
Abstract
Background
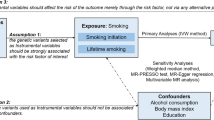
Striking changes in the demographic pattern of multiple sclerosis (MS) strongly indicate an influence of modifiable exposures, which lend themselves well to intervention. It is important to pinpoint which of the many environmental, lifestyle, and sociodemographic changes that have occurred over the past decades, such as higher smoking and obesity rates, are responsible. Mendelian randomization (MR) is an elegant tool to overcome limitations inherent to observational studies and leverage human genetics to inform prevention strategies in MS.
Methods
We use genetic variants from the largest genome-wide association study for smoking phenotypes (initiation: N = 378, heaviness: N = 55, lifetime smoking: N = 126) and body mass index (BMI, N = 656) and apply these as instrumental variables in a two-sample MR analysis to the most recent meta-analysis for MS. We adjust for the genetic correlation between smoking and BMI in a multivariable MR.
Results
In univariable and multivariable MR, smoking does not have an effect on MS risk nor explains part of the association between BMI and MS risk. In contrast, in both analyses each standard deviation increase in BMI, corresponding to roughly 5 kg/m2 units, confers a 30% increase in MS risk.
Conclusion
Despite observational studies repeatedly reporting an association between smoking and increased risk for MS, MR analyses on smoking phenotypes and MS risk could not confirm a causal relationship. This is in contrast with BMI, where observational studies and MR agree on a causal contribution. The reasons for the discrepancy between observational studies and our MR study concerning smoking and MS require further investigation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is an autoimmune disease of the central nervous system, with both genetic and environmental factors implicated in its etiology [1]. Genome-wide association studies (GWASs) have identified more than 200 independent associations mediating disease risk [2,3,4,5,6], but these genetic factors explain < 50% of variance in MS risk between individuals [3].
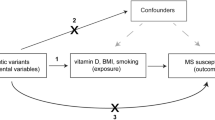
Environmental risk factors associated with MS in observational studies include Epstein-Barr virus infection, low serum vitamin D [25(OH)D3], obesity and smoking [7]. Unlike genetic risk factors, environmental and lifestyle factors can be modified, with potential for prevention. Behavioural, socioeconomic, and physiological factors are strongly interrelated, which can lead to residual confounding in observational studies [8, 9]. Furthermore, causal inference from observational studies is impeded by reverse causation, selection and recall bias. To overcome this, a Mendelian randomization (MR) analysis uses genetic variants that are a proxy for environmentally modifiable exposures as an instrumental variable (IV) to assess the presence of a causal relationship between environmental factors and an outcome [10]. For three of the environmental risk factors suggested by observational studies (low serum vitamin D, obesity and smoking), sufficiently strong IVs for use in MR studies are now available. Such analyses have repeatedly supported a causal association of low serum 25(OH)D3 and obesity with increased MS susceptibility [11,12,13,14,15]. However, a causal relationship between smoking and MS risk remains underinvestigated.
In this study, we make use of the largest GWAS summary statistics for smoking to date [16] and apply a two-sample MR analysis to the most recent meta-analysis GWAS summary statistics for MS from the International Multiple Sclerosis Genetics Consortium (IMSGC) [3].
Methods
Genetic datasets
Genome-wide significant (p < 5 × 10−8) genetic variants associated with smoking initiation, a binary phenotype indicating whether an individual has ever smoked regularly, were obtained from Supplementary Table 4 of the GWAS and Sequencing Consortium of Alcohol and Nicotine use (GSCAN) study for smoking initiation [16], with the largest sample size to date involving 1,232,091 individuals. The 378 single nucleotide polymorphisms (SNPs) explain 2.3% of the phenotypic variation in smoking initiation [16]. As secondary phenotypes, we included two measures reflecting a dose- and time-effect for smoking. A total of N = 55 genetic variants associated with smoking heaviness, measured by cigarettes per day in 337,334 individuals, were obtained from the same study, and explain ~ 1% of phenotypic variation [16]. For other variants than those reaching genome-wide significance, full summary statistics for smoking phenotypes were downloaded (Supplementary Table 1), but are only available for up to 632,807 individuals, excluding 23andMe participants. An IV reflecting lifetime smoking has been constructed in 462,690 individuals of European ancestry from the UK Biobank by integrating information on smoking status (current, former, never), age at initiation, age at cessation and number of cigarettes smoked per day. This IV is based on 126 independent, genome-wide significant SNPs and captures 0.36% of the variance [17].
Data from The Genetic Investigation of Anthropometric Traits (GIANT) Consortium meta-analysis for body mass index (BMI) in 681,275 individuals [18] (Supplementary Table 1) were used for investigating the causality between smoking phenotypes and BMI and for correcting the effect of smoking-associated SNPs on BMI and vice versa. All primary and secondary genome-wide significant SNPs (N = 941) explain 6% of variance in BMI in a cohort of 8852 individuals [18]. We include the N = 656 primary associations listed in the study as IVs. Corresponding effects of the smoking- and BMI-associated SNPs on MS susceptibility were derived from the discovery cohorts of the latest IMSGC meta-analysis, including up to 41,505 participants (14,802 MS, 26,703 controls) [3]. For the reverse analysis of genetically predicted MS risk on smoking initiation, cigarettes per day and lifetime smoking, N = 138 primary SNP associations with MS were derived from Supplementary Table 7 of the latest IMSGC meta-analysis for multiple sclerosis risk [3].
