
Abstract
Polyglutamine (polyQ) tract expansion leads to proteotoxic misfolding and drives a family of nine diseases. We study spinal and bulbar muscular atrophy (SBMA), a progressive degenerative disorder of the neuromuscular system caused by the polyQ androgen receptor (AR). Using a knock-in mouse model of SBMA, AR113Q mice, we show that E3 ubiquitin ligases which are a hallmark of the canonical muscle atrophy machinery are not induced in AR113Q muscle. Similarly, we find no evidence to suggest dysfunction of signaling pathways that trigger muscle hypertrophy or impairment of the muscle stem cell niche. Instead, we find that skeletal muscle atrophy is characterized by diminished function of the transcriptional regulator Myocyte Enhancer Factor 2 (MEF2), a regulator of myofiber homeostasis. Decreased expression of MEF2 target genes is age- and glutamine tract length-dependent, occurs due to polyQ AR proteotoxicity, and is associated with sequestration of MEF2 into intranuclear inclusions in muscle. Skeletal muscle from R6/2 mice, a model of Huntington disease which develops progressive atrophy, also sequesters MEF2 into inclusions and displays age-dependent loss of MEF2 target genes. Similarly, SBMA patient muscle shows loss of MEF2 target gene expression, and restoring MEF2 activity in AR113Q muscle rescues fiber size and MEF2-regulated gene expression. This work establishes MEF2 impairment as a novel mechanism of skeletal muscle atrophy downstream of toxic polyglutamine proteins and as a therapeutic target for muscle atrophy in these disorders.
Similar content being viewed by others


Introduction
Muscle atrophy is a hallmark of patients with spinal and bulbar muscular atrophy (SBMA), a degenerative disorder of the neuromuscular system. SBMA is caused by a CAG/polyglutamine (polyQ) tract expansion in the androgen receptor (AR), placing it into a family of nine degenerative disorders with similar mutations, including Huntington disease, dentatorubro-pallidoluysian atrophy, and six autosomal dominant spinocerebellar ataxias (type 1, 2, 3, 6, 7, and 17) [37, 62]. PolyQ tract expansion leads to both loss of normal AR function as a transcription factor and ligand-dependent proteotoxicity. These changes ultimately lead to dysregulation of pathways critical for normal cellular function [36, 46, 50, 54, 64, 70].
The neuromuscular degeneration that occurs in SBMA patients downstream of polyQ AR is characterized by loss of lower motor neurons from the brainstem and spinal cord [2, 72]. Clinically, signs of neuron dysfunction are seen including tremor and fasciculations, and progressive limb muscle weakness often necessitates the use of ambulatory assistance devices [5]. Additionally, SBMA patients frequently develop signs of sensory neuron dysfunction [34, 56, 60], suggesting that nervous system pathology is not isolated to the neuromuscular system.
While neuronal dysfunction in SBMA remains an active field of study, several lines of evidence have established skeletal muscle tissue as a key contributor to disease pathogenesis. SBMA patients develop progressive muscular weakness and concomitantly display signs of muscle toxicity, including evidence of myopathy on muscle biopsy and elevated serum creatine kinase levels above what is found in diseases of pure denervation [5, 65, 74]. Isolated skeletal muscle satellite cells show impairments in fusion to form myotubes, demonstrating cell-autonomous toxicity in muscle [42]. Knock-in mice expressing polyQ AR under the endogenous mouse promotor develop myopathy months before spinal cord pathology, with atrophy of both type 1 and type 2 muscle fibers and polyQ AR intranuclear aggregates in skeletal muscle within the first 3–4 months of age [14, 26, 66, 83]. Aggregates within spinal motor neurons of the anterior horn are visualized by 24 months of age [83]. Transgenic mice overexpressing wild-type (WT) AR only in skeletal muscle show hormone-dependent myopathy and motor axon loss; similar effects are seen in mice overexpressing polyQ AR only in muscle [49, 63]. Additionally, overexpression of insulin-like growth factor-1 (IGF-1) in muscle ameliorates the phenotypic severity of SBMA transgenic mice [57]. The contribution of muscle to the SBMA phenotype is further corroborated by studies demonstrating that knockdown of peripheral polyQ AR or conditional deletion of polyQ AR only in skeletal muscle rescues disease in mice [17, 38].
While skeletal muscle is known to be an important contributor to pathogenesis, little is known about the mechanisms driving muscle atrophy in SBMA. Atrophy is often triggered by induction of a specific program involving upregulation of the E3 ubiquitin ligases MuRF1, Atrogin-1, and MuSA1 as well as the proteasome [7, 8, 31]. However, recent analysis of a gene targeted mouse model of SBMA demonstrates age-dependent impairment of proteasome function in muscle, suggesting that polyQ AR-mediated skeletal muscle atrophy may occur through alternative mechanisms [53].
Here, we use a combination of RNAseq analysis and targeted functional assays to explore the pathway leading to skeletal muscle atrophy in SBMA. We identify impaired function of the transcription factor Myocyte Enhancer Factor 2 (MEF2), a well-established modulator of myofiber homeostasis and hypertrophy [51], as a novel contributor to muscle atrophy caused by polyQ AR. We demonstrate that changes in MEF2 function are age- and hormone-dependent and occur as a result of ligand-mediated proteotoxicity. We find that impairment of MEF2 also occurs in skeletal muscle of R6/2 mice, a model of Huntington disease where muscle atrophy is prominent. Finally, we demonstrate that SBMA patient muscle biopsies lose expression of MEF2 target genes, and that restoring MEF2 activity rescues mouse muscle fiber size in vivo. These studies establish MEF2 as a novel therapeutic target which mediates skeletal muscle atrophy in polyglutamine disease.
Materials and methods
Mice
AR21Q and AR113Q knock-in mice were generated using exon 1 specific targeting as previously described [1, 83, 84]. Specific pathogen-free housing was maintained on a 12 h light/dark cycle. Chow and water were administered ad libitum. Genotyping was performed with the following primers on ear samples: AR forward: 5′-CCAGAATCTGTTCCAGAGCGTG-3′ (MilliporeSigma, 6-FAM labeled); AR reverse: 5′-TGTTCCCCTGGACTCAGATG-3′ (Invitrogen). AR-targeted ASOs were injected subcutaneously (50 mg/kg body weight) from 6 to 26 weeks of age once per week. ASOs are made of a 16-mer, 2′,4′-constrained ethyl gapmer contain the following sequence: AAGTTGTAGTAGTCGC which is complementary to human and mouse AR transcripts and was detailed previously [38]. All procedures involving mice were approved by the University of Michigan Committee on Use and Care of Animals (PRO00008133) and conducted in accordance with institutional and federal ethical guidelines for animal testing and research.
For R6/2 mice, animal care and procedures were performed in compliance with United Kingdom Home Office regulations (Animals and Scientific Procedures Act 1986) and were approved by the University College London Ethical Review Process Committee. Hemizygous R6/2 mice [43] were bred by backcrossing R6/2 males to (CBA/Ca × C57BL/6J) F1 females (B6/CBAF1/OlaHsd Envigo, UK). Animals were kept on a 12 h light and dark cycle and room temperature was maintained at 21 °C ± 1 °C. All mice had access to moderate environmental enrichment (play tube, wooden chew sticks). Cages were provided with an unlimited supply of water and a high protein chow diet. Mice were genotyped and the HTT CAG repeat length was quantified as previously described [69]. The CAG repeat sizes were 188 ± 1.2 (SD) at 4 weeks, 188 ± 3.06 at 8 weeks and 185 ± 3.37 at 12 weeks of age.
Patient samples
Anonymized control and patient biopsy samples were obtained from the Neuromuscular Bank of Tissues and DNA samples, Telethon Network of Genetic Biobanks, and the EuroBioBank network. Information regarding the age, muscle, clinical status of controls, and Q tract length of SBMA patients is provided (Table S6).
Orchiectomy
Surgical castration was performed at 5–6 weeks as previously described [14]. In brief, an incision no longer than 5 mm was made in the abdomen at the level of the hind legs. Following incision, the vas deferens and spermatic cord were ligated using nonabsorbable sutures. The testes were removed and the incision was closed using absorbable sutures. Mice were aged to 14 weeks, at which point they were euthanized for further analysis.
qPCR
Mouse tissues were lysed in TRIzol for RNA extraction according to manufacturer’s instructions. 2 μg of RNA were reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR reactions used FastStart Taqman Probe Master Mix (Roche), a 7500 Real-Time PCR SDS System (Applied Biosystems), and FAM-labeled TaqMan primer/probe mixes specific to the gene of interest (Applied Biosystems). Gene expression in mouse studies was normalized to Cpsf2-Vic multiplexed within the same well.
