
Abstract
This paper summarises the five presentations at the First International Workshop on Systems Radiation Biology that were concerned with mechanistic models for carcinogenesis. The mathematical description of various hypotheses about the carcinogenic process, and its comparison with available data is an example of systems biology. It promises better understanding of effects at the whole body level based on properties of cells and signalling mechanisms between them. Of these five presentations, three dealt with multistage carcinogenesis within the framework of stochastic multistage clonal expansion models, another presented a deterministic multistage model incorporating chromosomal aberrations and neoplastic transformation, and the last presented a model of DNA double-strand break repair pathways for second breast cancers following radiation therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Five presentations at the First International Workshop on Systems Radiation Biology held at GSF on 14–16 February 2007 were concerned with mechanistic models for radiation carcinogenesis. These presentations are summarized in this manuscript. Of these five presentations, three (given by Heidenreich, Little, and Moolgavkar) dealt with the theme of multistage carcinogenesis within the framework of stochastic multistage clonal expansion models. The presentation by Schöllnberger discussed a deterministic multistage model incorporating chromosomal aberrations and neoplastic transformation. Finally, Thomas presented a model of DNA double-strand break (DSB) repair pathways for second breast cancers following radiation therapy.
Stochastic multistage clonal expansion models for carcinogenesis
Stochastic multistage clonal expansion models are extensions of the two-stage clonal expansion (TSCE) model of carcinogenesis generally attributed to Moolgavkar and Venzon [1] and Moolgavkar and Knudson [2] and sometimes referred to as the MVK model. In this, its simplest, incarnation, the model assumes that initiated cells, the hallmark of which is a slight growth advantage over normal neighbours, arise from stem cells according to a non-homogeneous Poisson process. Once created, initiated cells undergo a stochastic birth–death–mutation process with the birth and death process giving rise to clones of initiated cells and the mutation process leading to the conversion of an initiated cell into a fully malignant cell. Multistage extensions of this basic model have been proposed by various investigators. A couple of these extensions are discussed later in this report. The introduction of one or more stages with stochastic clonal expansion in multistage cancer models has profound implications for inferences drawn from the models.
Clonal expansion and carcinogenesis (Moolgavkar)
Clonally expanding cell populations are important in carcinogenesis and modelling their dynamics stochastically rather than deterministically has interesting consequences. We give some examples here.
Gestational mutations and cancer
Frank and Nowak [3] suggested that mutations at critical loci during the exponential phase of growth during gestation could have important implications not only for childhood, but also for adult cancers. Mutations occurring early in gestation would give rise to jackpots (i.e. clones) of cells with mutations at critical loci increasing the probability of cancer in an individual carrying these mutations. The quantitative consequences of gestational mutations were investigated by Meza et al. [4]. They used the Luria–Delbruck model for jackpots in tandem with the Luebeck–Moolgavkar model [5] for colon cancer and concluded that, depending on the rates of gestational mutations, between 2 and 20% of colon cancers in the surveillance epidemiology and end results (SEER) registry in the year 2000 arose from stem cells that had sustained one or more critical mutations during gestation. Meza et al. [4] also investigated the cancer risks associated with radiation exposure during gestation and concluded that radiation during the last trimester of pregnancy carried the highest risk, consistent with the epidemiological data.
Clonal expansion and cancer incidence rates
Stochastic clonal expansion of intermediate cells on the pathway to cancer also has important implications for the incidence of cancer in human populations. Some of these flow from the fact that stochasticity of carcinogenesis introduces heterogeneity of cancer risk. Even if one starts off with a completely homogeneous population, with the passage of time, the population becomes increasingly heterogeneous with respect to cancer risk as a consequence of the stochasticity of the carcinogenic process. Much of this heterogeneity arises from the fact that clonal expansion of intermediate cells is modelled by stochastic birth and death processes, leading to distributions of cells in intermediate stages on the pathway to carcinogenesis. Unfortunate individuals in the upper tails of these distributions tend to develop cancer earlier than their more fortunate brethren who lie in the lower tails. An immediate consequence of heterogeneity is the widely observed departure of cancer incidence rates below the log–log incidence predicted by deterministic versions of many multistage models.