Selection of instrumental variables
Clumping and data harmonization were implemented in R v3.6.1 using the TwoSampleMR package (v0.5.1) [19]. For each genetic variant, alleles were aligned and matched so that their effects correspond to an increase in the corresponding exposure to which they are associated. The odds ratios (ORs) and p values of the summary statistics from the IMSGC were transformed into β coefficients and standard errors for subsequent analyses. SNPs with OR of exactly one were excluded. As the BMI phenotype in the GIANT meta-analysis was normalized, β coefficients correspond to standard deviations (SD) of BMI, with one SD equaling a mean of 4.70 BMI units (kg/m2) among cohorts in the GIANT consortium [18]. For smoking, the scale of β is on the unit of the SD of the phenotype, with a one SD increase in genetically predicted smoking initiation and heaviness corresponding to a 10% increased risk of smoking and three additional daily cigarettes, respectively [16]. Individuals who have never smoked and have no smoking exposure have a lifetime smoking score of zero, and a one SD increase is equivalent to an individual smoking 20 cigarettes a day for 15 years and stopping 17 years ago or an individual smoking 60 cigarettes a day for 13 years and stopping 22 years ago [17]. To prevent result bias by strongly correlated SNPs, SNPs were excluded from analyses if their measured linkage disequilibrium (LD) is r2 > 0.05 in the European samples of 1000 Genomes. For SNPs in LD with r2 > 0.05, the SNP with the lowest p value is retained. Furthermore, to prevent strand ambiguity issues and as minor allele frequencies were not available for all summary statistics, only non-palindromic SNPs were retained. For exposure-associated variants not directly ascertained in the outcome datasets in the univariable analysis, we looked for proxy SNPs in high linkage disequilibrium (r2 > 0.9) using LDlinkR package in R (v3.6.1). Supplementary Table 2 contains an overview of the number of included and excluded SNPs.
In the univariable MR analysis for the effect of exposures on MS risk, a total of N = 297 SNPs, N = 38 SNPs, N = 111 SNPs and N = 529 SNPs were included for smoking initiation (Supplementary Table 3), cigarettes per day (Supplementary Table 4), lifetime smoking (Supplementary Table 5) and BMI (Supplementary Table 6), respectively. In the multivariable MR analysis, N = 576 SNPs associated with either smoking initiation or BMI (Supplementary Table 7) were retained. To investigate the effect of BMI on smoking initiation and the reverse, N = 530 SNPs (Supplementary Table 8) and N = 266 SNPs (Supplementary Table 9) were included, respectively. Finally, to examine causality between BMI and cigarettes per day, N = 531 SNPs were used (Supplementary Table 10), and for the reverse analysis N = 29 SNPs (Supplementary Table 11).
For the reverse analysis of genetically predicted MS risk on smoking initiation, cigarettes per day and lifetime smoking, a total of N = 118 SNPs were included (Supplementary Table 12, 13 and 14, respectively).
Statistical analyses
MR analyses were implemented in R v3.6.1 using the TwoSampleMR package (v0.5.1) [19].
Effects of IVs on outcomes were estimated using the traditional MR approach, that is a multiplicative random-effects inverse-variance weighted (IVW) analysis [19, 20]. Sensitivity tests more robust to potential pleiotropy but potentially less powered included MR Egger [21], weighted median regression [22] and the mode-based estimator (simple and weighted) [23]. We considered as MR results suggestive of causal effects those that were concordant in direction across multiple MR approaches and pass nominal significance in IVW MR.
The Cochran Q test and I2 statistic [24] were calculated to measure the degree of heterogeneity across the individual effect estimates derived from each genetic variant [25]. Horizontal pleiotropy was evaluated based on the intercept obtained from the MR Egger analysis being significantly different from zero [21, 26] and by visual inspection of the funnel plot, where asymmetry is indicative of horizontal pleiotropy [19].
BMI and smoking are genetically correlated traits [16]. Among the IVs for smoking initiation, 5% (15/297) are overlapping or highly correlated (r2 ≥ 0.8) with the IVs for BMI. Hence, we performed a multivariable MR (MVMR) to estimate the independent effects of smoking initiation and BMI on MS risk. The IVW linear regression model with multiplicative random effects was used for multivariable MR.