For patient samples, total RNA was extracted with TRIzol (Thermo Fisher Scientific), and RNA was reverse transcribed using the iScript Reverse Transcription Supermix (1708841 Bio-Rad) following the manufacturer’s instructions. Gene expression was measured by RT-qPCR using the SsoAdvanced Universal Sybr green supermix (1725274 Bio-rad) and the C1000 Touch Thermal Cycler–CFX96 Real-Time System (Bio-Rad). Human gene expression was normalized to Actin. Mouse TaqMan Primer/Probes and human primer sequences used are listed below:
Mouse qPCR TaqMan primer/probes | |
|---|---|
Gene | TaqMan primer/probe |
MuRF1 (Trim63) | Mm01185221_m1 |
Atrogin-1(FBXO32) | Mm00499523_m1 |
MuSA1 (FBXO30) | Mm01191299_m1 |
MSTN | Mm01254559_m1 |
Pax7 | Mm01354484_m1 |
Myf5 | Mm00435125_m1 |
Myog | Mm00446194_m1 |
PGAM2 | Mm01187768_m1 |
CLCN1 | Mm00658624_m1 |
PFKM | Mm01309576_m1 |
Myl1 | Mm00659043_m1 |
Myoz1 | Mm00469642_m1 |
Ttn | Mm00621005_m1 |
Mef2a | Mm01318991_m1 |
Mef2c | Mm01340842_m1 |
Cpsf2-VIC | Mm00489754_m1 |
Human qPCR primers | ||
|---|---|---|
Gene | Forward | Reverse |
h_CLCN1 n1 | CCCTGGAGGAGCTACAGAA | TTCGAGTTGAAGTCGTGTTCC |
h_PFKM n1 | CAAAGCCATTGCTGTCTTAACC | GTGAAGATACCAACTCGAACCA |
h_Myoz1 n3 | CTGATCATGGAACTCACTGGAG | GACAGTTCCTCCAACATCACA |
h_Ttn n1 | TCGCCCATAAGACACTCC | TGAGCAATGGAGACCTAACA |
h_Actin | GGACTTCGAGCAAGAGATGG | AGCACTGTGTTGGCGTACAG |
Protein isolation and western blot
Tissues were digested in RIPA buffer (Teknova) containing cOmplete Mini Protease Inhibitor Cocktail (Roche) and 5 mM NEM (MilliporeSigma). Tissue was lysed mechanically using a benchtop tissue homogenizer. Muscle lysates were incubated on a rocker at 4 °C for 1 h to allow for complete digestion, then cleared by spinning at 12,000 g for 10 min at 4 °C. Protein concentration was calculated using the DC assay (Bio-Rad). Equal amounts of protein were loaded into NuPAGE 4–12%, 10-well gels (Invitrogen) and run in 1 × MOPS buffer (Invitrogen). Gels were transferred to PVDF membranes using a Semi-Dry Transfer System (Bio-Rad). Blots were blocked for 1 h in 5% nonfat dry milk in TBS containing 0.1% Tween and placed into primary antibodies in blocking solution at 4 °C overnight. Goat anti-rabbit HRP (Bio-Rad) and goat anti-mouse HRP (Bio-Rad) were diluted at 1:5,000 in 5% nonfat dried milk and incubated for 1 h at room temperature. Blots were quantified using ImageStudio Lite (Licor).
Skeletal muscle processing
Muscle was harvested from euthanized mice and placed in a vessel containing OCT. The OCT vessel was frozen in methylbutane cooled by dry ice. Tissue was cryosectioned using a Leica CM1900 Cryostat (Leica Biosystems) at 20 μm thickness and slides were stored at − 80 °C.
Immunofluorescence
Slides were removed and thawed at room temperature. Tissue slices were outlined with an ImmunoPen (ImmEdge) and fixed in ice cold methanol for 4 min. For muscle fiber quantification, slides were washed three times in PBS and covered in 10 μg/mL labeled wheat germ agglutinin (Thermo) for 10 min at room temperature. For colocalization studies, slides were washed three times in PBS containing 0.1% triton-X (PBS-T) and blocked for 1 h at room temperature in 10% Normal Goat Serum in PBS. Slides were then incubated in primary antibody in blocking solution overnight at 4 °C. Slides were washed three times in PBS-T and incubated in secondary antibody for 1 h at room temperature in blocking solution. Slides were again washed three times in PBS-T and mounted using Prolong Gold with DAPI. Images were taken on a Nikon A1 confocal microscope. Percent of colocalization and muscle fiber size was quantified using automated CellProfiler pipelines and a Zeiss Epifluorescent Microscope.
H&E staining
Slides were fixed in ice cold methanol for 4 min. Sections were placed into hematoxylin (modified Mayer’s solution) (Abcam) for 1.5 min at room temperature. Sections were dipped into distilled water 30 times or until no longer running. Sections were then placed into eosin for 5 min at room temperature. Sections were dipped again 30 times into water, followed by 30 times in 70%, 95%, and 100% ethanol. Finally, sections were mounted with DPX mounting solution and imaged. For H&E images, the white balance was adjusted per image on each whole image to ensure true white levels of background using Adobe Photoshop.
Electroporation of mouse muscle
Electroporation was performed according to previous studies [22]. In brief, mice at indicated ages were anesthetized using isoflurane in a VIP3000 Isoflurane Matrx by nose cone. Following deep inhaled anesthesia at 2% concentration, the hind limb was shaved and 30 μL of 0.8 U/mL hyaluronidase in sterile saline was injected into the tibialis anterior. Mice were then allowed to recover for 2 h, then re-anesthetized and the TA was injected with 50 μg total of plasmid DNA. The hindlimb was then coated in ultrasound gel (Medline). Ten 20 ms pulses of 175 V/cm were then administered to the hindlimb using an ECM 830 Electroporator (BTX Technologies).
Denervation
Under inhaled isoflurane anesthesia, the right sciatic nerve was exposed inferior to the sciatic notch as previously described [26]. The proximal and distal sides were ligated using monocryl 4–0 sutures and 2 mm of nerve was cut in between these ligations, preventing axonal regeneration. Tissue was harvested 3 days after surgery for RNA isolation.
Cardiotoxin assay
Mice were injected with cardiotoxin (Millipore, 217503) at 19 weeks of age. One leg was injected with 30 μL of 12 μM cardiotoxin, while the contralateral leg was injected with saline. Mice were sacrificed at 7 days and 14 days post-injection and the TA muscle was harvested and frozen for processing.
Luciferase assay
Mice were electroporated as described above with a MEF2 promoter-luciferase plasmid and co-transfected with Renilla luciferase as previously described [71]. Muscle was harvested after electroporation, flash frozen in liquid nitrogen, and ground using a pestle and mortar. Luciferase activity was measured using the dual luciferase reporter assay system (Promega). The amount of luciferase activity was normalized to Renilla luciferase activity.
Co-immunoprecipitation
Co-immunoprecipitation was performed following previously established protocols [23]. For co-immunoprecipitation of polyQ AR and HA-MEF2C, a well-established PC12 model was used [77]. These cells express AR112Q under the control of the TetOn promoter. Cells were electroporated using a Neon Electroporator (Thermo) to express HA-MEF2. 24 h after transfection, cells were induced to express polyQ AR with 1 µg/mL of doxycycline and translocation and aggregation was induced by adding 10 nM of R1881, a synthetic testosterone. After 72 h, cells were lysed in CoIP buffer (0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, pH 7.4) by sonication. Aggregated species were pelleted by centrifugation at 15,000g for 15 min. Cleared supernatant was used as input. Input was precleared by incubating on a rotator with Protein A/G beads (Santa Cruz, sc-2003) for 30 min at 4 °C. Input was then incubated with androgen receptor antibody (Millipore, PG-21) or non-immune rabbit IgG (Santa Cruz) overnight at 4 °C on a rotator. Prewashed protein A-agarose beads were added, and samples were rotated for an additional 1 h at 4 °C. Protein-antibody-bead complexes were then washed six times in CoIP buffer. Proteins were eluted by boiling the complexes in SDS-loading buffer for 5 min at 95 °C. Samples were then resolved by western blot as detailed above.
For co-immunoprecipitation of HA-MEF2C and GFP-Q74 Htt, HEK-293 cells were transfected with 1 µg of each plasmid using lipofectamine 2000. After 24 h, cells were lysed in CoIP buffer (0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP-40, 5% glycerol, pH 7.4) and aggregates were pelleted by centrifugation at 15,000g for 15 min. Cleared lysates were treated as above, but instead incubated with either rabbit anti-HA antibody (Cell Signaling, 3724S) or non-immune rabbit IgG (Santa Cruz). Isolated protein–antibody–bead complexes were split in half and run using the western blot methods detailed above. Samples were analyzed in duplicate to allow for resolving both HA-MEF2 and GFP-Q74 Htt. The membrane was cut down the middle. One side was probed with chicken anti-GFP (abcam), and the other side was probed with mouse anti-HA.11 (Biolegend).