Heterogeneity of intermediate cell populations also plays an important role in the temporal evolution of cancer risk in a population exposed to a carcinogen, such as ionising radiation or cigarette smoke. When smokers quit, the incidence rates of lung cancer fall below those among continuing smokers and gradually over a period of 15–20 years approach those among life-long non-smokers. While this decline to background rates has often been attributed to repair of smoke-induced damage of lung tissue, it can be explained by the heterogeneity in the population of intermediate cells among smokers. Within the framework of the TSCE model, the main effect of tobacco smoke is to stimulate clonal expansion of initiated cells (promotion). This leads to a distribution of initiated cells that is shifted to the right, i.e., to larger numbers, but overlaps the distribution of initiated cells among non-smokers. When smokers quit, individuals in the upper tail of the distribution develop lung cancer at a higher rate than the non-smokers. Eventually, however, the high-risk individuals are depleted, and the risk among ex-smokers is now determined by individuals in the lower tail of the distribution, which overlaps the distribution among non-smokers.
The so-called inverse exposure-rate effect for high-LET radiation has also been explained in terms of a promotional effect of high-LET radiation in carcinogenesis, first postulated as a result of an analysis of the Colorado Plateau miners’ data [6] and subsequently confirmed by analysis of experimental data [7]. More recently, it has been suggested that low-LET radiation may also have some promoting activity [8]. The promoting action of radiation is discussed in more detail (section of Heidenreich) later in this paper. With an inverse exposure-rate effect, protraction of the time over which a given exposure is administered has an interesting consequence, namely, one observes crossing incidence functions, a phenomenon that is difficult to detect and address using conventional epidemiological methods. Methods of analyses based on ideas of multistage carcinogenesis are much better suited to addressing issues of this type.
Analyses of pre-malignant lesions
The stochastic treatment of clonal expansion allows also for the development of expressions for the distributions of numbers and sizes of pre-malignant lesions, such as altered foci arising in rodent hepatocarcinogenesis experiments, and adenomatous polyps in the human colon [9, 10], which can then be used for data analyses [11, 12]. Jeon et al. [10] developed expressions for the distribution of the number and sizes of adenomatous polyps based on the Luebeck–Moolgavkar model using a combination of simulation and mathematical techniques. They used these expressions to devise optimal screening schedules for colon cancer [10].
Epidemiological data and models of radiation carcinogenesis (Heidenreich)
Unfortunately, cancer biologists have not yet worked out the details of how cancer evolves in an organ [13]. Therefore, fairly crude simplifications of these processes have to be used when trying to connect the most relevant biological processes with the observed risk patterns due to ageing or radiation. When extracting information about the cancer process, or the action of radiation from epidemiological or experimental cancer incidence data, it is crucial:
-
To understand what quantities are identifiable in the models. Ideally, the signals to look for are selected before any analysis is done
-
To choose the best available data (power, quality) for the addressed question
-
Alternatively, at least to verify that the tested models make different predictions for the chosen data.
This type of research requires cooperation of epidemiologists, biologists and modellers (usually physicists or mathematicians). Depending on the definition, it may thus be considered as systems radiation biology.
Here solid cancers are considered, and the action of radiation is studied. To be specific, the following model-components are used:
-
Initiation: A Poisson process resulting in cells with a growth advantage
-
Promotion: Slow clonal expansion of initiated cells
-
Conversion: A rate-limiting step from initiated cells to malignant ones
-
Lag time: Deterministic growth of one malignant cell to an observed tumour.
Each of these components could be radiation-dependent and could consist of sub-steps.
Radiation action on initiation
According to a conventional view, radiation only influences the initiating event of the multi-step carcinogenic process [14]. With this assumption, 50 mGy gamma-rays were estimated to be about as efficient in initiation as 1 year of normal life [15, 16]. If part of the effects in the atomic bomb survivors data is due to other radiation actions, than this number may become larger [17]. In the lung cancer induction of mice, a dose rate of about 10 mGy/day was estimated to double the spontaneous initiation rate [18]. High LET radiation was also found to be acting on initiation [19, 20].
Radiation action on promotion
Radiation effects on the effective clonal expansion rate of initiated cells (promotion) have been found for radon and lung cancer [19–21], alpha-particles from Thorotrast in liver [22], alpha-particles from radium in bone cancer [23], as well as for neutrons and gamma-rays on lung cancer in mice [18].
The most direct evidence for a promoting action of radiation comes from protraction effects. As long as effects are ignored that are typically observable only in old age, the following holds:
-
Initiating action is additive: two exposures with a sufficient time between them give (approximately) an excess risk, which is the sum of the single exposure excess risks.
-
Promoting action is multiplicative: two exposures with a sufficient time between them give (approximately) a relative risk, which is the product of the single exposure relative risks.
Fractionated exposure with equal fractions may be best to test between these possibilities, as there is no freedom due to varying dose rate.
