochizuki M, Nakasone A, Ishizuka N, Murashima A (2016) Pregnancy outcomes after exposure to tocilizumab: a retrospective analysis of 61 patients in Japan. Mod Rheumatol 26(5):667–671
Sammaritano LR, Bermas BL, Chakravarty EE, Chambers C, Clowse MEB, Lockshin MD et al (2020) 2020 American College of rheumatology guideline for the management of reproductive health in rheumatic and musculoskeletal diseases. Arthritis Rheumatol 72(4):529–556
National Institute of health. Drugs and Lactation Database Eculizumab (July 2020). https://www.ncbi.nlm.nih.gov/books/NBK501921/. Accessed 24 Aug 2020
Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S et al (2010) Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 28(11):1203–1207
Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125(Pt 7):1450–1461
Soltys J, Liu Y, Ritchie A, Wemlinger S, Schaller K, Schumann H et al (2019) Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J Clin Investig 129(5):2000–2013
Geis C, Ritter C, Ruschil C, Weishaupt A, Grunewald B, Stoll G et al (2015) The intrinsic pathogenic role of autoantibodies to aquaporin 4 mediating spinal cord disease in a rat passive-transfer model. Exp Neurol 265:8–21
Bennett JL, Owens GP (2017) Neuromyelitis optica: deciphering a complex immune-mediated astrocytopathy. J Neuroophthalmol 37(3):291–299
Schatz-Jakobsen JA, Zhang Y, Johnson K, Neill A, Sheridan D, Andersen GR (2016) Structural basis for eculizumab-mediated inhibition of the complement terminal pathway. J Immunol 197(1):337–344
Pardo S, Giovannoni G, Hawkes C, Lechner-Scott J, Waubant E, Levy M (2019) Editorial on: Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 33:A1–A2. https://doi.org/10.1016/j.msard.2019.07.001
Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF et al (2013) Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 12(6):554–562
Issa E, Salloum T, Tokajian S (2020) From normal flora to brain abscesses: a review of Streptococcus intermedius. Front Microbiol 11:826. https://doi.org/10.3389/fmicb.2020.00826
Sarno L, Tufano A, Maruotti GM, Martinelli P, Balletta MM, Russo D (2019) Eculizumab in pregnancy: a narrative overview. J Nephrol 32(1):17–25
Hallstensen RF, Bergseth G, Foss S, Jaeger S, Gedde-Dahl T, Holt J et al (2015) Eculizumab treatment during pregnancy does not affect the complement system activity of the newborn. Immunobiology 220(4):452–459
Kelly RJ, Hochsmann B, Szer J, Kulasekararaj A, de Guibert S, Roth A et al (2015) Eculizumab in pregnant patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 373(11):1032–1039
Duineveld C, Wijnsma KL, Volokhina EB, van den Heuvel LPB, van de Kar NCAJ, Wetzels JFM (2019) Placental passage of eculizumab and complement blockade in a newborn. Kidney Int 95(4):996
Sabatino JJ Jr, Probstel AK, Zamvil SS (2019) B cells in autoimmune and neurodegenerative central nervous system diseases. Nat Rev Neurosci 20(12):728–745
Sellebjerg F, Blinkenberg M, Sorensen PS (2020) Anti-CD20 monoclonal antibodies for relapsing and progressive multiple sclerosis. CNS Drugs 34(3):269–280
Ciron J, Audoin B, Bourre B, Brassat D, Durand-Dubief F, Laplaud D et al (2018) Recommendations for the use of Rituximab in neuromyelitis optica spectrum disorders. Rev Neurol (Paris) 174(4):255–264
Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C (2005) An open label study of the effects of rituximab in neuromyelitis optica. Neurology 64(7):1270–1272
Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ et al (2008) Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol 65(11):1443–1448
Bedi GS, Brown AD, Delgado SR, Usmani N, Lam BL, Sheremata WA (2011) Impact of rituximab on relapse rate and disability in neuromyelitis optica. Mult Scler 17(10):1225–1230
Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ (2013) A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol 70(9):1110–1117
Zephir H, Bernard-Valnet R, Lebrun C, Outteryck O, Audoin B, Bourre B et al (2015) Rituximab as first-line therapy in neuromyelitis optica: efficiency and tolerability. J Neurol 262(10):2329–2335