Antibodies and reagents
The following antibodies were used: Phospho-S6RP (Cell Signaling, 4858), S6RP (Cell Signaling, 2217), Deptor (Cell Signaling, 11816S), REDD1 (Protein Tech, 10638-1-AP), Vinculin (Sigma, V9131), Phospho-SMAD3 (Cell Signaling, 9520S), SMAD3 (Cell Signaling, 9513), MEF2AC (abcam, ab197070), p62 (03-GP62-C), Androgen Receptor (Millipore, PG-21), Phospho-SMAD3 (abcam, ab52903), Actin (Sigma, A2228), HA (Cell Signaling, 3724S), HA.11 (Biolegend, 901533), GFP (abcam, ab13970), Wheat Germ Agglutinin Alexa Fluor 594 (Thermo Scientific, W11262), Wheat Germ Agglutinin Alexa Fluor 488 (W11261).
Transcriptome data
Sequencing data were used from the following previously published GEO datasets analyzed as stated: SBMA LABC: GSE106521 [53], SBMA Quadriceps: GSE60691 [13], R6/2 Quadriceps: GSE81367 [10], reanalyzed by DeSeq2 (counts > 2 in at least 5 samples, B&H FDR < 0.05, FC > 1.5), MEF2 KD: GSE61207 [78], and Mrf4 KD: GSE67069 [51].
Plasmids
MEF2-VP16 (CA-MEF2) and MEF2ΔDBD-VP16 (DBD-MEF2) were previously characterized and kindly provided by Michael Greenberg [24]. MEF2-Luciferase (RSRF-Luc-2wt) was a gift from Astar Winoto (Addgene plasmid # 31818) [80]. Tk-Renilla plasmid was from Promega. pcDNA3.1-MEF2C-HA was a gift from Andrew Lassar (Addgene plasmid # 32515) [30]. pEGFP-Q74 (pEGFP-Q74 Htt) was a gift from David Rubinsztein (Addgene plasmid # 40262) [52].
Statistics
Statistical analysis was performed in GraphPad Prism using two-tailed unpaired student’s t test for comparisons of two groups and one-way ANOVA with Tukey’s multiple comparison test for groups of three or more. α < 0.05 was set as the threshold for significance.
Results
AR113Q muscle atrophy is independent of ubiquitin–proteasome machinery induction
Skeletal muscle atrophy occurring downstream of denervation, fasting, immobilization, and motor neuron degeneration involves induction of several key E3 ubiquitin ligases and the proteasome [31, 32, 67]. Upregulation of degradative machinery targets sarcomeric proteins to the proteasome and results in fiber size reduction [15]. Three of the best characterized ubiquitin ligases involved in skeletal muscle atrophy are MuRF1, Atrogin-1, and MUSA1 [8, 15, 68]. We confirmed induction of these genes after denervation by comparing mRNA expression levels in control and denervated WT mouse gastrocnemius 3 days following sciatic nerve transection (Fig. 1a).

Atrophy-related E3 ubiquitin ligases are not induced in AR113Q skeletal muscle. a Relative mRNA expression of FBXO32/Atrogin-1, MuRF1, and MuSA1 in WT control (ctrl) and denervated (den) mouse gastrocnemius 3 days following sciatic nerve transection (n = 3/group). b Relative mRNA expression of FBXO32/Atrogin-1, MuRF1, and MuSA1 in AR113Q control (ctrl) and denervated (den) mouse gastrocnemius 3 days following sciatic nerve transection (n = 3/group). c–e Relative mRNA expression of (c) MuRF1, (d) FBXO32/Atrogin-1, (e) MuSA1 in WT or AR113Q mice in quadriceps (Quad) at 14, 26 and 52 weeks (left, n = 6/group) and in LABC at 14 weeks (right, n = 3/group). Data are shown as mean ± SEM. ns not significant (p > 0.05), *p < 0.05; **p < 0.01; ***p < 0.001 by unpaired t test (quadriceps) or by one-way ANOVA with Tukey’s (LABC)
Next, we examined AR113Q mice, a model of SBMA generated by gene targeting, to determine whether mutant males also exhibit induction of the E3 ubiquitin ligases associated with muscle atrophy. AR113Q mice express a humanized form of AR in which much of the coding sequence of mouse AR exon 1 is replaced by human sequence containing 113 CAG repeats [83, 84]. These mice express endogenous levels of polyQ AR under the control of the mouse AR gene promoter and transcriptional machinery. AR113Q males develop a progressive neuromuscular phenotype which mimics important aspects of the SBMA phenotype (Supplemetary Fig. 1) [83]. Because the AR gene is on the X chromosome, males are hemizygous for either the mutant or WT allele. Using AR113Q mice, we performed the same denervation experiment at 26 weeks and examined the gastrocnemius 3 days post-denervation (Fig. 1b). While AR113Q mice also showed significant induction of Atrogin-1 and MuSA1, MuRF1 was not significantly induced, and the amount of induction of all three genes was blunted compared to WT mice. This indicates that SBMA mice are impaired in their ability to induce the E3 ubiquitin ligases responsible for atrophy following denervation.
We next examined expression of these E3 ubiquitin ligases in AR113Q mice across multiple time points. In contrast to denervation, we found no significant induction of any of these E3 ubiquitin ligases in AR113Q quadriceps at 14, 26, and 52 weeks (Fig. 1c–e), despite the occurrence of progressive weakness and muscle atrophy (Supplementary Fig. 1). In fact, AR113Q males display age-dependent suppression of the MuRF1 and Atrogin-1 genes (Fig. 1c, d). These data complement a recent study from our group demonstrating diminished proteasome expression and function in AR113Q muscle and show that SBMA atrophy has several distinctions from other forms of atrophy [53].
Similar to quadriceps muscle, induction of MuRF1, Atrogin-1, and MuSA1 does not occur in the levator ani/bulbocavernosus muscle (LABC), a pelvic floor muscle which undergoes severe atrophy due to high levels of AR expression (Fig. 1c–e, right). We observed no induction of E3 ubiquitin ligases in comparison to either WT or AR21Q males. (AR21Q males were generated using the same gene targeting strategy but express a normal CAG repeat length [1] and thereby control for effects of humanizing the mouse AR gene). Previous work has demonstrated that AR21Q mice are phenotypically similar to WT, establishing that the neuromuscular phenotype of AR113Q mice is due solely to polyglutamine tract expansion above the pathogenic threshold [83]. Collectively, these analyses demonstrate that AR113Q mice lack induction of the classical, ubiquitin–proteasome system (UPS)-based muscle atrophy machinery and suggest that alternative mechanisms underlie muscle atrophy in this disease.
Hypertrophic signaling is increased in SBMA muscle
We next sought to determine whether impaired signaling to stimulate muscle hypertrophy contributes to AR113Q atrophy. Two major signaling pathways, regulated by mTOR and myostatin, are well established in modulating skeletal muscle hypertrophy (Fig. 2a) [71]. As reduced mTOR activity is a well-established hallmark of muscle atrophy [9, 15, 55], we hypothesized that mTOR signaling may be impaired in SBMA and contributes to reduction of muscle fiber size. We examined phosphorylation of S6RP, a well-established mTORC1 target, in 14-week LABC. In contrast to expectation, we found significant upregulation of mTOR signaling in AR113Q muscle (Fig. 2b), similar to a previous report [66]. This signal was ablated by administration of rapamycin for 2 weeks, demonstrating specificity for mTOR activation (Fig. 2b). We next sought to determine if upstream regulators of mTOR were differentially affected in AR113Q mice. Two well-characterized mTOR inhibitors are Deptor [59] and Redd1 [11, 73]. AR113Q muscle displays significant reduction in levels of both Deptor and Redd1 proteins, demonstrating that loss of inhibitors coincides with significant mTOR activation (Fig. 2c). These results demonstrate that mTOR signaling is activated to favor hypertrophy in AR113Q muscle and led us to examine whether other effectors of muscle hypertrophy are dysregulated in disease.

