
Abstract
Glioblastoma IDH-wildtype presents with a wide histological spectrum. Some features are so distinctive that they are considered as separate histological variants or patterns for the purpose of classification. However, these usually lack defined (epi-)genetic alterations or profiles correlating with this histology. Here, we describe a molecular subtype with overlap to the unique histological pattern of glioblastoma with primitive neuronal component. Our cohort consists of 63 IDH-wildtype glioblastomas that harbor a characteristic DNA methylation profile. Median age at diagnosis was 59.5 years. Copy-number variations and genetic sequencing revealed frequent alterations in TP53, RB1 and PTEN, with fewer gains of chromosome 7 and homozygous CDKN2A/B deletions than usually described for IDH-wildtype glioblastoma. Gains of chromosome 1 were detected in more than half of the cases. A poorly differentiated phenotype with frequent absence of GFAP expression, high proliferation index and strong staining for p53 and TTF1 often caused misleading histological classification as carcinoma metastasis or primitive neuroectodermal tumor. Clinically, many patients presented with leptomeningeal dissemination and spinal metastasis. Outcome was poor with a median overall survival of only 12 months. Overall, we describe a new molecular subtype of IDH-wildtype glioblastoma with a distinct histological appearance and genetic signature.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma is the most common malignant brain tumor among adults. At present, median overall survival after standard of care treatment, which includes resection followed by radio- and chemotherapy, is only 15 months [15]. This tumor type is predominantly found in the elderly, with a peak of incidence at 75–84 years [23]. Compared with other solid tumors, metastases are rare, and patients mainly die from neurological complications [31]. However, leptomeningeal dissemination and spinal metastasis, which are more common in patients with embryonal tumors such as medulloblastoma, may occur in a minor fraction of glioblastoma patients [19].
Morphologically, glioblastoma may present with different histological features. Presence of microvascular proliferation and/or necrosis, often accompanied by perinecrotic palisades, are mandatory for the histological diagnosis of glioblastoma. Irrespective of these characteristics, glioblastoma may harbor histological patterns of small cells, primitive neuronal cells, oligodendroglial components, gemistocytes, granular cells, lipidized cells, multinucleated giant cells and sarcomatous components. Such histological patterns occurring in small areas within an otherwise ‘typical’ case are separated from true histological variants, which are defined by the clear predominance of a given pattern in one tumor. Giant-cell glioblastoma, gliosarcoma and epithelioid glioblastoma are listed as variants in the WHO classification of tumors of the central nervous system (CNS) from 2016. Except for giant-cell glioblastoma, which is reported to have a tendency towards a better prognosis, different histological features are not associated with difference in survival [18].
In 2009, Perry et al. described 53 brain tumors with combined features of malignant gliomas and primitive neuroectodermal tumors. Common findings were loss of GFAP expression, p53 and synaptophysin expression, highly elevated Ki-67 index, MYC/MYCN amplification in the primitive neuronal component, a poor clinical prognosis and increased risk of leptomeningeal dissemination. A fluorescence in-situ hybridization (FISH) analysis revealed evidence that this group of tumors generally represents conventional gliomas from which embryonal-like elements with neuronal immunophenotypes secondarily emerged [25]. The group was included in 2016 WHO classification of tumors of the CNS and is now referred to glioblastomas with primitive neuronal component.
Besides histological patterns, glioblastomas can be divided into different methylation subgroups. Isocitrate dehydrogenase (IDH)-wildtype glioblastoma diagnosed in adults mainly fall into one of the following subgroups: mesenchymal, or receptor tyrosine kinase (RTK) 1 or 2 [5, 33]. DNA methylation-based classification was shown to correlate with previous molecular classes of glioblastoma described by the The Cancer Genome Atlas (TCGA) [22, 36] and is a helpful tool to define a precise diagnosis, especially if histology is not compelling or material is limited [5, 6, 8]. For some entities such as medulloblastoma and meningioma, methylation profiling is also of prognostic relevance [21, 29]. In addition, copy-number profiles calculated from DNA-methylation profiling can help to define a precise diagnosis [27]. Based on cIMPACT-NOW update 3, diffuse IDH-wildtype gliomas that are histologically lower grade (WHO grade 2 or 3) can be diagnosed as IDH-wildtype glioblastoma based upon presence of epidermal growth factor receptor (EGFR)-amplification and/or promoter mutation in telomerase reverse-transcriptase (TERT) and/or a combination of gain of complete chromosome 7 and loss of complete chromosome 10 [4].