At Pacific North–West National Laboratory (PNNL), rats were exposed for 2–700 days at constant exposure rate. The excess relative risk (ERR) per 100 working-level months (WLM) radon for fatal lung tumours based on the fitted background was estimated [24] (see Fig. 1). Initiation only predicts a decrease of ERR/WLM with duration, due to age-at-exposure effects.

Estimate of the lifetime excess relative risk per nominal exposure unit of rats exposed to radon for various periods of time with a constant exposure rate (100 WL). The dashed line represents model estimates with only an initiating radiation action (I-model), and the solid line one with initiation and promotion (IP-model) (based on model fits carried out by Heidenreich et al. [24])
The radiation action that induces promotion is not clear. At least one mechanism was proposed [25]. If (spontaneously) initiated cells can replace neighbouring cells killed by radiation faster than normal cells, then there is a promoting effect of radiation.
Radiation action on conversion
If conversion is radiation-sensitive, then an acute irradiation induces a peak in risk after the lag time. This may be smeared out if the assumed deterministic growth of malignant cells is distributed over a period of time.
In the JANUS experiment, a large number of mice were exposed to acute gamma-rays and neutrons up to high doses [18]. The lung cancer cases are plotted in Fig. 2. There is not a single lung cancer case up to 400 days after exposure. The lag time should definitely be shorter than this.

Number of lung cancer cases in mice after acute exposure to gamma-rays and neutrons (based on model fits carried out by Heidenreich et al. [18])
In various other analyses, the radiation-induced conversion rate relative to the baseline one was found to be a factor of at least ten smaller than that for initiation [21, 24]. This agrees qualitatively with radio-biological understanding: candidate mutations for late events may be mostly induced spontaneously.
Radiation action on lag time
In neutron-exposed mice, the estimated lag time to lung cancer was found to be significantly smaller than in gamma-exposed mice [18]. Possibly neutron-induced instability gives more aggressive cancer-subtypes than occur spontaneously or after gamma-ray exposures.
Distribution of risk due to the stochastic cancer process
The considered cancer model is a stochastic one. The mathematical description does not use the expectation of the number of initiated cells, but their distribution. As the number of initiated cells is proportional to the risk after the lag time, this allows one to calculate a distribution of risk at any given age. Under the assumptions of the present clonal expansion model it was found that:
-
The estimated baseline risk for liver cancer of more than 95% of a population at age 40 is less then 10% of the population risk, while it is more than 10-fold for the top percentile in risk [26].
-
After the injection of Thorotrast, the estimated relative risk for the low baseline risk person is more than 10 times larger than for the top percentile in baseline risk [27]. For the high baseline risk group, cancer is not a rare disease. Therefore, the population-based estimate of relative risks can be an underestimation for most persons most of the time.
A (thorny) path to better risk estimates at low doses
Integrated projects such as the EU-project RISC-RAD [28] should demonstrate that productive interactions between cancer modelling and radio-biology are possible. This may help in going from a more detailed understanding of radiation actions observed in experimental work to quantitative risk estimates. This in turn may be the only way to improve the estimates of small risks after radiation beyond direct epidemiology.
Multistage models for cancer incorporating genomic instability (Little)
There are many biological data suggesting that the initiating lesion in the multistage process leading to cancer might be one involving a destabilization of the genome resulting in elevation of mutation rates, reviewed by Morgan [29, 30]. Loeb [31, 32] has presented evidence that an early step in carcinogenesis is mutation in a gene controlling genome stability. Stoler et al. [33] showed that there are 11,000 mutations per carcinoma cell for a number of different cancer types, again implying that genomic destabilization is an early event in carcinogenesis. In particular, there is strong evidence of such an early genomic destabilization event for colon cancer [31–33]. However, the question of whether chromosomal instability is the initiating event in carcinogenesis, in particular colon cancer, is controversial. Tomlinson and Bodmer [34] argue that cancer is an evolutionary process, and that the observed accumulation of chromosomal and other damage in colon cancers may simply be the result of selection for cells with growth advantage.
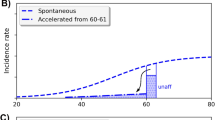
Recently, two papers have appeared proposing formulations of stochastic carcinogenesis models that incorporate genomic instability (GI) [35, 36], both applied to colon cancer. In contrast, Luebeck and Moolgavkar [5] have recently proposed a four-stage stochastic model positing inactivation of the adenomatous polyposis coli (APC) gene followed by a high frequency event, possibly positional in nature, an extension of the two-stage clonal expansion model of Moolgavkar and Venzon [1] and Knudson [37] (the so-called MVK model); this model does not assume GI. The paper of Little and Wright [36] proposed a general class of carcinogenesis models that includes, as special cases, the models proposed by Luebeck and Moolgavkar [5] and Nowak et al. [35], illustrated in Fig. 3. The model of Little and Wright [36] also generalizes the class of so-called generalized MVK models developed by Little [38], and which in turn therefore generalizes the two-mutation model of Moolgavkar and Venzon [1] and Knudson [37]. The model is close in spirit to the model of Mao et al. [39].