Activity of signaling pathways that influence fiber size in AR113Q muscle. a Schematic of signaling pathways which regulate muscle regeneration and hypertrophy (inhibitory in magenta, stimulatory in cyan). b Levels of phospho-S6RP in WT vs AR113Q LABC at 14 weeks, following 2-week treatment with vehicle (Veh) or rapamycin (Rap) I.P. (n = 3–4 mice/group) *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA with Tukey’s post-hoc test (c) Relative levels of Deptor and REDD1 proteins in WT vs AR113Q LABC muscle at 14 weeks (n = 3/group). Data are mean ± SEM. **p < 0.01 by unpaired t test. d, e Relative mRNA expression of myostatin (MSTN) in WT or AR113Q quadriceps at 14, 26, and 52 weeks (d, n = 6/group) and LABC at 14 weeks (e, n = 3/group). f Levels of phospho-SMAD3 in WT and AR113Q quadriceps at 26 weeks (n = 3/group). d Data are mean ± SEM. ns not significant (p > 0.05), *p < 0.05, **p < 0.01 by unpaired t test. e Data are mean ± SEM. *p < 0.05, **p < 0.01 by one-way ANOVA with Tukey’s post hoc test. f Data are mean ± SEM. *p < 0.05 by unpaired t test
Myostatin is known to be a potent inhibitor of skeletal muscle hypertrophy [45]. To determine whether myostatin induction plays a role in AR113Q muscle atrophy, we performed qPCR on 14, 26, and 52 week quadriceps. We observed no change in myostatin expression in the quadriceps at early time points but significant suppression of myostatin expression in 52 week quadriceps (Fig. 2d) and 14 week LABC (Fig. 2e). To support the notion that myostatin signaling is diminished in AR113Q muscle, we examined the level of phospho-SMAD3, a downstream effector of myostatin. We detected an age-dependent, significant decrease in phospho-SMAD3 in AR113Q muscle, supporting the conclusion that myostatin signaling is suppressed (Fig. 2f, Supplementary Fig. 2A, 2B). As both myostatin and mTOR signaling changed to favor hypertrophy rather than atrophy in mutant muscle, we conclude that alterations in these pathways do not underlie AR113Q skeletal muscle pathology.
Muscle regeneration is intact in SBMA muscle
We next asked whether the cells that replenish muscle, satellite cells and myoblasts, are dysfunctional in AR113Q males. We performed qPCR for markers of muscle satellite cells, Pax7 and Myf5 [18, 27], and the myoblast marker myogenin (Myog) (Fig. 3a). For all these markers, we saw increases in expression, with Myf5 and Myog increasing as early as 14 weeks, suggesting that these cell populations are present in AR113Q skeletal muscle (Fig. 3b). Moreover, the induction of Pax7 and Myf5 expression was significant compared to AR21Q (Fig. 3c), demonstrating that upregulation of satellite cell markers is dependent on glutamine tract expansion.

Myoregeneration is intact in AR113Q muscle. a Schematic of examined markers of muscle satellite cells (PAX7, Myf5) and myoblasts/myocytes (MyoG). b, c Relative mRNA expression of PAX7 (top), Myf5 (middle) and MyoG (bottom) in WT and AR113Q quadriceps at 14, 26, and 52 weeks (b, n = 6/group) and in WT, AR21Q, and AR113Q LABC at 14 weeks (c, n = 3/group). d–f Tibialis anterior of WT or AR113Q mice injected with cardiotoxin (CTX) or saline at 19 weeks and examined by H&E stain at 7 days (d) or 14 days (e) post-injection. Scale bar = 20 µm. (f, Left) Muscle fibers were stained with Wheat Germ Agglutinin (WGA) to delineate muscle fiber membranes and DAPI at 14 days post-injection. Fiber size and nuclei/fiber were quantified (f, right) at 14 days post-injection and are displayed as a violin plot (n = 3 mice/group, 3 images/mouse, > 100 fibers/mouse). Scale bar = 50 µm. ns not significant (p > 0.05), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by unpaired t test (b), one-way ANOVA with Tukey’s post hoc test (c), and two-way ANOVA with Tukey’s post hoc test (f, left and right panel)
We sought to determine whether satellite cell dysfunction and impaired regeneration after injury contributes to AR113Q muscle atrophy. To accomplish this, we injected cardiotoxin (CTX) into the tibialis anterior muscle, leading to severe muscle damage and fiber destruction [6, 58]. In WT mice, muscle fibers are replenished to their original size by satellite cells 12–14 days after CTX administration [28, 58]. Significant destruction of muscle fibers was present in both WT and AR113Q mice at 7 days post-CTX injection (Fig. 3d), and both WT and AR113Q mice successfully regenerated muscle fibers by 14 days post-injection (Fig. 3e). Following regeneration, fibers trended larger for WT mice and were slightly, but significantly larger (6%) for AR113Q mice than their respective original sizes (Fig. 3f, left), demonstrating that AR113Q mouse satellite cells remain functional. Although fibers successfully regenerated to their original size, subtle changes in satellite cell function were suggested by the reduction in nuclei per fiber in SBMA mouse muscle (Fig. 3f, right). Similar changes have been reported in primary satellite cell cultures from SBMA patients [42]. As these defects do not prevent fiber regeneration in vivo post-injury by the muscle stem cell population, we conclude that they are unlikely to significantly contribute to AR113Q atrophy. Having determined that UPS-based degradation, impaired hypertrophic signaling, and regenerative capability do not account for reduced muscle fiber size in AR113Q mice, we sought out alternative factors which may contribute to muscle atrophy in disease.
MEF2 target expression is decreased in AR113Q and SBMA patient skeletal muscle
Using an RNAseq data set generated from AR113Q mouse quadriceps at 14 weeks [14] and GO Term: Cellular Component analysis of differentially expressed genes, we identified that all of the top five enriched categories are related to sarcomere structural genes (Table 1). We performed transcription factor-binding site analysis using ToppGene to nominate transcription factors that could account for this loss of sarcomere gene transcription. Strikingly, we found that binding sites for the transcription factor Myocyte Enhancer Factor 2 (MEF2) account for 5 of the top 5 and 4 of the top 5 hits when analyzing differentially expressed genes in the quadriceps and LABC, respectively (Table S1-S2). Gene Set Enrichment Analysis (GSEA) also showed MEF2-binding sites accounting for 5 of the top 15 transcription factor-binding sites, and as the only transcription factor with multiple hits (Table S3). These results were unexpected, as MEF2 mRNA expression (Supp Fig. 3a) and protein level (Supplementary Fig. 3B) were not altered. RNAseq data also showed no significant change in expression of any Mef2 family gene (Table S4). To test whether MEF2 activity is impaired in AR113Q muscle, we electroporated a plasmid containing a MEF2 promoter site upstream of luciferase into mouse hindlimb muscle. Luciferase activity was examined 48 h post-electroporation and normalized to co-transfected, constitutively expressed Renilla luciferase. We found significant reduction of MEF2 activity in AR113Q muscle (Fig. 4a) without a difference in Renilla luciferase activity (Supplementary Fig. 3C), consistent with impaired MEF2 activity in vivo. We next compared differentially expressed genes in 14 week AR113Q quadriceps and LA/BC to a published data set of MEF2A targets in C2C12 cells which were identified by RNAseq and confirmed by MEF2A ChIP-Exo [78]. Strikingly, ~ 20% of identified MEF2 targets were altered in AR113Q quadriceps and ~ 40% of targets in AR113Q LABC (Fig. 4b). We further compared differentially expressed genes in 14 week AR113Q quadriceps and LA/BC muscles to another data set in which the MEF2 inhibitor Mrf4 was knocked down in rat tibialis anterior muscle by electroporation at 6 weeks of age and expression analysis was performed in the context of activated MEF2 [51]. We found that ~ 10% of all genes that changed upon Mrf4 knockdown were altered in AR113Q quadriceps; similarly, ~ 30% were altered in AR113Q LABC (Fig. 4c).

Diminished MEF2 activity in AR113Q muscle. a Tibialis anterior of 26-week WT and AR113Q mice were co-transfected with MEF2-luciferase and constitutively expressed Renilla luciferase. MEF2-luciferase activity was measured and normalized to renilla luciferase activity (n = 4/group). Data are mean ± SEM. *p < 0.05 by unpaired t test. b Differentially expressed genes (AR113Q vs WT) from 14 week LA/BC and quadriceps of AR113Q mice were compared to a published set of MEF2 target genes identified by gene knockdown (KD) and validated by ChIP-exo in C2C12 cells. p = 6.21e26 by hypergeometric test for Quad overlap with MEF2 KD and p = 2.67e27 by hypergeometric test for LABC overlap with MEF2 KD. c Differentially expressed genes (AR113Q vs WT) from 14 week LA/BC and quadriceps of AR113Q mice were compared to a published set of gene expression changes downstream of knockdown of Mrf4, a MEF2 repressor protein, in rat tibialis anterior at 6 weeks of age. p = 2.12e−68 for LABC overlap with Mrf4 KD and p = 5.20e−36 for Quad overlap with Mrf4 KD d Heatmap showing fold change of 161 overlapping genes between AR113Q Quad and Mrf4 KD. Three comparisons to WT are displayed: AR113Q Quad, AR113Q Quad from mice treated with antisense oligonucleotides, and tibialis anterior from WT mice following Mrf4 KD. e Relative expression of MEF2 targets PFKM, Ryr1, Myl1, CLCN1, Myoz1 and PGAM2 was measured in WT and AR113Q mice at 14 (left), 26 (middle), and 52 (right) weeks in quadriceps (n = 5–6/group). f SBMA patient and control (Ctrl) muscle biopsies were examined for expression of MEF2 targets CLCN1 (n = 3v6), Myoz1 (n = 3v4), PFKM (n = 3v6), and TTN (n = 3v6) by qPCR. Data are shown as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001 by unpaired t test
We also analyzed genes significantly altered in both the quadriceps and LABC of 14 week AR113Q mice (Supplementary Fig. 4A). Of these 552 genes, we found that ~ 15% of published genes that changed with MEF2 knockdown and were validated by ChIP-Exo in C2C12 cells were part of this subset (Supplementary Fig. 4B). Additionally, ~ 20% of these 552 genes overlap with targets responsive to Mrf4 knockdown in 6-week rat tibialis anterior (Supplementary Fig. 4C).