Nosadini M, Alper G, Riney CJ, Benson LA, Mohammad SS, Ramanathan S et al (2016) Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol Neuroimmunol Neuroinflamm 3(1):e188
Yang CS, Yang L, Li T, Zhang DQ, Jin WN, Li MS et al (2013) Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis optica. Neurology 81(8):710–713
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ (2011) Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 68(11):1412–1420
Pellkofer HL, Krumbholz M, Berthele A, Hemmer B, Gerdes LA, Havla J et al (2011) Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology 76(15):1310–1315
Radaelli M, Moiola L, Sangalli F, Esposito F, Barcella V, Ferre L et al (2016) Neuromyelitis optica spectrum disorders: long-term safety and efficacy of rituximab in Caucasian patients. Mult Scler 22(4):511–519
Durozard P, Rico A, Boutiere C, Maarouf A, Lacroix R, Cointe S et al (2020) Comparison of the response to rituximab between myelin oligodendrocyte glycoprotein and aquaporin-4 antibody diseases. Ann Neurol 87(2):256–266
Trewin BP, Adelstein S, Spies JM, Beadnall HN, Barton J, Ho N et al (2020) Precision therapy for neuromyelitis optica spectrum disorder: a retrospective analysis of the use of class-switched memory B-cells for individualised rituximab dosing schedules. Mult Scler Relat Disord 43:102175
Chen D, Gallagher S, Monson NL, Herbst R, Wang Y (2016) Inebilizumab, a B cell-depleting anti-CD19 antibody for the treatment of autoimmune neurological diseases: insights from preclinical studies. J Clin Med 5(12):107. https://doi.org/10.3390/jcm5120107
Schiopu E, Chatterjee S, Hsu V, Flor A, Cimbora D, Patra K et al (2016) Safety and tolerability of an anti-CD19 monoclonal antibody, MEDI-551, in subjects with systemic sclerosis: a phase I, randomized, placebo-controlled, escalating single-dose study. Arthritis Res Ther 18(1):131
Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K et al (2019) Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult Scler 25(2):235–245
Palmeira P, Quinello C, Silveira-Lessa AL, Zago CA, Carneiro-Sampaio M (2012) IgG placental transfer in healthy and pathological pregnancies. Clin Dev Immunol 2012:985646
Breedveld F, Agarwal S, Yin M, Ren S, Li NF, Shaw TM et al (2007) Rituximab pharmacokinetics in patients with rheumatoid arthritis: B-cell levels do not correlate with clinical response. J Clin Pharmacol 47(9):1119–1128
Chakravarty EF, Murray ER, Kelman A, Farmer P (2001) Pregnancy outcomes after maternal exposure to rituximab. J Am Soc Hematol 117(5):1499–1506
Smith JB, Hellwig K, Fink K, Lyell DJ, Piehl F, Langer-Gould A (2020) Rituximab, MS, and pregnancy. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000000734
Das G, Damotte V, Gelfand JM, Bevan C, Cree BAC, Do L et al (2018) Rituximab before and during pregnancy: a systematic review, and a case series in MS and NMOSD. Neurol Neuroimmunol Neuroinflamm 5(3):e453
Munger KC, Samkoff LM (2020) Initiation of rituximab therapy for new onset neuromyelitis optica spectrum disorder during pregnancy. Mult Scler Relat Disord 37:101442. https://doi.org/10.1016/j.msard.2019.101442
Bragnes Y, Boshuizen R, de VA, Lexberg A, Ostensen M (2017) Low level of Rituximab in human breast milk in a patient treated during lactation. Rheumatology (Oxford) 56(6):1047–1048
Krysko KM, LaHue SC, Anderson A, Rutatangwa A, Rowles W, Schubert RD et al (2020) Minimal breast milk transfer of rituximab, a monoclonal antibody used in neurological conditions. Neurol Neuroimmunol Neuroinflamm. https://doi.org/10.1212/NXI.0000000000000637
National Institute of health. Rtuximab. Drugs and lactation Database (lactMed).https://www.ncbi.nlm.nih.gov/books/NBK501798/. Accessed 19 May 2020
Silbermann E, Bourdette D (2019) A new era for neuromyelitis optica spectrum disorder. Lancet 394(10206):1304–1305
Stellmann JP, Krumbholz M, Friede T, Gahlen A, Borisow N, Fischer K et al (2017) Immunotherapies in neuromyelitis optica spectrum disorder: efficacy and predictors of response. J Neurol Neurosurg Psychiatry 88(8):639–647
Salzer J, Svenningsson R, Alping P, Novakova L, Bjorck A, Fink K et al (2016) Rituximab in multiple sclerosis: a retrospective observational study on safety and efficacy. Neurology 87(20):2074–2081