Schematic diagram of generalized MVK model with k cancer-stage mutations and m destabilizing mutations, as developed by Little and Wright [36]
Little and Li [40] have recently compared the goodness of fit to US SEER [41] colon cancer data of three models developed by Little and Wright [36] with the ones recently proposed by Nowak et al. [35] and Luebeck and Moolgavkar [5]. The best fitting models were the two-stage model of Nowak et al. [35] and the two-stage model of Little and Wright [36], with the four stage model of Luebeck and Moolgavkar [5] not markedly inferior; model fits are illustrated in Fig. 4 (taken from Little and Li [40]). The fits of the three-stage and five-stage models were somewhat worse (P < 0.05), the five-stage model fitting particularly poorly (P < 0.01). Both optimal genomic instability models predicted cellular mutation rates that are at least 10,000 times higher after genomic destabilization, for both sexes [40]. These large elevations in mutation frequency are not inconsistent with a variety of biological data [31, 40, 42–44]. In particular, Loeb and colleagues [31, 42] derive estimates of numbers of gross chromosomal abnormalities in human tumours, which are of the order of 10,000–100,000-fold elevated compared with normal tissue. Mutation rates 1,000–100,000-fold elevated have been observed in bacteria with defects in the dnaQ (DNA polymerase coding) gene [43, 44].

Cancer hazards predicted by models of Nowak et al. [35] (with two cancer-stage mutations and one destabilizing mutation), Luebeck and Moolgavkar [5] (with four cancer-stage mutations and no destabilizing mutations), and Little and Wright [36] (with two cancer-stage mutations and one destabilizing mutation, three cancer-stage mutations and one destabilizing mutation, five cancer-stage mutations and two destabilizing mutations), with stem cell population fixed to 108 cells, refitted to SEER [41] colon cancer data, and observed data (with 95% CI, adjusted for overdispersion) (reproduced from Little and Li [40])
The work of Little and Li [40] suggests that analyses of data that contain information only in relation to the age distribution of cancer does not possess the power to discriminate between models and hence to confirm or to falsify the hypothesized involvement of GI in colon cancer. It is possible that additional analysis, incorporating, for example, quantitative information on exposure to various mutagenic agents (e.g. ionising radiation) would have the power to discriminate between these models.
State-Vector Model for in vitro neoplastic transformation (Schöllnberger)
The State-Vector Model is a deterministic multistage model for in vitro neoplastic transformation and was tested on data that show a U-shaped dose–response curve at low doses of γ-radiation [45]. In the model, initiation results from DSBs induced by radiation and endogenous processes. Promotion results from a disruption of intercellular communication and a compensatory proliferation of initiated cells [46]. Cell death is allowed via radiation-induced necrosis and also at low doses via bystander-induced apoptosis. This pathway has been hypothesized [47] to be responsible for the observed decrease of the in vitro neoplastic transformation frequency below the spontaneous level [45, 48, 49]. The protective apoptosis-mediated bystander effect is implemented via rate constant k ap. At first, the model with k ap = 0 is fitted to the control and high dose data points for immediate and delayed plating. The model with k ap as the only free parameter was then fitted to the whole data set, with the other parameter values as fixed inputs [50, 51]. Figure 5 shows the data of Redpath et al. [45] and the different model contributions. Since the publication of these studies [50, 51], 95% confidence intervals (CI) were calculated for the best-estimated values using the residuals and the Jacobi matrix obtained in local model fits that follow the global search of the parameter space. For delayed plating, the best estimated value for k ap is 0.054/day [51] (95% CI: 0.031–0.078) and for immediate plating k ap = 0.022/day (95% CI: 0.007–0.036).