Notably, knockdown of AR113Q in vivo using peripherally administered AR-targeted antisense oligonucleotides rescues changes in MEF2 target genes (Fig. 4d, Table S5) [38]. Importantly, this intervention diminishes expression of polyQ AR in peripheral tissues but not CNS [38], indicating that MEF2 impairment occurs independent of polyQ AR actions in lower motor neurons. To validate changes in MEF2-regulated genes in an independent cohort of mice, we examined expression of several MEF2 targets (genes found in MSigDB, MEF2-ChIP-Seq, or Mrf4 KD data sets) known to play an important role in skeletal muscle in AR113Q quadriceps at 14, 26, and 52 weeks. These studies demonstrated a significant decrease in target gene expression that often progressed with age (Fig. 4e).
We next sought to determine whether changes in expression of MEF2 target genes also occur in SBMA patients. To accomplish this, we performed qPCR on a cohort of SBMA skeletal muscle biopsies (Table S6). We found significant reduction of these targets in patients versus controls (Fig. 4f) without loss of MEF2 expression (Supplementary Fig. 4D), suggesting that MEF2 impairment is conserved between mouse models of disease and SBMA patients.
Diminished MEF2 target expression is caused by hormone- and polyglutamine length-dependent gain of function
Expansion of the AR’s polyQ tract leads to partial loss of its normal function as a transcriptional regulator. In addition, the mutant protein undergoes hormone-dependent misfolding and aggregation resulting in gain of function changes crucial to the neuromuscular phenotype. To determine whether loss of MEF2 targets is dependent on the polyglutamine tract expansion, we compared gene expression in WT, AR21Q, and AR113Q muscle. AR21Q mice are phenotypically similar to WT mice, demonstrating that the neuromuscular phenotype is solely due to the polyQ expansion [1]. We found that loss of MEF2 target gene expression, like the neuromuscular phenotype, is dependent on the expanded glutamine tract and not the humanized sequence of exon 1 (Fig. 5a).

AR113Q muscle shows hormone- and Q tract length-dependent MEF2 impairment and sequestration. a–c Relative mRNA expression of MEF2 targets was measured in the LABC of a WT, AR21Q, and AR113Q mice at 14 weeks of age. (n = 3/group), b WT mice at 14 weeks of age compared to WT mice castrated at 6 weeks and allowed to age to 14 weeks (WT C). (n = 3–5/group), and c AR113Q mice at 14 weeks of age compared to AR113Q mice castrated at 6 weeks and allowed to age to 14 weeks (AR113Q C) (n = 3–5/group) d Co-localization of p62 and MEF2A/C in LABC of WT and AR113Q mice at 14 weeks of age, quantified at right (n = 3 mice/group). Data are shown as mean ± SEM. Scale bar = 10 µm. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001 by unpaired t test (a, b, d) or one-way ANOVA with Tukey’s (c)
We next sought to establish whether impaired MEF2 function is due to loss of AR activity or hormone-dependent proteotoxicity. To determine this, we castrated WT males at 5–6 weeks and allowed them to age to 14 weeks. This manipulation eliminates testosterone production and abrogates AR signaling. We found that castration does not alter expression of MEF2-regulated genes (Fig. 5b) suggesting that loss of AR signaling does not impair MEF2 function. This conclusion is corroborated by the observation that ASO treatment rescues expression of MEF2 targets (Fig. 4d). As ASOs diminish AR levels, changes due to loss of function are exacerbated by treatment, while those due to gain of function proteotoxicity are ameliorated. The finding that MEF2 targets are rescued by ASO administration supports the notion that loss of MEF2 function is due to polyglutamine-mediated toxicity.
Next, we castrated AR113Q males. Castration prevents hormone-dependent misfolding and aggregation of polyQ AR and abrogates the neuromuscular phenotype of AR113Q mice [83]. We found that castration also rescues expression of MEF2 target genes (Fig. 5c) indicating that, like the motor phenotype, MEF2 impairment results from hormone-dependent proteotoxicity. Together, these results demonstrate that MEF2 impairment occurs in a gain of function, polyQ tract-dependent manner.
AR113Q misfolding is associated with the formation of intranuclear inclusions in skeletal muscle nuclei [83]. We performed immunofluorescence using a MEF2A/C antibody to determine whether MEF2 is also mislocalized in AR113Q muscle. While MEF2 signal was diffuse and intranuclear in WT muscle, we identified robust colocalization of MEF2 with intranuclear p62 positive puncta, a marker of inclusions formed in AR113Q muscle (Fig. 5d). Similar staining was observed using antibodies against MEF2A and MEF2C (Supplementary Fig. 3E–F). These data demonstrate MEF2 sequestration into intranuclear inclusions in mutant muscle.
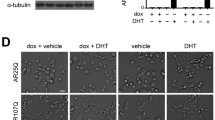
Having found that MEF2 is sequestered into aggregates in AR113Q muscle, we next asked whether there are properties of MEF2 that would suggest a high propensity for co-aggregation. Intrinsic disorder is a necessary property of many proteins and protein regions, as native disorder allows for responses to external stimuli and post-translational modifications [29]. However, intrinsic disorder in a protein is also a key modulator of aggregation [20], with intrinsically disordered proteins implicated in many neurodegenerative diseases characterized by protein misfolding and aggregation [76]. Using prediction pipelines (DisEMBL, FoldINDEX, DISpro, and PONDR-FIT), the sequence of MEF2C was found to contain regions of significant predicted disorder in the transactivating domain (Supplementary Fig. 4G). These findings suggest that MEF2 has structural features that confer an increased likelihood for co-aggregation. Moreover, co-immunoprecipitation experiments demonstrated interaction of soluble polyQ AR with MEF2 (Supplementary Fig. 4H–I) indicating that both soluble and aggregated polyQ species interact with MEF2.
Polyglutamine huntingtin sequesters MEF2 in skeletal muscle and decreases MEF2 target gene expression
As our findings suggest that polyQ expansion underlies MEF2 impairment and sequestration, we hypothesized that these mechanisms are similarly active in other models of polyQ disease that exhibit skeletal muscle atrophy. To test this hypothesis, we studied R6/2 mice. These mice express an N-terminal fragment of polyQ huntingtin (HTT), develop age-dependent skeletal muscle atrophy, and form intranuclear inclusions in skeletal muscle [10, 12]. We utilized a previously published RNAseq data set of R6/2 quadriceps at 12 weeks of age to determine whether polyQ HTT also alters MEF2 target gene expression [10]. This analysis revealed 2715 differentially expressed genes between WT and R6/2 muscle. We found significant overlap between differentially expressed genes from R6/2 quadriceps at 12 weeks and effects of MEF2 knockdown in C2C12 cells (Fig. 6a) and Mrf4 (Fig. 6b) knockdown in 6 week rat tibialis anterior.

Diminished MEF2 function and sequestration in R6/2 muscle. a Differentially expressed genes from 12 week quadriceps of R6/2 mice and 14 week quadriceps of AR113Q mice were compared to a published set of MEF2 target genes identified by gene knockdown (KD) and validated by ChIP-exo in C2C12 cells. p = 9.27e−07 for MEF2 KD vs R6/2 overlap b Differentially expressed genes from 12 week quadriceps of R6/2 mice and 14 week quadriceps from AR113Q mice were compared to a published set of gene expression changes downstream of knockdown of Mrf4, a MEF2 repressor protein, in rat tibialis anterior at 6 weeks of age. p = 6.69e−09 for Mrf4 KD vs R6/2 overlap. c Heatmap showing fold change of 280 overlapping genes in 12 week R6/2 Quad and Mrf4 KD TA. (D) R6/2 mice were examined for gene expression changes in quadriceps at 4, 8, and 12 weeks of age (n = 8–10/group for 4, 8 weeks, n = 9 WT 12 week, n = 7 R6/2 12 week). e Co-localization of p62 and MEF2A/C in quadriceps of WT and R6/2 mice at 12 weeks of age, quantified at right. Data are shown as mean ± SEM. Scale bar = 10 µm. *p < 0.05; **p < 0.01; ***p < 0.001, ****p < 0.0001 by unpaired t test
To examine conserved changes between both polyQ models, we additionally compared 14 week AR113Q quadriceps with 12 week R6/2 quadriceps and identified 481 genes differentially expressed in both (Supplementary Fig. 5A). We then compared these 481 genes to the published set of MEF2 targets validated by MEF2 knockdown and ChIP-Exo in C2C12 cells and found ~ 10% of these genes overlapped (Supplementary Fig. 5B). Additionally, when compared to genes that change with knockdown of the negative regulator of MEF2, Mrf4, in 6 week rat tibialis anterior, ~ 20% of the 481 genes overlapped (Supplementary Fig. 5C). Together, these analyses indicate significant dysregulation of MEF2 targets in polyQ muscle.