Here, we report a novel methylation subgroup of IDH-wildtype glioblastoma that differs from known molecular subgroups in terms of methylation and copy-number profile and is linked to the histological variant of glioblastomas with primitive neuronal component.
Material and methods
Tissue samples
Samples were collected from neuropathology departments from multiple medical centers. Case selection was performed through t-distributed stochastic neighbor embedding (t-SNE) analysis of genome-wide DNA methylation data in a cohort of more than 75,000 tumors. Group formation was based on similarities in DNA methylation profiles. Tissue and data collection were performed in accordance with local ethics regulations and approval.
DNA methylation and t-SNE analysis
DNA extraction was performed as previously described [35]. DNA methylation profiles were generated using the Infinium HumanMethylation450 (450 k) or Infinium MethylationEPIC (850 k) BeadChip array (Illumina, San Diego, USA) according to the manufacturer’s instructions. The data were processed as previously described [5]. The t-SNE plot was calculated using the 15,000 most variable CpG sites according to standard deviation, 3,000 iterations and a perplexity value of 10. Reference cases were selected based on a high classifier score (> 0.9) in the brain tumor classifier. Methylation of the O6-alkylguanine DNA alkyltransferase (MGMT) promoter region was calculated as described previously [35].
Copy-number plots
To generate copy-number variation (CNV) profiles from the methylation array data we used the ‘conumee’ package in R (http://bioconductor.org/packages/release/bioc/html/conumee.html, https://github.com/dstichel/conumee) with additional baseline correction. By condensing multiple copy-number plots, summary CNV plots were created.
Gene sequencing and mutational burden
Next-generation sequencing was performed on a NextSeq 500 sequencer (Illumina) as described previously [28]. Exonic and splicing indels and nonsynonymous single-nucleotide variants (SNVs) with a frequency of ≤ 0.001 in the 1000 genomes database (https://www.internationalgenome.org/) were identified after subtracting low-quality calls. Telomerase reverse-transcriptase (TERT)-promoter mutations were extracted and evaluated separately due to the often low number of reads at this position.
Immunohistochemistry
Immunohistochemistry was performed on 3 µm thick FFPE tissue sections attached on StarFrost Advanced Adhesive slides (Engelbrecht, Kassel, Germany), followed by drying at 80 °C for 15 min and conducted on a BenchMark Ultra immunostainer (Ventana Medical Systems, Tucson, USA). Slides were pretreated with Cell Conditioning Solution CC1 (Ventana Medical Systems) for 32 min at room temperature. Primary antibodies were incubated at 37 °C for 32 min. Afterwards we used Ventana standard signal amplification, UltraWash, counter-staining with one drop of hematoxylin for 4 min, and one drop of bluing reagent for 4 min. For visualization ultraView Universal DAB Detection Kit (Ventana Medical Systems) was used. Primary antibodies were diluted as followed: Glial fibrillary acidic protein (GFAP, 1:2000, Cell signalling, Promega), cytokeratin AE1/3 (RTU, DCS Hamburg, Germany), thyroid transcription factor 1 (TTF1, 1:50, clone EP229, Cell Marque, Rocklin, USA and 1:50, clone 8G7G3/1, Dako, Santa Clara, USA), tumor protein 53 (p53, 1:50, Novocastra, Wetzlar, Germany), synaptophysin (1:50, Cell Marque), retinoblastoma protein (Rb, 1:50, clone Ab-780, Merck, St. Louis, USA), NeuN (1:100, Milipore, Burlington, USA), neuron-specific enolase (NSE, 1:4, Linaris, Dossenheim, Germany), Ki67 (1:100, Dako). Stained slides were scanned on the Aperio AT2 Scanner (Aperio Technologies, Vista, USA) and digitalized using Aperio ImageScope software v12.3.2.8013.