Transformation frequency for CGL1 cells irradiated with γ-radiation [45]. Error bars represent 95% CI. a Data for immediate plating and SVM fit showing the three different contributions (dash line direct, dotted line bystander, solid line total). The direct contribution relates to a model fit with k ap = 0 to control and high dose data for delayed and immediate plating [51]. The contribution of the bystanders was calculated as the difference between the direct and the total contribution. b Data for delayed plating and SVM fit showing the three different contributions (dash line direct, dotted line bystander, solid line total). For the SVM fits denoted as “total”, the 95% CI are shown for the low doses (dash–dot). Insets low-dose range with the x-axis units in Gy (reproduced from Schöllnberger et al. [51])
The relevance of in vitro neoplastic transformation for cancer induction in humans has caused discussion [45]. Cells transformed in vitro cause sarcoma after inoculation into host animals [52]. The relative risks calculated from human and mouse in vitro data [45, 48, 49] are remarkably similar to those from human epidemiological studies, particularly for breast cancer and leukaemia [45]. Mitchel (personal communication) points out the importance of in vitro neoplastic transformation and argues that if a cell in vivo is “near transformation” for whatever reason and would eventually develop into a cancer, and if radiation protects against and slows/reverses that process as demonstrated [53–58], then this would be critically important. Protective mechanisms such as homologous recombination (HR), an error-free DSB repair mechanism that could prove to be crucially important in the observed reduction of the neoplastic transformation frequency below the spontaneous level, are evolutionarily conserved [59]. The conservation of HR reaches from prokaryotes to lower and higher eukaryotes including humans [57] and neoplastic transformation of human and other mammalian cells facilitates the study of such mechanisms.
Modelling DNA double-strand break repair pathways for second breast cancers following radiotherapy (Thomas)
Ataxia–telengiactasia (A–T) is a rare recessive disorder caused by homozygous truncating mutations in ataxia-telangiectasia mutated gene (ATM), with one of its features being extreme radiation sensitivity. It has been hypothesized that ATM heterozygotes have increased risk of cancer, particularly breast [60, 61] and that this susceptibility interacts with ionising radiation exposure [62]. Gatti et al. [63] suggested that the increased cancer risk is associated with missense mutations rather than the truncating mutations that cause A–T. In any event, it is well established that ATM plays a central role in two pathways for repair of DSBs, the HR repair and the nonhomologous end-joining repair pathways.
The Women’s Environment, Cancer, and Radiation Epidemiology (WECARE) study was established to test the hypothesis that women who are carriers of a mutant allele in the ATM or BRCA1/2 genes (or other DNA repair genes) are more susceptible to a second radiation-induced breast cancer than are non-carriers. A population-based, nested case–control study was conducted in a cohort of first breast cancer patients from five centres in the US and Denmark. A total of 708 cases of bilateral breast cancer were identified and individually matched on centre, age at and time since first cancer to 1,397 controls with unilateral breast cancer. Controls were also counter-matched 2:1 on whether or not they received radiotherapy as recorded in the cancer registries and doses to each of eight locations in the contralateral breast (the one at risk of a second cancer) were estimated by phantom dosimetry. The design and analysis of this study is fully described in Bernstein et al. [64]. All subjects were genotyped for ATM, BRCA1 and BRCA2, using a staged approach consisting of DHPLC followed by direct sequencing [65]. Mutation detection in other DNA repair genes is currently underway.
We have developed a hierarchical modelling strategy [66] to analyse the effects of the numerous rare variants in ATM, as well as to develop a comprehensive model for all these genes and six others currently being tested. The first level of the model is a standard conditional logistic regression model of the form \( {\text{logit Pr(}}Y_{i} {\kern 1pt} {\text{ = }}{\kern 1pt} {\text{1)}}{\kern 1pt} {\kern 1pt} {\text{ = }}{\kern 1pt} \alpha {\kern 1pt} {\text{ + }}{\kern 1pt} \Sigma _{j} b_{j} X_{{ij}} {\kern 1pt} {\text{ + }}{\kern 1pt} {\text{ $ \gamma $ Z}}_{i} \) where Y i denotes disease status for subject i, X ij is an indicator for variant j, and Z i is a vector of fixed covariates (including the offset term needed to allow for the countermatching). In the second level, the log relative risk coefficients (b j ) are in turn regressed on a vector of “prior covariates” W j describing characteristics of the variants, such as indicators for type of variant, SIFT score, etc. Preliminary analyses found no significant effects of any of these prior covariates, but the risk estimates for individual variants were considerably stabilized by the second-level model, particularly for the rarer variants.
We are currently conducting a series of functional assays on cell lines from a random sample of five subjects with each of the ten most common variants and all individuals with rare variants. Each cell line will be assayed (with two replicates each) for ATM expression levels, ATM kinase activity, cell cycle checkpoint activation, and colony survival after ionising radiation exposure. These assays can then be used in the hierarchical modelling strategy to improve the prediction of the effects of individual variants and test hypotheses about differences between subgroups of variants with similar functional characteristics.