Notably, knockdown of Mrf4 shows changes opposite to those observed in R6/2 quadriceps, supporting the concept of impaired MEF2 activity downstream of polyQ HTT (Fig. 6c, Table S7). We confirmed diminished expression of MEF2 target genes in an independent cohort of R6/2 mice by qPCR. We found progressive changes, with three targets showing diminished expression at 4 weeks, four targets at 8 weeks, and all six targets at 12 weeks of age (Fig. 6d). Moreover, using immunofluorescence we found significant sequestration of MEF2 into skeletal muscle inclusions, with MEF2 colocalizing to ~ 85% of p62 positive inclusions (Fig. 6e). Co-immunoprecipitation demonstrated interaction between GFP-74Q Htt and HA-MEF2C, indicating that MEF2 interacts not only with soluble polyQ AR but also with soluble polyQ Htt (Supplementary Fig. 5D–F).
Restoring MEF2 activity rescues AR113Q muscle atrophy.
To determine the extent to which loss of MEF2 function contributes to AR113Q muscle atrophy, we tested whether adding back constitutively active MEF2 (CA-MEF2) would rescue muscle fiber size [24, 44, 51]. This CA mutant consists of the MEF2 DNA-binding domain fused to the VP16 transactivating domain and estrogen receptor hormone binding domain; nuclear translocation and activity of this mutant is controlled by tamoxifen (Supplementary Fig. 6A) [24, 51]. In addition to its constitutively active nature, this construct has much less predicted disorder, suggesting less propensity for co-aggregation (Supplementary Fig. 6B). Consistent with this, when transiently expressed in cell culture HA-MEF2 localized to intranuclear polyQ AR aggregates (Supplementary Fig. 6C), whereas CA-MEF2 did not (Supplementary Fig. 6D). At 19 weeks of age, after AR113Q, mice show significant muscle atrophy (Supplementary Fig. 1, Fig. 3f), we electroporated CA-MEF2 into tibialis anterior (TA) muscle of one hind limb. The contralateral TA was electroporated with VP16-MEF2 containing a deletion in the DNA-binding domain (DBD-MEF2), thereby preventing transcriptional activity [24, 51]. Importantly, DBD-MEF2 does not alter muscle fiber size [51]. Following electroporation, mice were administered tamoxifen every other day for 7 days prior to harvesting muscle. We found a 24% increase in muscle fiber size owing to expression of CA-MEF2 in AR113Q muscle (Fig. 7a–d). Electroporated CA-MEF2 was predominantly diffuse within the nucleus of skeletal muscle and did not colocalize with p62 + intranuclear inclusions (Supplementary Fig. 6E). Notably, this rescue of muscle fiber size was accompanied by increased expression of MEF2 target genes, including many of the same genes reduced in SBMA patient muscle biopsies (Fig. 7e). Strikingly, restoration of MEF2 activity also rescued expression of critical genes mediating metabolic dysfunction in SBMA, an important disease phenotype characterized by diminished carbohydrate metabolism (Fig. 7f) [26, 66]. Taken together, these results demonstrate rescue of muscle atrophy by restoring MEF2 activity and target gene expression in AR113Q mice.

CA-MEF2 rescues AR113Q muscle atrophy and target gene expression. a–d AR113Q mice were transfected with CA-MEF2 in one leg and DBD-MEF2 in the contralateral leg. Each plasmid was co-transfected with GFP for fiber identification. After 24 h, mice were injected with tamoxifen every other day for 7 days.a Tibialis anterior was harvested and imaged for GFP (green), Wheat Germ Agglutinin (WGA) (red) and DAPI (blue). Scale bar = 50 μM. b Tibialis anterior was stained using Hematoxylin and Eosin (H&E). Scale bar = 50 μM. c, d Quantification of cross sectional area of transfected fibers (n = 3mice/group, 3 images/mouse, n > 50 fibers/mouse) is shown as a histogram (c) and mean ± SEM (d), ****p < 0.0001 by unpaired t test. e, f AR113Q muscle expressing DBD-MEF2 and CA-MEF2 was examined for expression levels of e MEF2 targets found to be altered in SBMA patients Ttn, MyoZ1, Pfkm, Clcn1 and fgenes that regulate metabolism including Gapdh, Hk2, and Nur77 (n = 6 mice/group). Data are mean ± SEM. *p < 0.05; ***p < 0.001 by unpaired t test
Discussion
We demonstrate a novel mechanism of skeletal muscle atrophy mediated by toxic proteins with expanded polyQ tracts, resulting in impaired function of the transcriptional regulator MEF2. The MEF2 family of transcription factors consists of four members, with Mef2a, Mef2c, and Mef2d most abundantly expressed in skeletal muscle (Table S4). Prior research demonstrates that MEF2A, -C, and -D bind the same consensus sequence [82]. Additionally, MEF2A, -C, and -D are capable of compensating for knockout of one but not all 3 Mef2 genes in satellite cells, suggesting redundant function of MEF2 family members in skeletal muscle [39]. Similar compensation of MEF2 family members has been demonstrated in the CNS, suggesting that functional redundancy is conserved between organ systems [41]. Mef2c expression is known to make important contributions to sarcomere integrity and maintenance [61]. Supporting this notion, MEF2 proteins regulate the expression of a large number of genes encoding structural proteins of the sarcomere [51, 78], and skeletal muscle-specific deletion of Mef2c leads to rapid deterioration of myofibers and disorganized sarcomeres [61]. Additional studies have demonstrated that postnatal growth is impaired by skeletal muscle-specific Mef2c deletion in mice [3]. Moreover, activation of MEF2 by silencing of the repressor Mrf4 or by overexpression of the constitutively active form, drives skeletal muscle hypertrophy in vivo, defining MEF2 as a key regulator of muscle hypertrophy [51]. In support of this role, knockout of another repressor of MEF2 activity, NCoR1, in mouse skeletal muscle also leads to MEF2 activation and skeletal muscle hypertrophy [81]. Additional studies have confirmed a role for MEF2 in cardiac [19] and smooth muscle hypertrophy [33], suggesting a conserved role between muscle types. Together, these studies have established MEF2 as a critical regulator of muscle homeostasis.
Here, we show that diminished MEF2 function occurs in atrophic muscles of SBMA mice (Figs. 4, 5), Huntington disease mice (Fig. 6), and SBMA patients (Fig. 4f). Evidence supporting this conclusion includes diminished activity of a MEF2-luciferase reporter in AR113Q muscle (Fig. 4a) and decreased expression of MEF2-regulated genes in AR113Q (Fig. 4b–e) and R6/2 mice (Fig. 6a–d) as well as in SBMA patients (Fig. 4f). Expression of CA-MEF2 rescues AR113Q muscle atrophy and target gene expression (Fig. 7b–e), indicating the importance of these changes to disease pathogenesis and suggesting that targeting MEF2 function may be a novel and unexplored therapeutic avenue. Notably, impaired MEF2 activity occurs without concurrent alterations in the activity of the canonical UPS-mediated atrophy pathway (Fig. 1) [53], disruption of signaling pathways that influence muscle hypertrophy (Fig. 2), or significant impairment of the muscle stem cell niche (Fig. 3). As such, toxic polyglutamine proteins trigger muscle atrophy through a distinct and previously uncharacterized mechanism.