Statistical analysis
Sample sizes (n) are indicated in figures and figure legends. Kaplan–Meier curves were created and log-rank tests calculated using SPSS (IBM, Armonk, USA). For Fig. 1b, an image from Servier Medical Art (http://smart.servier.com/) was used with little modifications licensed under a Creative Commons Attribution 3.0 Unported License.

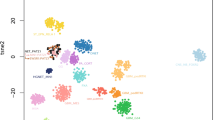
Glioblastomas with primitive neuronal component have a distinct methylation profile. a t-SNE analysis of 63 glioblastomas with primitive neuronal component and 311 reference cases. Distribution of primary localization b, age c, MGMT promoter methylation status d and gender e in glioblastomas with primitive neuronal component. GBM PNC glioblastoma with primitive neuronal component, MB medulloblastoma, NB neuroblastoma, HGNET high-grade neuroepithelial tumor, ANA PA anaplastic astrocytoma with piloid features, PXA pleomorphic xanthoastrocytoma, (un)meth (un)methylated
Results
Glioblastomas with primitive neuronal component have a distinct methylation profile
In a t-SNE analysis with more than 75,000 tumor samples, mainly including brain tumors and sarcomas, a distinct methylation class consisting of 63 samples was found. The group was located separate, yet close to the large but heterogeneous group of IDH-wildtype glioblastoma (data not shown). A more focused t-SNE analysis with selected cases of several reference comparison groups was conducted, highlighting the distinctiveness of the new molecular group (Fig. 1a). By looking at clinical data, we discovered that more than half of the tumors were localized in the temporal lobe (Fig. 1b). Patients were mostly older adults, with a median age of 59.5 years [5–81 years] (Fig. 1c). For 55.7% (34/61) of samples the promoter region of the MGMT gene was methylated (Fig. 1d). There was a predominance of males [1.7:1] (Fig. 1e). Almost all samples were obtained at first surgery for the brain tumor; whereas, one sample was diagnosed as a recurrence of an IDH-wildtype glioblastoma. Interestingly, the primary tumor of this case as well as two recurrent tumors from another case clustered within known methylation groups of IDH-wildtype glioblastoma (Suppl. Figure 1).
Frequent alterations in TP53, PTEN and RB1
Closer inspection of the copy-number profiles revealed loss of chromosome 10 in more than 90 percent of cases; whereas, a gain of chromosome 7 was only detected in half of the samples (Fig. 2a). Twelve cases (19%) presented with homozygous phosphatase and tensin homolog (PTEN) deletion (Fig. 2b). Loss of chromosome 13 was detected in 54.0% (34/63) of samples. Focal homozygous retinoblastoma tumor suppressor gene 1 (RB1) deletion was present in seven cases (11.1%). Interestingly, half of the cases showed a gain of chromosome 1. MDM2 and MDM4 were each focally amplified in 6.3% of cases (4/63). MYCN amplification was present in 14.3% (9/63) with two cases amplified for MYC (3.2%). Homozygous cyclin-dependent kinase inhibitor (CDKN) 2A/B deletion was only detected in 15.9% (10/63).

Frequent alterations in TP53, PTEN and RB1. a Summation copy-number plot of 63 glioblastomas with primitive neuronal component. b Copy-number plot of a glioblastoma with primitive neuronal component showing homozygous PTEN deletion. c Mutations and copy-number alterations in selected genes detected in 20 samples
Panel sequencing was performed for 19 cases. Another case was retrieved from TCGA with exome-sequencing data available. TERT-promoter mutations were found in 17 of 20 samples. In one case without TERT-promoter mutation an alpha thalassemia/mental retardation syndrome, X-linked (ATRX)-frameshift insertion was detected. In 80% of samples (16/20) at least one TP53 mutation was found. Interestingly, of nine cases where only one TP53 mutation was detected, seven showed a clear enrichment of the mutated allele; whereas, only two of these samples presented with a heterozygous loss of the TP53 locus (suggesting copy-neutral LOH as a prominent mechanism for loss of the wildtype allele). The four cases without detected TP53 mutations or deletions all showed homozygous deletion of the CDKN2A/B locus and two cases additionally had a clear amplification of MDM4. Seven cases showed a PTEN mutation, accompanied by heterozygous chromosomal deletion of the PTEN locus in five of these cases. RB1 mutations were detected in five samples, with four of these cases displaying a heterozygous deletion of the genetic locus (Fig. 2c).