We are also currently genotyping six other genes involved in DSB repair (CHEK2, TP53BP1, MDC1, MRE11, RAD50 and NBS1) and will expand our hierarchical model to include these genes and their interactions with each other and with radiation dose. We are also hoping to carry out a genome-wide association scan as well. In this way, we aim to build a comprehensive model for DNA damage from ionising radiation and competency of the various repair pathways.
Conclusion
Systems biology promises better understanding of cancer induction through understanding and modelling properties of cells, and the signalling mechanisms between them. The works presented here demonstrate different approaches of dealing with this issue.
References
Moolgavkar SH, Venzon DJ (1979) Two-event model for carcinogenesis: incidence curves for childhood and adult tumors. Math Biosci 47:55–77
Moolgavkar SH, Knudson AG (1981) Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst 66:1037–1052
Frank SA, Nowak MA (2003) Developmental predisposition to cancer. Nature 422:494
Meza R, Luebeck EG, Moolgavkar SH (2005) Gestational mutations and carcinogenesis. Math Biosci 197:188–210
Luebeck EG, Moolgavkar SH (2002) Multistage carcinogenesis and the incidence of colorectal cancer. Proc Natl Acad Sci USA 99:15095–15100
Moolgavkar SH, Luebeck EG, Krewski D, Zielinski JM (1993) Radon, cigarette smoke, and lung cancer: a reanalysis of the Colorado Plateau uranium miners’ data. Epidemiology 4:204–217
Luebeck EG, Curtis SB, Cross FT, Moolgavkar SH (1996) Two-stage model of radon-induced malignant lung tumors in rats: effects of cell killing. Radiat Res 145:163–173
Hazelton WD, Moolgavkar SH, Curtis SB, Zielinski JM, Ashmore JP, Krewski D (2006) Biologically based analysis of lung cancer incidence in a large Canadian occupational cohort with low-dose ionizing radiation exposure, and comparison with Japanese atomic bomb survivors. J Toxicol Environ Health 69:1013–1038
Dewanji A, Venzon DJ, Moolgavkar SH (1989) A stochastic two-stage model for cancer risk assessment II: the number and size of premalignant clones. Risk Anal 9:179–186
Jeon J, Meza R, Moolgavkar SH, Luebeck EG (2007) Analyses of intermediate lesions in multistage carcinogenesis. Math Biosci (submitted)
Moolgavkar SH, Luebeck EG, de Gunst M, Port RE, Schwarz M (1990) Quantitative analysis of enzyme-altered foci in rat hepatocarcinogenesis experiments-I. Single agent regimen. Carcinogenesis 11:1271–1278
Moolgavkar SH, Luebeck EG, Buchmann A, Bock KW (1996) Quantitative analysis of enzyme-altered foci in rats initiated with diethylnitrosamine and promoted with 2,3,7,8-tetrachlorodibenzo-p-dioxin or 1,2,3,4,6,7,8-heptachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol 138:31–42
Gatenby RA, Maini PK (2003) Cancer summed up. Nature 421:321
National Research Council, Committee to assess health risks from exposure to low levels of ionizing radiation (2006) Health risks from exposure to low levels of ionizing radiation. BEIR VII Phase 2. National Academy Press, Washington DC
Heidenreich WF, Jacob P, Paretzke HG (1997) Exact solutions of the clonal expansion model and their application to the incidence of solid tumors of atomic bomb survivors. Radiat Environ Biophys 36:45–58
Heidenreich WF, Luebeck EG, Hazelton WD, Paretzke HG, Moolgavkar SH (2002) Multistage models and the incidence of cancer in the cohort of atomic bomb survivors. Radiat Res 158:607–614
Heidenreich WF (2002) Signals for a promoting action of radiation in cancer incidence data. J Radiol Prot 22:A71–A74
Heidenreich WF, Carnes BA, Paretzke HG (2006) Lung cancer risk in mice: analysis of fractionation effects and neutron RBE with a biologically motivated model. Radiat Res 166:794–801
Kaiser JC, Heidenreich WF, Monchaux G, Morlier JP, Collier CG (2004) Lung tumour risk in radon-exposed rats from different experiments: comparative analysis with biologically based models. Radiat Environ Biophys 43:189–201
Heidenreich WF, Tomášek L, Rogel A, Laurier D, Tirmarche M (2004) Studies of radon-exposed miner cohorts using a biologically based model: comparison of current Czech and French data with historic data from China and Colorado. Radiat Environ Biophys 43:247–256
