
Abstract
Neurometabolic disorders are often inherited and complex disorders that result from abnormalities of enzymes important for development and function of the nervous system. Recently, biallelic mutations in NAXE (APOA1BP) were found in patients with an infantile, lethal, neurometabolic disease. Here, exome sequencing was performed in two affected sisters and their healthy parents. The best candidate, NAXE, was tested for replication in exome sequencing data from 4351 patients with neurodevelopmental disorders. Quantitative RT-PCR, western blot and form factor analysis were performed to assess NAXE expression, protein levels and to analyze mitochondrial morphology in fibroblasts. Vitamin B3 was administered to one patient. Compound heterozygous missense (c.757G>A: p.Gly253Ser) and splicing (c.665-1G>A) variants in NAXE were identified in both affected sisters. In contrast to the previously reported patients with biallelic NAXE variants, our patients showed a milder phenotype with disease onset in early adulthood with psychosis, cognitive impairment, seizures, cerebellar ataxia and spasticity. The symptoms fluctuated. Additional screening of NAXE identified three novel homozygous missense variants (p.Lys245Gln, p.Asp218Asn, p.Ile214Val) in three patients with overlapping phenotype (fluctuating disease course, respiratory insufficiency, movement disorder). Lastly, patients with the c.665-1G>A splicing variant showed a significant reduction of NAXE expression compared to control fibroblasts and undetectable NAXE protein levels compared to control fibroblasts. Based on the metabolic pathway, vitamin B3 and coenzyme Q treatment was introduced in one patient in addition to antiepileptic treatment. This combination and avoidance of triggers was associated with continuous motor and cognitive improvement. The NAXE variants identified in this study suggest a loss-of-function mechanism leading to an insufficient NAD(P)HX repair system. Importantly, symptoms of patients with NAXE variants may improve with vitamin B3/coenzyme Q administration.