Embryonal morphology, lack of GFAP expression and TTF1 positivity are common features
From 45 cases with known histological diagnosis, 28 cases were initially diagnosed as glioblastoma, five cases as gliosarcoma, 11 cases as primitive neuroectodermal tumor (PNET) and one case as ganglioneuroblastoma. Additional primitive neuronal component was described for seven of the histologically defined glioblastomas and five of the gliosarcomas. For three cases, neuroendocrine metastasis was considered as the diagnosis. Tumors showed undifferentiated, embryonal cells with high nuclear–cytoplasmatic ratio and high mitotic activity. However, none of the cases showed Homer Wright rosettes or clear cell wrapping that are characteristic features of medulloblastoma and other embryonal neoplasms. Some cases showed a biphasic pattern with additional glial tumor parts separated from embryonal tumor parts. For those samples that showed additional glial parts, these were mostly sharply separated from primitive parts; whereas, a minority of cases showed intermixed sections. Negative GFAP staining in embryonal appearing tumor cells was shown for 84% (21/25) of cases (Fig. 3). We additionally performed DNA-methylation profiling from only GFAP positive glial appearing tumor components of eight cases. Interestingly, three of the cases did not cluster with their GFAP-negative counterparts but to other methylation groups of IDH-wildtype glioblastoma (Suppl. Figure 1). Copy-number profiles of GFAP positive and negative tumor parts were mostly congruent. One case presented with a gain of chromosomal arm 1p only in the GFAP negative part. Another case harbored an amplification of the MYCN locus in GFAP negative areas; whereas, GFAP positive areas were MYCN wildtype. Almost all cases (27/29) presented with strong nuclear staining for p53 in the embryonal tumor component. Synaptophysin was positive for 85.7% (12/14) in embryonal tumor parts (Suppl. Figure 3). Tumors that stained negative for Synaptophysin showed positive staining for at least one other neuronal marker (NeuN or neuron specific enolase, NSE). All tumors tested showed positivity for NSE (9/9); whereas, only four out of eleven tumors were positive for NeuN (36.4%). We also stained for RB1 expression. However, none of the cases showed a clear loss of nuclear expression (Suppl. Figure 3). Surprisingly, 77.8% (14/18) cases showed positive staining of the nuclei of tumor cells for TTF1 using the EP229 clone (Fig. 3) in embryonal, GFAP-negative tumor components only. We additionally stained 20 conventional glioblastomas, 10 of which were small cell variants, that were all negative for the TTF1 EP229 clone. However, using the TTF1 8G7G3/1 clone only one of the samples showed nuclear positivity (1/15, 6.6%, Suppl. Figure 3). Potentially, further differences may also apply for other clones, and for findings on RB1 positivity, respectively.

Histology of glioblastoma with primitive neuronal component. Cases present with high nuclear-to-cytoplasmic ratio and frequent mitoses in the areas with embryonal phenotype (a, d, g). GFAP expression is lacking in these tumor regions (b, e). Glial areas with retained GFAP expression b, e and lower proliferation index c can be sharply demarcated from GFAP negative areas. Lacking cytokeratin expression in GFAP negative areas (f), but nuclear positivity for p53 (h) and TTF1 (clone EP229) (i). Scale bar 200 µm
Leptomeningeal dissemination is frequent and survival is poor
Survival data were available for 24 patients. Median overall survival was only 12 months (360 days, Fig. 4). There was no difference in survival regarding MGMT promoter methylation status (Suppl. Figure 2a). Progression-free survival was available for 23 patients and median PFS was 8 months (240 days, Suppl. Figure 2b). Interestingly, for 10 patients with clinical data available, leptomeningeal dissemination with spinal metastasis was reported in four patients. For one patient, the spinal metastasis was passed through DNA methylation analysis and showed a very similar methylation and copy-number profile as the primary tumor (Suppl. Figure 1). Three additional patients showed intracerebral metastases. Most patients were treated according to the Stupp protocol with fractionated irradiation and temozolomide.