Luebeck EG, Heidenreich WF, Hazelton WD, Paretzke HG, Moolgavkar SH (1999) Biologically based analysis of the data for the Colorado uranium miners cohort: age, dose and dose-rate effects. Radiat Res 152:339–351
Heidenreich WF, Nyberg U, Hall P (2003) A biologically based model for liver cancer risk in the Swedish Thorotrast patients. Radiat Res 159:656–662
Heidenreich WF, Müller WA, Paretzke HG, Rosemann M (2005) Bone cancer risk in mice exposed to 224Ra: protraction effects from promotion. Radiat Environ Biophys 44:61–67
Heidenreich WF, Jacob P, Paretzke HG, Cross FT, Dagle GE (1999) Two-step model for the risk of fatal and incidental lung tumors in rats exposed to radon. Radiat Res 151:209–217
Heidenreich WF, Atkinson M, Paretzke HG (2001) Radiation-induced cell inactivation can increase the cancer risk. Radiat Res 155:870–872
Heidenreich WF (2005) Heterogeneity of cancer risk due to stochastic effects. Risk Anal 25:1589–1594
Heidenreich WF (2006) Heterogeneity of cancer risk due to stochastic effects: emphasis on radiation-induced effects. Radiat Environ Biophys 45:33–37
RISC-RAD (2007) http://www.riscrad.org
Morgan WF (2003) Non-targeted and delayed effects of exposure to ionizing radiation: I. Radiation-induced genomic instability and bystander effects in vitro. Radiat Res 159:567–580
Morgan WF (2003) Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res 159:581–596
Loeb LA (1991) Mutator phenotype may be required for multistage carcinogenesis. Cancer Res 51:3075–3079
Loeb LA (2001) A mutator phenotype in cancer. Cancer Res 61:3230–3239
Stoler DL, Chen N, Basik M, Kahlenberg MS, Rodriguez-Bigas MA, Petrelli NJ, Anderson GR (1999) The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci USA 96:15121–15126
Tomlinson I, Bodmer W (1999) Selection, the mutation rate and cancer: ensuring that the tail does not wag the dog. Nature Med 5:11–12
Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih I-M, Vogelstein B, Lengauer C (2002) The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci USA 99:16226-16231
Little MP, Wright EG (2003) A stochastic carcinogenesis model incorporating genomic instability fitted to colon cancer data. Math Biosci 183:111–134
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820-823
Little MP (1995) Are two mutations sufficient to cause cancer? Some generalizations of the two-mutation model of carcinogenesis of Moolgavkar, Venzon, and Knudson, and of the multistage model of Armitage and Doll. Biometrics 51:1278-1291
Mao JH, Lindsay KA, Balmain A, Wheldon TE (1998) Stochastic modelling of tumorigenesis in p53 deficient mice. Br J Cancer 77:243–252
Little MP, Li G (2007) Stochastic modelling of colon cancer: is there a role for genomic instability? Carcinogenesis 28:479–487
Surveillance Epidemiology and End Results (SEER) Registry public-use data, 1973–2002 (2005) Cancer Statistics Branch, Surveillance Research Program, Division of Cancer Control and Population Sciences, National Cancer Institute, Bethesda (http://www.seer.cancer.gov/)
Jackson AL, Loeb LA (1998) On the origin of multiple mutations in human cancers. Semin Cancer Biol 8:421–429
Degnen GE, Cox EC (1974) Conditional mutator gene in Escherichia coli: isolation, mapping, and effector studies. J Bacteriol 117:477–487
Lancy ED, Lifsics MR, Kehres DG, Maurer R (1989) Isolation and characterization of mutants with deletions in dnaQ, the gene for the editing subunit of DNA polymerase III in Salmonella typhimurium. J Bacteriol 171:5572–5580
Redpath JL, Liang D, Taylor TH, Christie C, Elmore E (2001) The shape of the dose-response curve for radiation-induced neoplastic transformation in vitro: evidence for an adaptive response against neoplastic transformation at low doses of low-LET radiation. Radiat Res 156:700–707
Fleishman L, Crawford-Brown DJ, Hofmann W (2007) A computational model for radiation-induced cellular transformation to in vitro irradiation of cells by acute doses of X-rays. Math Biosci (submitted)
Portess DI, Bauer G, Hill MA, O’Neill P (2007) Low-dose irradiation of nontransformed cells stimulates the selective removal of precancerous cells via intercellular induction of apoptosis. Cancer Res 67:1246–1253