Similar content being viewed by others


References
Spiegel R, Shaag A, Shalev S (2016) Homozygous mutation in the APOA1BP is associated with a lethal infantile leukoencephalopathy. Neurogenetics 17:187–190
Van Bergen NJ, Guo Y, Rankin J, Paczia N, Becker-Kettern J, Kremer LS, Pyle A, Conrotte JF, Ellaway C, Procopis P, Prelog K, Homfray T, Baptista J, Baple E, Wakeling M, Massey S, Kay DP, Shukla A, Girisha KM, Lewis LES, Santra S, Power R, Daubeney P, Montoya J, Ruiz-Pesini E, Kovacs-Nagy R, Pritsch M, Ahting U, Thorburn DR, Prokisch H, Taylor RW, Christodoulou J, Linster CL, Ellard S, Hakonarson H (2019) NAD(P)HX dehydratase (NAXD) deficiency: a novel neurodegenerative disorder exacerbated by febrile illnesses. Brain 142(1):50–58. https://doi.org/10.1093/brain/awy3105255623
Kremer L, Danhauser K, Herebian D (2016) NAXE mutations disrupt the cellular NAD(P)HX. Am J Hum Genet 99:894–902
Pronicka E, Piekutowska-Abramczuk D, Ciara E, Trubicka J, Rokicki D, Karkucinska-Wieckowska A, Pajdowska M, Jurkiewicz E, Halat P, Kosinska J, Pollak A, Rydzanicz M, Stawinski P, Pronicki M, Krajewska-Walasek M, Ploski R (2016) New perspective in diagnostics of mitochondrial disorders: two years' experience with whole-exome sequencing at a national paediatric centre. J Transl Med 14(1):174. https://doi.org/10.1186/s12967-016-0930-910.1186/s12967-016-0930-9
Elhassan YS, Philp AA, Lavery GG (2017) Targeting NAD+ in metabolic disease: new insights into an old molecule. J Endocr Soc 1(7):816–835. https://doi.org/10.1210/js.2017-00092JS_201700092
Ng YS, Turnbull DM (2016) Mitochondrial disease: genetics and management. J Neurol 263(1):179–191. https://doi.org/10.1007/s00415-015-7884-310.1007/s00415-015-7884-3
Schondorf DC, Ivanyuk D, Baden P, Sanchez-Martinez A, De Cicco S, Yu C, Giunta I, Schwarz LK, Di Napoli G, Panagiotakopoulou V, Nestel S, Keatinge M, Pruszak J, Bandmann O, Heimrich B, Gasser T, Whitworth AJ, Deleidi M (2018) The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and Fly models of Parkinson's disease. Cell Rep 23(10):2976–2988. https://doi.org/10.1016/j.celrep.2018.05.009
Groza T, Kohler S, Moldenhauer D, Vasilevsky N, Baynam G, Zemojtel T, Schriml LM, Kibbe WA, Schofield PN, Beck T, Vasant D, Brookes AJ, Zankl A, Washington NL, Mungall CJ, Lewis SE, Haendel MA, Parkinson H, Robinson PN (2015) The human phenotype ontology: semantic unification of common and rare disease. Am J Hum Genet 97(1):111–124. https://doi.org/10.1016/j.ajhg.2015.05.020S0002-9297(15)00234-7
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14):1754–1760. https://doi.org/10.1093/bioinformatics/btp324btp324
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, Weiss ME, Koster J, Marais A, Paknia O, Schroder R, Garcia-Aznar JM, Werber M, Brandau O, Calvo Del Castillo M, Baldi C, Wessel K, Kishore S, Nahavandi N, Eyaid W, Al Rifai MT, Al-Rumayyan A, Al-Twaijri W, Alothaim A, Alhashem A, Al-Sannaa N, Al-Balwi M, Alfadhel M, Rolfs A, Abou Jamra R (2017) Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet 25(2):176–182. https://doi.org/10.1038/ejhg.2016.146ejhg2016146
Trinh J, Vilarino-Guell C, Donald A, Shah B, Yu I, Szu-Tu C, Aasly JO, Wu RM, Hentati F, Rajput AH, Rajput A, Farrer MJ (2013) STX6 rs1411478 is not associated with increased risk of Parkinson's disease. Parkinsonism Relat Disord 19(5):563–565. https://doi.org/10.1016/j.parkreldis.2013.01.019S1353-8020(13)00054-0
Grunewald A, Voges L, Rakovic A, Kasten M, Vandebona H, Hemmelmann C, Lohmann K, Orolicki S, Ramirez A, Schapira AH, Pramstaller PP, Sue CM, Klein C (2010) Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE 5(9):e12962. https://doi.org/10.1371/journal.pone.0012962e12962
Gasperi V, Sibilano M, Savini I, Catani MV (2019) Niacin in the central nervous system: an update of biological aspects and clinical applications. Int J Mol Sci 20(4):974. https://doi.org/10.3390/ijms20040974ijms20040974
Nikiforov A, Kulikova V, Ziegler M (2015) The human NAD metabolome: functions, metabolism and compartmentalization. Crit Rev Biochem Mol Biol 50(4):284–297. https://doi.org/10.3109/10409238.2015.1028612
Becker-Kettern J, Paczia N, Conrotte JF, Zhu C, Fiehn O, Jung PP, Steinmetz LM, Linster CL (2018) NAD(P)HX repair deficiency causes central metabolic perturbations in yeast and human cells. FEBS J 285(18):3376–3401. https://doi.org/10.1111/febs.14631
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, Walkiewicz M, Bi W, Xiao R, Ding Y, Xia F, Beaudet AL, Muzny DM, Gibbs RA, Boerwinkle E, Eng CM, Sutton VR, Shaw CA, Plon SE, Yang Y, Lupski JR (2017) Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med 376(1):21–31. https://doi.org/10.1056/NEJMoa1516767
Trinh J, Kandaswamy KK, Werber M, Weiss MER, Oprea G, Kishore S, Lohmann K, Rolfs A (2019) Novel pathogenic variants and multiple molecular diagnoses in neurodevelopmental disorders. J Neurodev Disord 11(1):11. https://doi.org/10.1186/s11689-019-9270-4
Pique-Duran E, Perez-Cejudo JA, Cameselle D, Palacios-Llopis S, Garcia-Vazquez O (2012) Pellagra: a clinical, histopathological, and epidemiological study of 7 cases. Actas Dermosifiliogr 103(1):51–58. https://doi.org/10.1016/j.adengl.2011.05.003
Salih MA, Bender DA, McCreanor GM (1985) Lethal familial pellagra-like skin lesion associated with neurologic and developmental impairment and the development of cataracts. Pediatrics 76(5):787–793
Baron DN, Dent CE, Harris H, Hart EW, Jepson JB (1956) Hereditary pellagra-like skin rash with temporary cerebellar ataxia, constant renal amino-aciduria, and other bizarre biochemical features. Lancet 271(6940):421–428. https://doi.org/10.1016/s0140-6736(56)91914-6
Acknowledgements
This research was supported by the Foundation of the University Medical Center Schleswig Holstein “Gutes Tun!” (A.M.). Funding has been obtained from the Hermann and Lilly Schilling Foundation, the Alexander von Humboldt Foundation, Canadian Institutes of Health Research, and the Joachim Herz Stiftung.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical standards
The authors hereby declare that the research documented in the submitted manuscript has been carried out in accordance with the above stated ethical standards.
Additional information
Joanne Trinh, Sophie Imhoff and Marija Dulovic-Mahlow are joint first authors and equally contributed. Katja Lohmann and Norbert Brüggemann are shared last authors and equally contributed.
Electronic supplementary material
Below is the link to the electronic supplementary material.
415_2019_9640_MOESM2_ESM.mp4
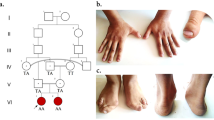
The clinical picture was initially characterized by severe generalized spasticity, epileptic seizures and intermittent fever of unknown source as shown on two pictures from October 2015 when the patient underwent 72-hour EEG. In the second part of the video (recorded in February 2018), the tracheostomy was removed and the patient was able to eat in addition to PEG infusions. Attention, memory and other cognitive functions were improved and the patient was able to speak, to follow instructions and to have conversations. The video shows perioral dyskinesias, dystonic laterocollis, jerky head movements probably related to dystonia, and generalized spasticity impairing fine finger movements. She had divergent strabism, bilateral adductor paresis, vertical gaze-evoked nystagmus when looking upward and horizontal gaze-evoked nystagmus. There was also a positive relative afferent pupil defect of the right eye and reduced visual acuity (right 0.1, left 0.2) (not shown) (MP4 3205 kb)
The video demonstrates patient II.4 at the age of 21 years with reduced concentration and memory problems, impairment of executive function including an abnormal Luria test, finger myoclonus when holding out the arms in front of her, dysmetric and ataxic finger-to-finger tests, positive rebound phenomenon, wide-based ataxic gait and inability to perform the tandem gait independently (MOV 16491 kb)
Over the disease course, patient II.4 developed psychotic symptoms including anxiety, persecutory delusions and bizarre behaviour. She additionally presented with mild gait ataxia (MOV 21358 kb)
Rights and permissions
About this article

Cite this article
Trinh, J., Imhoff, S., Dulovic-Mahlow, M. et al. Novel NAXE variants as a cause for neurometabolic disorder: implications for treatment. J Neurol 267, 770–782 (2020). https://doi.org/10.1007/s00415-019-09640-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-019-09640-2