Overall survival in glioblastoma with primitive neuronal component. Median survival is 360 days
Discussion
In this work, we present a new molecular type that is linked to a specific histological pattern of IDH-wildtype glioblastoma. The tumors present with a highly undifferentiated morphology, frequent leptomeningeal dissemination and occasional sarcomatous features. Lack of GFAP expression and nuclear positivity for TTF1 are common in embryonal components which may cause misinterpretation as carcinoma although these tumors do not express cytokeratins. Clinical outcome is very poor and in line with other IDH-wildtype glioblastomas. For their focal resemblance we provisionally named these tumors glioblastomas with primitive neuronal component.
Glioblastomas with primitive neuronal component harbor a unique methylation profile that is clearly distinguishable from methylation (sub-)groups known so far in the v11b4 version of the brain tumor classifier [5]. In a t-SNE analysis with more than 75,000 reference cases the group clusters close to IDH-wildtype glioblastomas. One case was a recurrent tumor of an IDH-wildtype glioblastoma, subtype mesenchymal. Another case recurred as IDH-wildtype glioblastoma, subtype RTK2. For three samples, GFAP-positive areas within the same tumor also clustered to distinct subtypes of IDH-wildtype glioblastoma. These results show that the new molecular entity described in this work clearly belongs to the type of IDH-wildtype glioblastoma as also discovered by the FISH analyses performed by Perry et al. [25], but it also indicates that molecular subtypes among IDH-wildtype glioblastomas may vary upon progression and several subtypes might even be present in one manifestation of a tumor. This indicates a high degree of plasticity in glioblastoma cells which is not a common feature of other tumor types (where methylation profiles stay remarkably constant over time), as also recently suggested based on single-cell trancriptomic analyses [34]. Neftel et al. showed that IDH-wildtype glioblastoma consists of four different cellular states that are all capable of tumor initiation and transition into another state [20]. Our study demonstrates that plasticity in glioblastoma is not only detected in the transcriptome but can also be found on DNA methylation level, which was thought to be more robust [5].
According to the 2016 WHO classification of brain tumors of the central nervous system, IDH-wildtype glioblastomas show homozygous deletion of CDKN2A/B in approximately 60% [18]. CDKN2A encodes for two proteins, one is p16 that activates retinoblastoma protein (Rb) through inhibition of cyclin-dependent kinases (CDK) 4 and 6, the second protein is p14arf activating p53 by inhibiting MDM2 [17, 26]. In glioblastoma with primitive neuronal component, CDKN2A/B is deleted in only 15.9% of samples. The pathway seems to be affected further downstream by frequent RB1 deletions and mutations as well as TP53 mutations. Interestingly, TP53 mutations are usually linked to IDH-mutations in astrocytic tumors and only found in 25–30% of IDH-wildtype glioblastoma [7, 18, 24, 37]. About 80% of glioblastomas with primitive neuronal component, however, have TP53 mutations. Regarding samples with genetic sequencing data available, only the small fraction of tumors lacking TP53 mutations harbored homozygous CDKN2A/B deletion. TP53 mutations together with PTEN mutations were also found in a study recently performed by Xu et al. performing exon sequencing on 11 glioblastomas with primitive neuronal component. In addition, the authors found frequent mutations in PIK3CA and PIK3R1; whereas, an RB1 mutation was only found in one case [38].
Mutational inactivation of p53 correlates with higher proliferation, invasion and a more stem-like phenotype [2, 13]. In our cohort, the proliferation index of the tumors was generally very high and a substantial proportion of patients presented with intracranial or even spinal leptomeningeal dissemination, which is otherwise described to be a rare event in glioblastoma [19]. Therefore, regular radiological screening of the neuroaxis should be considered subsequent to detection of this tumor type.