MEF2 activity is influenced by post-translational modifications, including acetylation and phosphorylation, which can enhance DNA binding and activity [40, 48, 85]. Several proteins have been identified as inhibiting MEF2 activity, either by direct binding or localization to MEF2-binding sites, including HDAC4 [16], Mrf4 [51], and NCoR1 [81]. Interestingly, MEF2 expression and nuclear translocation are not altered in AR113Q muscle, but its nuclear localization is disrupted by interaction with soluble polyQ AR and sequestration into insoluble inclusions, suggesting that co-aggregation is a final consequence of aberrant interaction with polyQ AR. Similar findings are observed in R6/2 muscle (Fig. 6), where the HDAC4-myogenin axis has been shown to be re-expressed [47]. This sequestration may reflect the fact that the MEF2C transactivating domain is intrinsically disordered and prone to aggregation (Supplementary Fig. 4G). These findings align with our observation that MEF2 impairment is mediated by polyQ AR proteotoxicity rather than loss of function (Fig. 5). Whether sequestration of MEF2 into morphologically visible inclusions is necessary for functional impairment is uncertain; it is possible that its aberrant association with soluble polyQ species in the nucleus may also disrupt activity and that sequestration into protein aggregates is the end stage of this process. Moreover, the extent to which sequestration of MEF2 into polyQ AR aggregates occurs in SBMA patient muscle remains to be determined; access to human tissue to perform this analysis was hampered by COVID-19 triggered laboratory shutdowns and, therefore, these experiments remain a future priority.
Although MEF2 represents a promising therapeutic target, limitations remain in leveraging this pathway in patients. While MEF2 is able to restore fiber size in atrophic muscle of adult SBMA mice, whether this effect is sustained during long-term treatment is not yet known. Furthermore, because MEF2 plays key roles in skeletal [51], cardiac [19], and smooth muscle [33] as well as in the nervous system [4, 24, 25, 44], therapeutically targeting MEF2 specifically in skeletal muscle remains a significant challenge. To overcome these limitations, we propose that in addition to direct modulation of MEF2, a strategy by which mutant AR is targeted to prevent aggregation and sequestration of MEF2, such as by administration of antisense oligonucleotides [38] or modulation of the HSP70/90 chaperone machinery [23, 79] may be synergistic as a long-term therapeutic.
The sequestration and functional impairment of transcriptional regulators, including cAMP response element-binding protein (CBP) [75], specific protein-1 (Sp1) [21, 35], and TAFII130 [21], has been implicated in neuronal dysfunction in polyQ disease. Our data suggest that a similar mechanism disrupts activity of MEF2, a critical regulator of sarcomere maintenance, to trigger skeletal muscle pathology. As MEF2 is also strongly implicated in activity-dependent neuronal survival [4, 24, 25, 44], we hypothesize that this mechanism of MEF2 sequestration and loss of function may additionally contribute to CNS pathology in polyQ diseases. It is intriguing to speculate that disease-specific disruptions of certain transcriptional regulators may contribute to cell-type vulnerability. In this regard, our observation of impaired MEF2 function in skeletal muscle provides both new mechanistic insight and identification of a therapeutic target for polyQ-mediated muscle atrophy.
References
Albertelli MA, Scheller A, Brogley M, Robins DM (2006) Replacing the mouse androgen receptor with human alleles demonstrates glutamine tract length-dependent effects on physiology and tumorigenesis in mice. Mol Endocrinol 20:1248–1260. https://doi.org/10.1210/me.2006-0021
Amato AA, Prior TW, Barohn RJ, Snyder P, Papp A, Mendell JR (1993) Kennedy's disease: a clinicopathologic correlation with mutations in the androgen receptor gene. Neurology 43:791–794. https://doi.org/10.1212/wnl.43.4.791
Anderson CM, Hu J, Barnes RM, Heidt AB, Cornelissen I, Black BL (2015) Myocyte enhancer factor 2C function in skeletal muscle is required for normal growth and glucose metabolism in mice. Skelet Muscle 5:7. https://doi.org/10.1186/s13395-015-0031-0
Andzelm MM, Vanness D, Greenberg ME, Linden DJ (2019) A late phase of long-term synaptic depression in cerebellar purkinje cells requires activation of MEF2. Cell Rep 26(1089–1097):e1083. https://doi.org/10.1016/j.celrep.2019.01.004
Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M et al (2006) Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain 129:1446–1455. https://doi.org/10.1093/brain/awl096
Attia M, Maurer M, Robinet M, Le Grand F, Fadel E, Le Panse R et al (2017) Muscle satellite cells are functionally impaired in myasthenia gravis: consequences on muscle regeneration. Acta Neuropathol. https://doi.org/10.1007/s00401-017-1754-2
Bodine SC, Baehr LM (2014) Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am J Physiol Endocrinol Metab 307:E469–484. https://doi.org/10.1152/ajpendo.00204.2014
Bodine SC, Latres E, Baumhueter S, Lai VK, Nunez L, Clarke BA et al (2001) Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294:1704–1708. https://doi.org/10.1126/science.1065874
Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R et al (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3:1014–1019. https://doi.org/10.1038/ncb1101-1014
Bondulich MK, Jolinon N, Osborne GF, Smith EJ, Rattray I, Neueder A et al (2017) Myostatin inhibition prevents skeletal muscle pathophysiology in Huntington's disease mice. Sci Rep 7:14275. https://doi.org/10.1038/s41598-017-14290-3
Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E et al (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18:2893–2904. https://doi.org/10.1101/gad.1256804
Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW et al (1998) Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci USA 95:6480–6485. https://doi.org/10.1073/pnas.95.11.6480
Chua JP, Lieberman AP (2013) Pathogenic mechanisms and therapeutic strategies in spinobulbar muscular atrophy. CNS Neurol Disord Drug Targets 12:1146–1156
Chua JP, Reddy SL, Yu Z, Giorgetti E, Montie HL, Mukherjee S et al (2015) Disrupting SUMOylation enhances transcriptional function and ameliorates polyglutamine androgen receptor-mediated disease. J Clin Invest 125:831–845. https://doi.org/10.1172/JCI73214
Cohen S, Nathan JA, Goldberg AL (2015) Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14:58–74. https://doi.org/10.1038/nrd4467
Cohen TJ, Barrientos T, Hartman ZC, Garvey SM, Cox GA, Yao TP (2009) The deacetylase HDAC4 controls myocyte enhancing factor-2-dependent structural gene expression in response to neural activity. FASEB J 23:99–106. https://doi.org/10.1096/fj.08-115931
Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L et al (2014) Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron 82:295–307. https://doi.org/10.1016/j.neuron.2014.03.001
Crist CG, Montarras D, Buckingham M (2012) Muscle satellite cells are primed for myogenesis but maintain quiescence with sequestration of Myf5 mRNA targeted by microRNA-31 in mRNP granules. Cell Stem Cell 11:118–126. https://doi.org/10.1016/j.stem.2012.03.011
Dadson K, Turdi S, Hashemi S, Zhao J, Polidovitch N, Beca S et al (2015) Adiponectin is required for cardiac MEF2 activation during pressure overload induced hypertrophy. J Mol Cell Cardiol 86:102–109. https://doi.org/10.1016/j.yjmcc.2015.06.020
De Simone A, Kitchen C, Kwan AH, Sunde M, Dobson CM, Frenkel D (2012) Intrinsic disorder modulates protein self-assembly and aggregation. Proc Natl Acad Sci USA 109:6951–6956. https://doi.org/10.1073/pnas.1118048109
Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM et al (2002) Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science 296:2238–2243. https://doi.org/10.1126/science.1072613
Ebert SM, Dyle MC, Kunkel SD, Bullard SA, Bongers KS, Fox DK et al (2012) Stress-induced skeletal muscle Gadd45a expression reprograms myonuclei and causes muscle atrophy. J Biol Chem 287:27290–27301. https://doi.org/10.1074/jbc.M112.374777
Eftekharzadeh B, Banduseela VC, Chiesa G, Martinez-Cristobal P, Rauch JN, Nath SR et al (2019) Hsp70 and Hsp40 inhibit an inter-domain interaction necessary for transcriptional activity in the androgen receptor. Nat Commun 10:3562. https://doi.org/10.1038/s41467-019-11594-y
Flavell SW, Cowan CW, Kim TK, Greer PL, Lin Y, Paradis S et al (2006) Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311:1008–1012. https://doi.org/10.1126/science.1122511
Flavell SW, Kim TK, Gray JM, Harmin DA, Hemberg M, Hong EJ et al (2008) Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60:1022–1038. https://doi.org/10.1016/j.neuron.2008.11.029