Results
Smoking phenotypes not associated with MS risk
In an MR analysis, genetic predisposition to smoking initiation was not associated with MS risk using the random-effects inverse variance weighted (IVW) method [OR: 1.06, 95% confidence interval (CI) 0.92, 1.21, p = 0.42], and findings from the sensitivity tests were consistent (Fig. 1a). The scatter plot of the individual SNP estimates of smoking initiation and MS risk is shown in Supplementary Fig. 4a. The Cochran Q test and I2 statistic revealed moderate heterogeneity among the individual SNP effect estimates in the IVW analysis (Q = 443, p = 6.12 × 10−8; I2 = 33%). There was no evidence for directional pleiotropy from the MR Egger regression intercept [Egger intercept − 0.0008, 95% CI (− 0.012, 0.010), p = 0.89] and no asymmetry in the funnel plot (Fig. 3a).

Mendelian randomization (MR) estimates of smoking initiation and BMI with multiple sclerosis (MS) from the primary analysis (IVW) and sensitivity analyses Data are displayed as odds ratio (OR) and 95% confidence interval (CI) per SD increment in (a) genetically predicted smoking initiation (b) genetically predicted BMI (c) genetically predicted smoking initiation and BMI, respectively. IVW inverse-variance weighted method, MVMR multivariable Mendelian randomization. In the original GWASs, a SD increase in genetically predicted smoking initiation and BMI corresponds to a 10% increased risk of smoking and an increase of 4.7 kg/m2, respectively [16, 18]
Genetically predicted smoking heaviness, expressed as cigarettes per day, was similarly not associated with MS risk in the main analysis [OR: 1.03, 95% CI (0.77, 1.40), p = 0.83] nor in sensitivity tests (Fig. 2a, Supplementary Fig. 4b). The individual SNP effect estimates showed moderate heterogeneity (Q = 61, p = 0.007, I2 = 40%). MR-Egger regression analyses suggested that pleiotropy did not greatly influence the results of the MR analyses [Egger intercept − 0.0008, 95% CI (− 0.0143, 0.0126), p = 0.90], and neither did the funnel plot suggest directional pleiotropy (Fig. 3b).

Mendelian randomization (MR) estimates of cigarettes per day and the lifetime smoking index with multiple sclerosis (MS) from the primary analysis (IVW) and sensitivity analyses Data are displayed as odds ratio (OR) and 95% confidence interval (CI) per SD increment in (a) genetically predicted cigarettes per day (b) lifetime smoking index. IVW inverse-variance weighted method. In the original GWASs, a SD increase in genetically predicted cigarettes per day corresponds to three additional daily cigarettes [16]

Funnel plots for the effect of smoking phenotypes and BMI on risk of MS For each single-nucleotide polymorphism (SNP), the resulting Mendelian randomization (MR) estimate is plotted against the inverse of the standard error of the MR estimate. Symmetry noted in this plot provides evidence against the presence of directional horizontal pleiotropy. The vertical line represents the summary measure of the effect of (a) smoking initiation (b) cigarettes per day (c) lifetime smoking index (d) BMI on risk of multiple sclerosis (MS) on the log-odds ratio scale
Integrating different IVs into a measure reflecting lifetime smoking was not associated with MS risk in the main analysis [OR: 1.06, 95% CI (0.82, 1.38), p = 0.65] nor in sensitivity tests (Fig. 2b, Supplementary Fig. 4c). The individual SNP effect estimates showed only limited heterogeneity (Q = 138, p = 0.04, I2 = 21%). There was no evidence for directional pleiotropy from the MR Egger regression intercept [Egger intercept − 0.0042, 95% CI (− 0.0201, 0.0112), p = 0.60] (Fig. 3c).
MR provides the possibility to explore reverse causation, a concern in observational studies. In our data, genetically predicted MS risk was not associated with smoking initiation [OR = 1.00, 95% CI (0.99, 1.01), p = 0.96], or lifetime smoking [β = 0.002, 95% CI (− 0.003, 0.007), p = 0.51], with a possible trend seen only for smoking heaviness [β = 0.016, 95% CI (0.003, 0.028), p = 0.02].
Smoking phenotypes and BMI are correlated
As smoking and BMI are genetically correlated [16], we evaluated their relation in an MR approach. The IVW analysis of BMI and smoking initiation was highly significant and appeared bidirectional. Each genetically determined SD increase in BMI was associated with an increased likelihood of smoking initiation [OR: 1.21, 95% CI (1.17, 1.26), p = 8.64 × 10−21] (Supplementary Figs. 1a, 5a). Reversely, genetic predisposition to smoking was positively associated with BMI [β = 0.16, 95% CI (0.11, 0.21), p = 1.68 × 10−11] (Supplementary Figs. 1b, 5c). All sensitivity tests supported these findings (Supplementary Fig. 1), and there was no evidence for directional pleiotropy [Egger intercept − 0.0002, 95% CI (− 0.0020, 0.0016), p = 0.85 and Egger intercept − 0.0025, 95% CI (− 0.0061, 0.0011), p = 0.18] (Supplementary Fig. 2a, c). The individual SNP effect estimates did show substantial to considerable heterogeneity (Q = 1953, p = 5.29 × 10−162; I2 = 73% and Q = 4143, p < 1.0 × 10−200; I2 = 94%).