Azzam EI, de Toledo SM, Raaphorst GP, Mitchel REJ (1996) Low-dose ionizing radiation decreases the frequency of neoplastic transformation to a level below the spontaneous rate in C3H 10T1/2 cells. Radiat Res 146:369–373
Redpath JL, Antoniono RJ (1998) Induction of an adaptive response against spontaneous neoplastic transformation in vitro by low-dose gamma radiation. Radiat Res 149:517–520
Schöllnberger H, Eckl PM (2007) Protective bystander effects simulated with the State-Vector Model. Dose-Response 5:187–203
Schöllnberger H, Mitchel REJ, Redpath JL, Crawford-Brown DJ, Hofmann W (2007) Detrimental and protective bystander effects: a model approach. Radiat Res 168:614–626
Reznikoff CA, Bertram JS, Brankow DW, Heidelberger C (1973) Quantitative and qualitative studies of chemical transformation of cloned C3H mouse embryo cells sensitive to postconfluence inhibition of cell division. Cancer Res 33:3239–3249
Mitchel REJ, Jackson JS, McCann RA, Boreham DR (1999) The adaptive response modifies latency for radiation-induced myeloid leukemia in CBA/H mice. Radiat Res 152:273–279
Mitchel REJ, Dolling J-A, Misonoh J, Boreham DR (2002) Influence of prior exposure to low-dose adapting radiation on radiation-induced teratogenic effects in fetal mice with varying Trp53 function. Radiat Res 158:458–463
Mitchel REJ, Jackson JS, Morrison DP, Carlisle SM (2003) Low doses of radiation increase the latency of spontaneous lymphomas and spinal osteosarcomas in cancer-prone, radiation-sensitive Trp53 heterozygous mice. Radiat Res 159:320–327
Mitchel REJ, Jackson JS, Carlisle SM (2004) Upper dose thresholds for radiation-induced adaptive response against cancer in high-dose-exposed, cancer-prone, radiation-sensitive Trp53 heterozygous mice. Radiat Res 162:20–30
Mitchel REJ (2006) Low doses of radiation are protective in vitro and in vivo: evolutionary origins. Dose-Response 4:75–90
Mitchel REJ (2007) Low doses of radiation reduce risk in vivo. Dose-Response 5:1–10
Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T (2006) DNA repair and mutagenesis. ASM Press, Washington DC
Swift M, Sholman L, Perry M, Chase C (1976) Malignant neoplasms in the families of patients with ataxia-telangiectasia. Cancer Res 36:209–215
Inskip HM, Kinlen LJ, Taylor AMR, Woods CG, Arlett CF (1999) Risk of breast cancer and other cancers in heterozygotes for ataxia-telangiectasia. Br J Cancer 79:1304–1307
Swift M, Reitnauer PJ, Morrell D, Chase CL (1987) Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med 316:1289–1294
Gatti RA, Tward A, Concannon P (1999) Cancer risk in ATM heterozygotes: a model of phenotypic and mechanistic differences between missense and truncating mutations. Mol Genet Metab 68:419–423
Bernstein JL, Langholz B, Haile RW, Bernstein L, Thomas DC, Stovall M, Malone KE, Lynch CF, Olsen JH, Anton-Culver H, Shore RE, Boice JD Jr, Berkowitz GS, Gatti RA, Teitelbaum SL, Smith SA, Rosenstein BS, Børresen-Dale A-L, Concannon P, Thompson WD (2004) Study design: Evaluating gene–environment interactions in the etiology of breast cancer––the WECARE study. Breast Cancer Res 6:R199–R214
Bernstein JL, Teraoka S, Haile RW, Børresen-Dale A-L, Rosenstein BS, Gatti RA, Diep AT, Jansen L, Atencio DP, Olsen JH, Bernstein L, Teitelbaum SL, Thompson WD, Concannon P, WECARE Study Collaborative Group (2003) Designing and implementing quality control for multi-center screening of mutations in the ATM gene among women with breast cancer. Hum Mutat 21:542–550
Conti DV, Cortessis V, Molitor J, Thomas DC (2003) Bayesian modeling of complex metabolic pathways. Hum Hered 56:83–93
Acknowledgments
This work was funded partially by the European Commission under contracts FI6R-CT-2003-508842 (RISC-RAD) and FP6-036465 (NOTE) and by the Austrian Science Fund FWF (project P18055-N02). We thank Dr. Ronald Mitchel, of Atomic Energy of Canada Limited, for critically reading Dr. Schöllnberger’s contribution to the manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Little, M.P., Heidenreich, W.F., Moolgavkar, S.H. et al. Systems biological and mechanistic modelling of radiation-induced cancer. Radiat Environ Biophys 47, 39–47 (2008). https://doi.org/10.1007/s00411-007-0150-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00411-007-0150-z