Dunn N, Juto A, Ryner M, Manouchehrinia A, Piccoli L, Fink K et al (2018) Rituximab in multiple sclerosis: frequency and clinical relevance of anti-drug antibodies. Mult Scler 24(9):1224–1233
Holmoy T, Fogdell-Hahn A, Svenningsson A (2019) Serum sickness following rituximab therapy in multiple sclerosis. Neurol Clin Pract 9(6):519–521
Burmester GR, Choy E, Kivitz A, Ogata A, Bao M, Nomura A et al (2017) Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis 76(6):1078–1085
Frampton JE (2020) Eculizumab: a review in neuromyelitis optica spectrum disorder. Drugs 80(7):719–727
Koelman DLH, Brouwer MC, van de Beek D (2019) Targeting the complement system in bacterial meningitis. Brain 142(11):3325–3337
Alashkar F, Vance C, Herich-Terhurne D, Preising N, Duhrsen U, Roth A (2017) Serologic response to meningococcal vaccination in patients with paroxysmal nocturnal hemoglobinuria (PNH) chronically treated with the terminal complement inhibitor eculizumab. Ann Hematol 96(4):589–596
Muppidi S, Utsugisawa K, Benatar M, Murai H, Barohn RJ, Illa I et al (2019) Long-term safety and efficacy of eculizumab in generalized myasthenia gravis. Muscle Nerve 60(1):14–24
Luna G, Alping P, Burman J, Fink K, Fogdell-Hahn A, Gunnarsson M et al (2019) Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol 77(2):184–191
Yun H, Xie F, Delzell E, Levitan EB, Chen L, Lewis JD et al (2016) Comparative risk of hospitalized infection associated with biologic agents in rheumatoid arthritis patients enrolled in medicare. Arthritis Rheumatol 68(1):56–66
Mealy MA, Cook LJ, Pache F, Velez DL, Borisow N, Becker D et al (2018) Vaccines and the association with relapses in patients with neuromyelitis optica spectrum disorder. Mult Scler Relat Disord 23:78–82
Nazi I, Kelton JG, Larche M, Snider DP, Heddle NM, Crowther MA et al (2013) The effect of rituximab on vaccine responses in patients with immune thrombocytopenia. Blood 122(11):1946–1953
Funding
Open Access funding provided by University of Oslo (incl Oslo University Hospital). No targeted funding was received for this work. KMM and ØT are funded by Norwegian Research Council Grant #288164 (Neuro-SysMed).
Author information
Authors and Affiliations
Contributions
All authors participated in data collection and writing the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
Trygve Holmøy has received speakers’ honoraria and/or unrestricted research grants from Biogen, Roche, Sanofi and Merck, and has been principal investigator in studies sponsored by Biogen, Merck and Roche. Rune A. Høglund has received speakers’ honoraria and/or unrestricted grants from Biogen, Merck, Roche and Novartis, and participated as investigator in study sponsored by Roche. Zsolt Illes has received speakers’ honoraria and/or research grants from Biogen, Roche, Sanofi, Novartis and Merck, has been member of advisory boards at Alexion, Biogen, Sanofi, Merck, Roche, Novartis, was member of the adjudication relapse committee in the SAkuraStar and SAkuraSky trials, and has been principal investigator in studies sponsored by Biogen, Merck and Sanofi. Øivind Torkildsen has received speakers’ honoraria and/or unrestricted research grants from Biogen, Roche, Novartis, Merck and Sanofi and has been principal investigator in studies sponsored by Sanofi. Kjell-Morten Myhr has received unrestricted research grants to his institution; scientific advisory board, and speakers’ honoraria from Almirall, Biogen, Genzyme, Merck, Novartis, Roche, or Teva; and has participated in clinical trials organized by Biogen, Merck, Novartis, or Roche.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Consent to publish the figures has been granted by Kari C Toverud.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holmøy, T., Høglund, R.A., Illes, Z. et al. Recent progress in maintenance treatment of neuromyelitis optica spectrum disorder. J Neurol 268, 4522–4536 (2021). https://doi.org/10.1007/s00415-020-10235-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10235-5