For smoking heaviness, MR effects were one-directional. Genetically predicted BMI was positively associated with the number of cigarettes per day [β = 0.35, 95% CI (0.31, 0.40), p = 3.60 × 10−49], consistent across all sensitivity analyses (Supplementary Figs. 3a, 5b). Genetically predicted smoking heaviness, on the other hand, was not correlated with BMI [β = 0.04, 95% CI [− 0.04, 0.12), p = 0.31] (Supplementary Figs. 3b, 5d). For the latter, there was, however, considerable heterogeneity (Q = 454, p = 1.88 × 10−78, I2 = 94%) and evidence for directional pleiotropy [Egger intercept 0.0065, 95% CI (0.0020, 0.0109), p = 0.0088] (Supplementary Fig. 2d), and sensitivity tests accounting for pleiotropy indicated even a possible inverse relation (Supplementary Fig. 3b).
BMI associated with MS risk
Previously performed MR analyses have already demonstrated a causal positive association between BMI and MS risk [11, 13]. In our univariable analysis employing 529 BMI-associated SNPs as IVs to the largest MS genetics dataset, each genetically determined SD increase in adult BMI was associated with a 31% increased risk for MS [OR: 1.31, 95% CI (1.15, 1.49), p = 5.63 × 10−5] (Fig. 1b). The scatter plot of the individual SNP estimates of BMI and MS risk is shown in Supplementary Fig. 4d. The Cochran Q test and I2 statistic revealed moderate heterogeneity among the individual SNP effect estimates in the IVW analysis (Q = 947, p = 3.02 × 10−26, I2 = 44%). There was no evidence of significant unbalanced horizontal pleiotropy [Egger intercept − 0.0016, 95% CI (− 0.0075, 0.0042), p = 0.58], and neither did the funnel plot suggest directional pleiotropy (Fig. 3d), and all sensitivity tests provided similar estimates with overlapping confidence intervals (Fig. 1b).
Multivariable MR: BMI but not smoking affects MS risk
A multivariable MR analysis allows to simultaneously adjust for the genetically predicted effects of two correlated exposures, smoking initiation and BMI, using the weighted regression-based framework. This multivariable analysis provided estimates for MS risk that were consistent with and nearly identical to the univariable results (Fig. 1c). BMI [OR: 1.30, 95% CI (1.15, 1.48), p = 2.78 × 10−5] but not smoking initiation [OR: 1.08, 95% CI (0.89, 1.32), p = 0.69] conferred an increased risk of MS.
Discussion
In this MR study investigating the role of smoking in MS risk, we did not find evidence for causal effects of genetically predicted smoking initiation, smoking heaviness or lifetime smoking index on MS susceptibility.
For low serum vitamin D and increased BMI, MR analyses agree with observational studies and support a causal association with increased MS susceptibility [11,12,13,14,15]. For smoking, in contrast, our findings do not confirm the observational literature. Observational studies have repeatedly found an increased risk for MS in smokers versus non-smokers as described by subsequent meta-analyses [27,28,29]. They also observed that smoking correlates with MS risk in a dose-dependent manner, with both duration and intensity of smoking associated with an increased MS risk, and this effect is regardless of age at exposure [30]. No association was seen for conversion from clinically isolated syndrome (CIS) to MS [28]. The most recent meta-analysis finds little evidence for publication bias but reviews other limitations, which mostly do not reflect the quality of the studies but are inherent to the observational study design. Limitations listed are the retrospective case–control design that is prone to recall bias in a large majority of studies (89%), smoking status based on self-reporting which tends to lead to under-reporting in most studies (94%), and a low response/participation rate and/or a noticeably different response/participation rate between cases and controls in up to 25% of studies [28]. Despite overall evidence in observational studies for a dose–response relationship between smoking and MS risk, our MR study could not confirm a causal relation between genetically predicted smoking status (ever versus never smoker) or smoking heaviness (cigarettes smoked per day) and risk for MS.
MR studies are based on their own assumptions, which need to be checked adequately. Pleiotropy, that is an effect of a genetic variant on the outcome that is independent of the exposure, is an important concern. We addressed this by quantifying the MR Egger intercept as the average effect of IVs, independent of the exposure, by inspecting funnel plots and by applying four sensitivity analyses that are more robust to pleiotropy than the IVW analysis that is standard in MR. For all analyses of exposures on MS risk, there is no evidence of pleiotropy and results are consistent across the main analysis and sensitivity tests. Co-incident geographical variation in genotypes and health traits may bias genetic studies, including MR [31]. The GWAS studies that we used as basis for IVs only included individuals of European ancestry, and residual stratification was further corrected at the meta-analytic level with study-specific genomic controls. Moreover, latitude and ancestry were not associated with smoking in observational studies [29]. The use of the same study cohort to identify the genetic variants associated with an exposure and to apply them as IVs for association with an outcome may bias the MR results towards the exposure-outcome association from observational studies. For the relationship between BMI and smoking, the inclusion of UK Biobank results in a large overlap in individuals between the GSCAN and GIANT studies. Hence, the exact causal direction and effect size for BMI-smoking should be regarded with caution but we mainly wish to emphasize the correlation between the two traits as supported by different methods. The overlap in samples between exposures and MS is much less prominent, although it cannot be excluded or quantified precisely. Importantly, we observe a discrepancy with rather than a bias towards observational studies for the exposure of smoking.