In our cohort, TERT promoter mutations and a chromosome 7/10 signature in at least half of the samples are shared findings with conventional IDH-wildtype glioblastoma and further imply the belonging to this entity. However, aberrations in RB1 and MYC/MYCN as well as leptomeningeal dissemination are characteristics of embryonal tumors as medulloblastomas or atypical teratoid rhabdoid tumors. Histologically, half of the cases were diagnosed as either PNET or glioblastoma/gliosarcoma with primitive neuronal component. However, the diagnosis PNET itself became obsolete with the WHO classification of CNS tumors released in 2016 [18]. The latest WHO classification for CNS tumors which will be released in spring 2021 still recognizes glioblastomas with primitive neuronal component, and loss of GFAP expression, MYC/MYCN gene amplification, elevated Ki67 proliferation index and increased cerebrospinal fluid dissemination. The histological defined pattern has wide overlap with our molecular defined subtype and, therefore, we named our methylation group glioblastomas with primitive neuronal component [18, 25]. Gliosarcomas with primitive neuronal component have also been described before [12, 14, 30], and interestingly, glioblastomas with PNET phenotype have also been described in mouse models with inactivation of p53, Rb, and Pten in neural progenitors [10, 11]. The morphology can also be shared by IDH-mutant gliomas [32]. However, none of the tumors described in our study harbored a pathogenic IDH-mutation.
In addition, we detected positivity of tumor cell nuclei for TTF1 using the EP229 clone; whereas, conventional glioblastomas, including small cell variants, all stained negative for this clone. It is also worth noting that nuclear TTF1 expression was only detectable in embryonal patterns in our tumors; whereas, glial tumor components did not express the protein. TTF1 expression in glioblastoma has not been described for this TTF1 clone so far. In neuropathological routine, TTF1 immunohistochemistry is most often used to distinguish metastases of lung and thyroid cancers from primary brain tumors. Although nuclear TTF1 expression in glioblastoma is rare, reactivity may differ among different antibody clones [9]. We additionally stained glioblastomas with primitive neuronal component with the TTF1 8G7G3/1 clone shown to be negative in glioblastomas and only detected positive nuclear staining for TTF1 in one case. These findings indicate that not all TTF1 clones are equally applicable to diagnose glioblastomas with primitive neuronal component. TTF1 is known to be expressed during not only lung and thyroid morphogenesis but also during brain development [3, 16] which might lead to the assumption that positivity for this protein in glioblastomas might represent further evidence for a more primitive and undifferentiated phenotype. It might also be hypothesized that glioblastomas with primitive neuronal component have undergone “pan-cancer convergence” that was described by Balanis et al. for epithelial cancers that acquire TP53 and RB1 mutations [1]. However, TTF1 expression can also be found among low grade tumors as pituicytomas or spindle cell oncocytomas of the pituitary gland.
In conclusion we describe a molecular variant of glioblastoma with overlap to a histological pattern presented previously. Glioblastomas with primitive neuronal component are characterized by a distinct methylation profile in the component of embryonal phenotype. Our study extends the basis for molecular diagnostics of challenging diagnostic cases.