A possible reason for the discrepancy between observational and MR studies could be weak instrument bias, where the strength of the known genetic IVs associated with the exposure is insufficient. For smoking initiation, however, IVs explain 2.3% of variance in the trait, and this is similar to the strength of 97 IVs (2.7%) for BMI in earlier, positive MR studies in MS [11, 13]. Also, in contrast to MS, other MR studies including smoking initiation as exposure variable were in line with observational studies for traits such as the risk for ischemic stroke [32], coronary artery disease [33] and type 2 diabetes [34], but also for neurological diseases such as amyotrophic lateral sclerosis [35] and autoimmune diseases such as rheumatoid arthritis [36]. Hence, although weak instrument bias cannot be excluded completely, other explanations for the discrepancy between observational and MR studies specifically in MS should be considered.
Two MS risk factors suggested by observational studies, that is smoking initiation/heaviness and BMI, are genetically correlated traits (r = 0.12–0.19) as demonstrated by LD-score regression [16]. Using the most recent IVs and summary statistics for BMI and smoking phenotypes, we replicate earlier evidence for a bidirectional relationship between BMI and smoking initiation [37], and demonstrate a unidirectional relationship for BMI on smoking heaviness. Both in a univariable MR and a multivariable MR, each SD increase in BMI, corresponding to roughly 5 kg/m2 units, conferred a 30% increase in MS risk. This effect and its size are in line with earlier MR studies starting from 97 instead of the currently known 656 genetic variants as IVs [11, 13]. Our MR study indicates that smoking does not have a direct effect on MS risk nor explains part of the association between BMI and MS risk. This is in line with the absence of an overall genetic correlation between smoking and MS using LD-score regression [16]. The observational studies associating smoking with MS susceptibility show heterogeneity in adjusting for established risk factors and other sociodemographic factors [27,28,29]. The vast majority of studies (86%) accounted for age and/or gender but only 6% of studies controlled for potential confounding by BMI [28], which we have here demonstrated to be highly correlated with both smoking and MS risk. Any residual confounding by this and other factors should hence be excluded.
Although MR provides an elegant approach to the study of causal relationships for exposures, it has limitations in capturing specific aspects of such relationships which may assist in understanding the discrepancies between observational studies and MR. First, the risk of smoking on MS in observational studies seems time-dependent, as it is higher for current versus past smoking [38], and abates 5 years after smoking cessation [39]. MR, on the other hand, typically captures the genetic predisposition to an exposure during one’s life-time, such as ever having smoked regularly. MR approaches that better capture time-varying exposures are now being developed [40]. The lifetime smoking score applied here is an example of such an approach. It captures well the known time- and dose-varying causal association of smoking with lung cancer (OR > 4) and cardiovascular diseases, and is correlated with known smoking-induced demethylation at the AHRR locus [17, 33]. Importantly, all IVs for smoking initiation, heaviness and lifetime smoking provide essentially identical results for MS with no evidence for an association. Secondly, the strength of the BMI IVs may differ by smoking status, as a 20% increase in the effect of the BMI genetic score on the actually observed BMI was seen in current smokers compared to former or never smokers [37]. Novel MR approaches such as factorial MR allow to model interactions between BMI and smoking phenotypes or an effect modification of smoking on BMI [41]. These studies necessitate the availability of individual-level genetic data, which is typically limited for large-scale studies.
Striking changes in the demographic pattern of multiple sclerosis (MS), with increasing prevalence and incidence over time, strongly indicate an influence of modifiable exposures on the disease [42]. Modifiable exposures lend themselves well to intervention, but it is important to pinpoint which of the many environmental, lifestyle, and sociodemographic changes that have occurred over the past decades such as higher smoking and obesity rates are responsible. MR is an elegant tool to leverage human genetics to inform prevention strategies in MS [43]. Despite observational studies repeatedly reporting an association between smoking and MS, our MR analyses on smoking phenotypes and MS risk could not confirm a causal relationship. The reasons for the discrepancy between observational studies and our MR study require further investigation, along the lines discussed. In addition, it remains to be determined whether interventions such as smoking cessation after disease onset, when the patient comes to the attention of the neurologist, do have an effect. Factors shaping the disease course after onset appear to be different from factors triggering disease [44, 45], implying that dedicated observational and MR studies are required to investigate factors underlying disease evolution [43, 46].
Data availability
This study makes use of summary statistics of genome-wide association studies that are publicly available (for smoking and BMI) or that have been shared by the International Multiple Sclerosis Genetics Consortium (for MS), as described in detail under the heading Genetic Datasets in the Methods section.
References
Compston A, Coles A (2008) Multiple sclerosis. Lancet 372(9648):1502–1517. https://doi.org/10.1016/S0140-6736(08)61620-7
International Multiple Sclerosis Genetics Consortium (2018) Low-frequency and rare-coding variation contributes to multiple sclerosis risk. Cell 175(6):1679–1687.e7. https://doi.org/10.1016/j.cell.2018.09.049
International Multiple Sclerosis Genetics Consortium (2019) Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365(6460):eaav7188. https://doi.org/10.1126/science.aav7188
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium, International IBD Genetics Consortium (2013) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45(11):1353–1360. https://doi.org/10.1038/ng.2770
International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476(7359):214–219. https://doi.org/10.1038/nature10251
Moutsianas L, Jostins L, Beecham AH, Dilthey AT, Xifara DK, Ban M, Shah TS, Patsopoulos NA, Alfredsson L, Anderson CA, Attfield KE, Baranzini SE, Barrett J, Binder TMC, Booth D, Buck D, Celius EG, Cotsapas C, D'Alfonso S, Dendrou CA, Donnelly P, Dubois B, Fontaine B, Fugger L, Goris A, Gourraud PA, Graetz C, Hemmer B, Hillert J, International IBDGC, Kockum I, Leslie S, Lill CM, Martinelli-Boneschi F, Oksenberg JR, Olsson T, Oturai A, Saarela J, Sondergaard HB, Spurkland A, Taylor B, Winkelmann J, Zipp F, Haines JL, Pericak-Vance MA, Spencer CCA, Stewart G, Hafler DA, Ivinson AJ, Harbo HF, Hauser SL, De Jager PL, Compston A, McCauley JL, Sawcer S, McVean G (2015) Class II HLA interactions modulate genetic risk for multiple sclerosis. Nat Genet 47(10):1107–1113. https://doi.org/10.1038/ng.3395
Olsson T, Barcellos LF, Alfredsson L (2017) Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 13(1):25–36. https://doi.org/10.1038/nrneurol.2016.187
Smith GD, Lawlor DA, Harbord R, Timpson N, Day I, Ebrahim S (2007) Clustered environments and randomized genes: a fundamental distinction between conventional and genetic epidemiology. PLoS Med 4(12):e352. https://doi.org/10.1371/journal.pmed.0040352
Smith GD, Phillips AN (1992) Confounding in epidemiological studies: why “independent” effects may not be all they seem. BMJ 305(6856):757–759. https://doi.org/10.1136/bmj.305.6856.757
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G (2008) Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med 27(8):1133–1163. https://doi.org/10.1002/sim.3034
Gianfrancesco MA, Stridh P, Rhead B, Shao X, Xu E, Graves JS, Chitnis T, Waldman A, Lotze T, Schreiner T, Belman A, Greenberg B, Weinstock-Guttman B, Aaen G, Tillema JM, Hart J, Caillier S, Ness J, Harris Y, Rubin J, Candee M, Krupp L, Gorman M, Benson L, Rodriguez M, Mar S, Kahn I, Rose J, Roalstad S, Casper TC, Shen L, Quach H, Quach D, Hillert J, Baarnhielm M, Hedstrom A, Olsson T, Kockum I, Alfredsson L, Metayer C, Schaefer C, Barcellos LF, Waubant E, Network of Pediatric Multiple Sclerosis C (2017) Evidence for a causal relationship between low vitamin D, high BMI, and pediatric-onset MS. Neurology 88(17):1623–1629. https://doi.org/10.1212/WNL.0000000000003849
Mokry LE, Ross S, Ahmad OS, Forgetta V, Smith GD, Goltzman D, Leong A, Greenwood CM, Thanassoulis G, Richards JB (2015) Vitamin D and risk of multiple sclerosis: a Mendelian randomization study. PLoS Med 12(8):e1001866. https://doi.org/10.1371/journal.pmed.1001866
Mokry LE, Ross S, Timpson NJ, Sawcer S, Davey Smith G, Richards JB (2016) Obesity and multiple sclerosis: a Mendelian randomization study. PLoS Med 13(6):e1002053. https://doi.org/10.1371/journal.pmed.1002053
Rhead B, Baarnhielm M, Gianfrancesco M, Mok A, Shao X, Quach H, Shen L, Schaefer C, Link J, Gyllenberg A, Hedstrom AK, Olsson T, Hillert J, Kockum I, Glymour MM, Alfredsson L, Barcellos LF (2016) Mendelian randomization shows a causal effect of low vitamin D on multiple sclerosis risk. Neurol Genet 2(5):e97. https://doi.org/10.1212/NXG.0000000000000097
Harroud A, Morris JA, Forgetta V, Mitchell R, Smith GD, Sawcer SJ, Richards JB (2019) Effect of age at puberty on risk of multiple sclerosis: a Mendelian randomization study. Neurology 92(16):e1803–e1810. https://doi.org/10.1212/WNL.0000000000007325