References
Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, et al. (2019) Pan-cancer convergence to a small-cell neuroendocrine phenotype that shares susceptibilities with hematological malignancies. Cancer Cell 36:17-34.e17. https://doi.org/10.1016/j.ccell.2019.06.005
Bieging KT, Mello SS, Attardi LD (2014) Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer 14:359–370. https://doi.org/10.1038/nrc3711
Bingle CD (1997) Thyroid transcription factor-1. Int J Biochem Cell Biol 29:1471–1473. https://doi.org/10.1016/s1357-2725(97)00007-1
Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB, et al. (2018) cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV.” Acta Neuropathol 136:805–810. https://doi.org/10.1007/s00401-018-1913-0
Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. (2018) DNA methylation-based classification of central nervous system tumours. Nature 555:469–474. https://doi.org/10.1038/nature26000
Capper D, Stichel D, Sahm F, Jones DTW, Schrimpf D, Sill M, et al. (2018) Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol 136:181–210. https://doi.org/10.1007/s00401-018-1879-y
Comprehensive genomic characterization defines human glioblastoma genes and core pathways (2008). Nature 455:1061–1068. https://doi.org/10.1038/nature07385
Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, et al. (2017) Same-day genomic and epigenomic diagnosis of brain tumors using real-time nanopore sequencing. Acta Neuropathol 134:691–703. https://doi.org/10.1007/s00401-017-1743-5
Galloway M, Sim R (2007) TTF-1 staining in glioblastoma multiforme. Virchows Arch 451:109–111. https://doi.org/10.1007/s00428-007-0432-5
Henriquez NV, Forshew T, Tatevossian R, Ellis M, Richard-Loendt A, Rogers H, et al. (2013) Comparative expression analysis reveals lineage relationships between human and murine gliomas and a dominance of glial signatures during tumor propagation in vitro. Cancer Res 73:5834–5844. https://doi.org/10.1158/0008-5472.Can-13-1299
Jacques TS, Swales A, Brzozowski MJ, Henriquez NV, Linehan JM, Mirzadeh Z, et al. (2010) Combinations of genetic mutations in the adult neural stem cell compartment determine brain tumour phenotypes. Embo J 29:222–235. https://doi.org/10.1038/emboj.2009.327
Kaplan KJ, Perry A (2007) Gliosarcoma with primitive neuroectodermal differentiation: case report and review of the literature. J Neurooncol 83:313–318. https://doi.org/10.1007/s11060-007-9331-5
Kastenhuber ER, Lowe SW (2017) Putting p53 in context. Cell 170:1062–1078. https://doi.org/10.1016/j.cell.2017.08.028
Kepes JJ (2002) Gliosarcoma with areas of primitive neuroepithelial differentiation and extracranial metastasis. Clin Neuropathol 21:193–195 (author reply 195-196)
Koshy M, Villano JL, Dolecek TA, Howard A, Mahmood U, Chmura SJ, et al. (2012) Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol 107:207–212. https://doi.org/10.1007/s11060-011-0738-7
Lazzaro D, Price M, de Felice M, Di Lauro R (1991) The transcription factor TTF-1 is expressed at the onset of thyroid and lung morphogenesis and in restricted regions of the foetal brain. Development (Cambridge, England) 113:1093–1104
Li Y, Nichols MA, Shay JW, Xiong Y (1994) Transcriptional repression of the D-type cyclin-dependent kinase inhibitor p16 by the retinoblastoma susceptibility gene product pRb. Can Res 54:6078–6082
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. (2016) The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131:803–820. https://doi.org/10.1007/s00401-016-1545-1
Mandel JJ, Yust-Katz S, Cachia D, Wu J, Liu D, de Groot JF, et al. (2014) Leptomeningeal dissemination in glioblastoma; an inspection of risk factors, treatment, and outcomes at a single institution. J Neurooncol 120:597–605. https://doi.org/10.1007/s11060-014-1592-1
Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. (2019) An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell 178:835-849.e821. https://doi.org/10.1016/j.cell.2019.06.024
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. (2017) The whole-genome landscape of medulloblastoma subtypes. Nature 547:311–317. https://doi.org/10.1038/nature22973
Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. https://doi.org/10.1016/j.ccr.2010.03.017
Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS (2020) CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro-Oncol 22:iv1–iv96. https://doi.org/10.1093/neuonc/noaa200
Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. (2008) An integrated genomic analysis of human glioblastoma multiforme. Science 321:1807–1812. https://doi.org/10.1126/science.1164382
Perry A, Miller CR, Gujrati M, Scheithauer BW, Zambrano SC, Jost SC, et al. (2009) Malignant gliomas with primitive neuroectodermal tumor-like components: a clinicopathologic and genetic study of 53 cases. Brain Pathol 19:81–90. https://doi.org/10.1111/j.1750-3639.2008.00167.x
Rayess H, Wang MB, Srivatsan ES (2012) Cellular senescence and tumor suppressor gene p16. Int J Cancer 130:1715–1725. https://doi.org/10.1002/ijc.27316
Reuss DE, Sahm F, Schrimpf D, Wiestler B, Capper D, Koelsche C, et al. (2015) ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol 129:133–146. https://doi.org/10.1007/s00401-014-1370-3
Sahm F, Schrimpf D, Jones DT, Meyer J, Kratz A, Reuss D, et al. (2016) Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol 131:903–910. https://doi.org/10.1007/s00401-015-1519-8
Sahm F, Schrimpf D, Stichel D, Jones DTW, Hielscher T, Schefzyk S, et al. (2017) DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol 18:682–694. https://doi.org/10.1016/s1470-2045(17)30155-9
Shintaku M, Yoneda H, Hirato J, Nagaishi M, Okabe H (2013) Gliosarcoma with ependymal and PNET-like differentiation. Clin Neuropathol 32:508–514. https://doi.org/10.5414/np300624
Silbergeld DL, Rostomily RC, Alvord EC Jr (1991) The cause of death in patients with glioblastoma is multifactorial: clinical factors and autopsy findings in 117 cases of supratentorial glioblastoma in adults. J Neurooncol 10:179–185. https://doi.org/10.1007/bf00146880
Song X, Andrew Allen R, Terence Dunn S, Fung KM, Farmer P, Gandhi S, et al. (2011) Glioblastoma with PNET-like components has a higher frequency of isocitrate dehydrogenase 1 (IDH1) mutation and likely a better prognosis than primary glioblastoma. Int J Clin Exp Pathol 4:651–660
Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DT, Konermann C, et al. (2012) Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 22:425–437. https://doi.org/10.1016/j.ccr.2012.08.024
Suvà ML, Tirosh I (2020) The glioma stem cell model in the era of single-cell genomics. Cancer Cell 37:630–636. https://doi.org/10.1016/j.ccell.2020.04.001
Suwala AK, Stichel D, Schrimpf D, Kloor M, Wefers AK, Reinhardt A, et al. (2020) Primary mismatch repair deficient IDH-mutant astrocytoma (PMMRDIA) is a distinct type with a poor prognosis. Acta Neuropathol. https://doi.org/10.1007/s00401-020-02243-6
Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. (2010) Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 17:98–110. https://doi.org/10.1016/j.ccr.2009.12.020
Watanabe K, Sato K, Biernat W, Tachibana O, von Ammon K, Ogata N, et al. (1997) Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res: Off J Am Assoc Cancer Res 3:523–530
Xu G, Zheng H, Li JY (2019) Next-generation whole exome sequencing of glioblastoma with a primitive neuronal component. Brain Tumor Pathol 36:129–134. https://doi.org/10.1007/s10014-019-00334-1
Acknowledgements
This study was financially supported by the SFB 1389 UNITE by the German research foundation (DFG). AKS is a fellow of the Mildred Scheel Postdoctoral Fellowship Program of the German Cancer Aid. FS is a fellow of the Else Kröner Excellence Program of the Else Kröner-Fresenius Stiftung (EKFS). PS is a fellow of the Hertie Academy of Excellence in Clinical Neuroscience. LS is a fellow of the BIH‐Charité Clinical Scientist Program by the Charité and BIH and supported by a DKTK Young Investigator grant. Methylation profiling at NYU Langone Health was in part supported by grants from the Friedberg Charitable Foundation, the Making Headway Foundation and the Sohn Foundation (to M.S.). Methylation profiling at UCL/UCLH was partly supported by NIHR funding the UCLH BRC (to SB). We would like to thank Viktoria Zeller, Ulrike Vogel, Sabrina Sprengart, Lisa Kreinbihl, Laura Dörner, Moritz Schalles, Lea Hofmann, Ulrike Lass and Jochen Meyer for excellent assistance.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Suwala, A.K., Stichel, D., Schrimpf, D. et al. Glioblastomas with primitive neuronal component harbor a distinct methylation and copy-number profile with inactivation of TP53, PTEN, and RB1. Acta Neuropathol 142, 179–189 (2021). https://doi.org/10.1007/s00401-021-02302-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-021-02302-6