Liu M, Jiang Y, Wedow R, Li Y, Brazel DM, Chen F, Datta G, Davila-Velderrain J, McGuire D, Tian C, Zhan X, andMe Research T, Psychiatry HA-I, Choquet H, Docherty AR, Faul JD, Foerster JR, Fritsche LG, Gabrielsen ME, Gordon SD, Haessler J, Hottenga JJ, Huang H, Jang SK, Jansen PR, Ling Y, Magi R, Matoba N, McMahon G, Mulas A, Orru V, Palviainen T, Pandit A, Reginsson GW, Skogholt AH, Smith JA, Taylor AE, Turman C, Willemsen G, Young H, Young KA, Zajac GJM, Zhao W, Zhou W, Bjornsdottir G, Boardman JD, Boehnke M, Boomsma DI, Chen C, Cucca F, Davies GE, Eaton CB, Ehringer MA, Esko T, Fiorillo E, Gillespie NA, Gudbjartsson DF, Haller T, Harris KM, Heath AC, Hewitt JK, Hickie IB, Hokanson JE, Hopfer CJ, Hunter DJ, Iacono WG, Johnson EO, Kamatani Y, Kardia SLR, Keller MC, Kellis M, Kooperberg C, Kraft P, Krauter KS, Laakso M, Lind PA, Loukola A, Lutz SM, Madden PAF, Martin NG, McGue M, McQueen MB, Medland SE, Metspalu A, Mohlke KL, Nielsen JB, Okada Y, Peters U, Polderman TJC, Posthuma D, Reiner AP, Rice JP, Rimm E, Rose RJ, Runarsdottir V, Stallings MC, Stancakova A, Stefansson H, Thai KK, Tindle HA, Tyrfingsson T, Wall TL, Weir DR, Weisner C, Whitfield JB, Winsvold BS, Yin J, Zuccolo L, Bierut LJ, Hveem K, Lee JJ, Munafo MR, Saccone NL, Willer CJ, Cornelis MC, David SP, Hinds DA, Jorgenson E, Kaprio J, Stitzel JA, Stefansson K, Thorgeirsson TE, Abecasis G, Liu DJ, Vrieze S (2019) Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 51(2):237–244. https://doi.org/10.1038/s41588-018-0307-5
Wootton RE, Richmond RC, Stuijfzand BG, Lawn RB, Sallis HM, Taylor GMJ, Hemani G, Jones HJ, Zammit S, Davey Smith G, Munafo MR (2019) Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med. https://doi.org/10.1017/S0033291719002678
Yengo L, Sidorenko J, Kemper KE, Zheng Z, Wood AR, Weedon MN, Frayling TM, Hirschhorn J, Yang J, Visscher PM, Consortium G (2018) Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Hum Mol Genet 27(20):3641–3649. https://doi.org/10.1093/hmg/ddy271
Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, Tan VY, Yarmolinsky J, Shihab HA, Timpson NJ, Evans DM, Relton C, Martin RM, Davey Smith G, Gaunt TR, Haycock PC (2018) The MR-Base platform supports systematic causal inference across the human phenome. Elife 7:e34408. https://doi.org/10.7554/eLife.34408
Burgess S, Butterworth A, Thompson SG (2013) Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 37(7):658–665. https://doi.org/10.1002/gepi.21758
Bowden J, Davey Smith G, Burgess S (2015) Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 44(2):512–525. https://doi.org/10.1093/ije/dyv080
Bowden J, Davey Smith G, Haycock PC, Burgess S (2016) Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol 40(4):304–314. https://doi.org/10.1002/gepi.21965
Hartwig FP, Davey Smith G, Bowden J (2017) Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol 46(6):1985–1998. https://doi.org/10.1093/ije/dyx102
Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR (2016) Assessing the suitability of summary data for two-sample Mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int J Epidemiol 45(6):1961–1974. https://doi.org/10.1093/ije/dyw220
Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G (2016) Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr 103(4):965–978. https://doi.org/10.3945/ajcn.115.118216
Burgess S, Thompson SG (2017) Interpreting findings from Mendelian randomization using the MR-Egger method. Eur J Epidemiol 32(5):377–389. https://doi.org/10.1007/s10654-017-0255-x
Belbasis L, Bellou V, Evangelou E, Ioannidis JP, Tzoulaki I (2015) Environmental risk factors and multiple sclerosis: an umbrella review of systematic reviews and meta-analyses. Lancet Neurol 14(3):263–273. https://doi.org/10.1016/S1474-4422(14)70267-4
Degelman ML, Herman KM (2017) Smoking and multiple sclerosis: a systematic review and meta-analysis using the Bradford Hill criteria for causation. Mult Scler Relat Disord 17:207–216. https://doi.org/10.1016/j.msard.2017.07.020
Handel AE, Williamson AJ, Disanto G, Dobson R, Giovannoni G, Ramagopalan SV (2011) Smoking and multiple sclerosis: an updated meta-analysis. PLoS ONE 6(1):e16149. https://doi.org/10.1371/journal.pone.0016149
Hedstrom AK, Hillert J, Olsson T, Alfredsson L (2013) Smoking and multiple sclerosis susceptibility. Eur J Epidemiol 28(11):867–874. https://doi.org/10.1007/s10654-013-9853-4
Haworth S, Mitchell R, Corbin L, Wade KH, Dudding T, Budu-Aggrey A, Carslake D, Hemani G, Paternoster L, Smith GD, Davies N, Lawson DJ, Nicholas JT (2019) Apparent latent structure within the UK Biobank sample has implications for epidemiological analysis. Nat Commun 10(1):333. https://doi.org/10.1038/s41467-018-08219-1
Larsson SC, Burgess S, Michaelsson K (2019) Smoking and stroke: a Mendelian randomization study. Ann Neurol 86(3):468–471. https://doi.org/10.1002/ana.25534
Larsson SC, Mason AM, Back M, Klarin D, Damrauer SM, Million Veteran P, Michaelsson K, Burgess S (2020) Genetic predisposition to smoking in relation to 14 cardiovascular diseases. Eur Heart J. https://doi.org/10.1093/eurheartj/ehaa193
Yuan S, Larsson SC (2019) A causal relationship between cigarette smoking and type 2 diabetes mellitus: a Mendelian randomization study. Sci Rep 9(1):19342. https://doi.org/10.1038/s41598-019-56014-9
Zhan Y, Fang F (2019) Smoking and amyotrophic lateral sclerosis: a Mendelian randomization study. Ann Neurol 85(4):482–484. https://doi.org/10.1002/ana.25443
Qian Y, Zhang L, Wu DJH, Xie Z, Wen C, Mao Y (2020) Genetic predisposition to smoking is associated with risk of rheumatoid arthritis: a Mendelian randomization study. Arthritis Res Ther 22(1):44. https://doi.org/10.1186/s13075-020-2134-1
Taylor AE, Richmond RC, Palviainen T, Loukola A, Wootton RE, Kaprio J, Relton CL, Davey Smith G, Munafo MR (2019) The effect of body mass index on smoking behaviour and nicotine metabolism: a Mendelian randomization study. Hum Mol Genet 28(8):1322–1330. https://doi.org/10.1093/hmg/ddy434
Hernan MA, Jick SS, Logroscino G, Olek MJ, Ascherio A, Jick H (2005) Cigarette smoking and the progression of multiple sclerosis. Brain 128(Pt 6):1461–1465. https://doi.org/10.1093/brain/awh471
Hedstrom AK, Baarnhielm M, Olsson T, Alfredsson L (2009) Tobacco smoking, but not Swedish snuff use, increases the risk of multiple sclerosis. Neurology 73(9):696–701. https://doi.org/10.1212/WNL.0b013e3181b59c40
Cao Y, Rajan SS, Wei P (2016) Mendelian randomization analysis of a time-varying exposure for binary disease outcomes using functional data analysis methods. Genet Epidemiol 40(8):744–755. https://doi.org/10.1002/gepi.22013
Rees JMB, Foley CN, Burgess S (2019) Factorial Mendelian randomization: using genetic variants to assess interactions. Int J Epidemiol. https://doi.org/10.1093/ije/dyz161
Koch-Henriksen N, Sorensen PS (2010) The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol 9(5):520–532. https://doi.org/10.1016/S1474-4422(10)70064-8
Goris A, Dubois B (2019) Leveraging human genetics to inform intervention strategies for multiple sclerosis. Neurology 92(16):735–736. https://doi.org/10.1212/WNL.0000000000007298
Hilven K, Patsopoulos NA, Dubois B, Goris A (2015) Burden of risk variants correlates with phenotype of multiple sclerosis. Mult Scler 21(13):1670–1680. https://doi.org/10.1177/1352458514568174
Hilven K, Vandebergh M, Smets I, Mallants K, Goris A, Dubois B (2018) Genetic basis for relapse rate in multiple sclerosis: association with LRP2 genetic variation. Mult Scler 24(13):1773–1775. https://doi.org/10.1177/1352458517749894
Paternoster L, Tilling K, Davey Smith G (2017) Genetic epidemiology and Mendelian randomization for informing disease therapeutics: conceptual and methodological challenges. PLoS Genet 13(10):e1006944. https://doi.org/10.1371/journal.pgen.1006944
Acknowledgements
We gratefully thank all the studies and databases that provided publicly available GWAS summary data (GSCAN and GIANT consortia). We also thank the International Multiple Sclerosis Genetics Consortium for sharing summary statistics. MV is a PhD Fellow (11ZZZ19N) of the Research Foundation-Flanders (FWO-Vlaanderen). This study was supported by the Research Fund KU Leuven (C24/16/045), and the Research Foundation-Flanders (FWO G.07334.15). The computational resources and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by the Research Foundation-Flanders (FWO-Vlaanderen) and the Flemish Government (department EWI).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical statement
This study makes use of summary statistics that are publicly available or have been shared for analysis within the scope of the original genome-wide association studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vandebergh, M., Goris, A. Smoking and multiple sclerosis risk: a Mendelian randomization study. J Neurol 267, 3083–3091 (2020). https://doi.org/10.1007/s00415-020-09980-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-09980-4