Giorgetti E, Yu Z, Chua JP, Shimamura R, Zhao L, Zhu F et al (2016) Rescue of metabolic alterations in AR113Q skeletal muscle by peripheral androgen receptor gene silencing. Cell Rep 17:125–136. https://doi.org/10.1016/j.celrep.2016.08.084
Gunther S, Kim J, Kostin S, Lepper C, Fan CM, Braun T (2013) Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell Stem Cell 13:590–601. https://doi.org/10.1016/j.stem.2013.07.016
Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thepenier C et al (2016) Comparative study of injury models for studying muscle regeneration in mice. PLoS ONE 11:e0147198. https://doi.org/10.1371/journal.pone.0147198
Hsu CC, Buehler MJ, Tarakanova A (2020) The order-disorder continuum: linking predictions of protein structure and disorder through molecular simulation. Sci Rep 10:2068. https://doi.org/10.1038/s41598-020-58868-w
Kozhemyakina E, Cohen T, Yao TP, Lassar AB (2009) Parathyroid hormone-related peptide represses chondrocyte hypertrophy through a protein phosphatase 2A/histone deacetylase 4/MEF2 pathway. Mol Cell Biol 29:5751–5762. https://doi.org/10.1128/MCB.00415-09
Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J et al (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18:39–51. https://doi.org/10.1096/fj.03-0610com
Leger B, Vergani L, Soraru G, Hespel P, Derave W, Gobelet C et al (2006) Human skeletal muscle atrophy in amyotrophic lateral sclerosis reveals a reduction in Akt and an increase in atrogin-1. FASEB J 20:583–585. https://doi.org/10.1096/fj.05-5249fje
Li C, Vu K, Hazelgrove K, Kuemmerle JF (2015) Increased IGF-IEc expression and mechano-growth factor production in intestinal muscle of fibrostenotic Crohn's disease and smooth muscle hypertrophy. Am J Physiol Gastrointest Liver Physiol 309:G888–899. https://doi.org/10.1152/ajpgi.00414.2014
Li M, Sobue G, Doyu M, Mukai E, Hashizume Y, Mitsuma T (1995) Primary sensory neurons in X-linked recessive bulbospinal neuropathy: histopathology and androgen receptor gene expression. Muscle Nerve 18:301–308. https://doi.org/10.1002/mus.880180306
Li SH, Cheng AL, Zhou H, Lam S, Rao M, Li H et al (2002) Interaction of Huntington disease protein with transcriptional activator Sp1. Mol Cell Biol 22:1277–1287. https://doi.org/10.1128/mcb.22.5.1277-1287.2002
Lieberman AP, Harmison G, Strand AD, Olson JM, Fischbeck KH (2002) Altered transcriptional regulation in cells expressing the expanded polyglutamine androgen receptor. Hum Mol Genet 11:1967–1976
Lieberman AP, Shakkottai VG, Albin RL (2019) Polyglutamine repeats in neurodegenerative diseases. Annu Rev Pathol 14:1–27. https://doi.org/10.1146/annurev-pathmechdis-012418-012857
Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S et al (2014) Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep 7:774–784. https://doi.org/10.1016/j.celrep.2014.02.008
Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R et al (2014) Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci USA 111:4109–4114. https://doi.org/10.1073/pnas.1401732111
Ma K, Chan JK, Zhu G, Wu Z (2005) Myocyte enhancer factor 2 acetylation by p300 enhances its DNA binding activity, transcriptional activity, and myogenic differentiation. Mol Cell Biol 25:3575–3582. https://doi.org/10.1128/MCB.25.9.3575-3582.2005
Majidi SP, Reddy NC, Moore MJ, Chen H, Yamada T, Andzelm MM et al (2019) Chromatin environment and cellular context specify compensatory activity of paralogous MEF2 transcription factors. Cell Rep 29(2001–2015):e2005. https://doi.org/10.1016/j.celrep.2019.10.033
Malena A, Pennuto M, Tezze C, Querin G, D'Ascenzo C, Silani V et al (2013) Androgen-dependent impairment of myogenesis in spinal and bulbar muscular atrophy. Acta Neuropathol 126:109–121. https://doi.org/10.1007/s00401-013-1122-9
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C et al (1996) Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 87:493–506. https://doi.org/10.1016/s0092-8674(00)81369-0
Mao Z, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME (1999) Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science 286:785–790. https://doi.org/10.1126/science.286.5440.785
McPherron AC, Lawler AM, Lee SJ (1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 387:83–90. https://doi.org/10.1038/387083a0
Mhatre AN, Trifiro MA, Kaufman M, Kazemi-Esfarjani P, Figlewicz D, Rouleau G et al (1993) Reduced transcriptional regulatory competence of the androgen receptor in X-linked spinal and bulbar muscular atrophy. Nat Genet 5:184–188. https://doi.org/10.1038/ng1093-184
Mielcarek M, Toczek M, Smeets CJ, Franklin SA, Bondulich MK, Jolinon N et al (2015) HDAC4-myogenin axis as an important marker of HD-related skeletal muscle atrophy. PLoS Genet 11:e1005021. https://doi.org/10.1371/journal.pgen.1005021
Molkentin JD, Li L, Olson EN (1996) Phosphorylation of the MADS-Box transcription factor MEF2C enhances its DNA binding activity. J Biol Chem 271:17199–17204. https://doi.org/10.1074/jbc.271.29.17199
Monks DA, Johansen JA, Mo K, Rao P, Eagleson B, Yu Z et al (2007) Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc Natl Acad Sci USA 104:18259–18264. https://doi.org/10.1073/pnas.0705501104
Montie HL, Cho MS, Holder L, Liu Y, Tsvetkov AS, Finkbeiner S et al (2009) Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 18:1937–1950. https://doi.org/10.1093/hmg/ddp115
Moretti I, Ciciliot S, Dyar KA, Abraham R, Murgia M, Agatea L et al (2016) MRF4 negatively regulates adult skeletal muscle growth by repressing MEF2 activity. Nat Commun 7:12397. https://doi.org/10.1038/ncomms12397
Narain Y, Wyttenbach A, Rankin J, Furlong RA, Rubinsztein DC (1999) A molecular investigation of true dominance in Huntington's disease. J Med Genet 36:739–746. https://doi.org/10.1136/jmg.36.10.739
Nath SR, Yu Z, Gipson TA, Marsh GB, Yoshidome E, Robins DM et al (2018) Androgen receptor polyglutamine expansion drives age-dependent quality control defects and muscle dysfunction. J Clin Invest 128:3630–3641. https://doi.org/10.1172/JCI99042
Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie Z et al (2010) Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron 67:936–952. https://doi.org/10.1016/j.neuron.2010.08.034
Ohanna M, Sobering AK, Lapointe T, Lorenzo L, Praud C, Petroulakis E et al (2005) Atrophy of S6K1(−/−) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat Cell Biol 7:286–294. https://doi.org/10.1038/ncb1231
Olney RK, Aminoff MJ, So YT (1991) Clinical and electrodiagnostic features of X-linked recessive bulbospinal neuronopathy. Neurology 41:823–828. https://doi.org/10.1212/wnl.41.6.823
Palazzolo I, Stack C, Kong L, Musaro A, Adachi H, Katsuno M et al (2009) Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 63:316–328. https://doi.org/10.1016/j.neuron.2009.07.019
Park SY, Yun Y, Lim JS, Kim MJ, Kim SY, Kim JE et al (2016) Stabilin-2 modulates the efficiency of myoblast fusion during myogenic differentiation and muscle regeneration. Nat Commun 7:10871. https://doi.org/10.1038/ncomms10871
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM et al (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137:873–886. https://doi.org/10.1016/j.cell.2009.03.046
Polo A, Teatini F, D'Anna S, Manganotti P, Salviati A, Dallapiccola B et al (1996) Sensory involvement in X-linked spino-bulbar muscular atrophy (Kennedy's syndrome): an electrophysiological study. J Neurol 243:388–392. https://doi.org/10.1007/bf00868997
Potthoff MJ, Arnold MA, McAnally J, Richardson JA, Bassel-Duby R, Olson EN (2007) Regulation of skeletal muscle sarcomere integrity and postnatal muscle function by Mef2c. Mol Cell Biol 27:8143–8151. https://doi.org/10.1128/MCB.01187-07
Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP (2015) Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu Rev Pharmacol Toxicol 55:353–371. https://doi.org/10.1146/annurev-pharmtox-010814-124332
Ramzan F, McPhail M, Rao P, Mo K, Halievski K, Swift-Gallant A et al (2015) Distinct etiological roles for myocytes and motor neurons in a mouse model of kennedy's disease/spinobulbar muscular atrophy. J Neurosci 35:6444–6451. https://doi.org/10.1523/JNEUROSCI.3599-14.2015
Ranganathan S, Harmison GG, Meyertholen K, Pennuto M, Burnett BG, Fischbeck KH (2009) Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum Mol Genet 18:27–42. https://doi.org/10.1093/hmg/ddn310
Rhodes LE, Freeman BK, Auh S, Kokkinis AD, La Pean A, Chen C et al (2009) Clinical features of spinal and bulbar muscular atrophy. Brain 132:3242–3251. https://doi.org/10.1093/brain/awp258
Rocchi A, Milioto C, Parodi S, Armirotti A, Borgia D, Pellegrini M et al (2016) Glycolytic-to-oxidative fiber-type switch and mTOR signaling activation are early-onset features of SBMA muscle modified by high-fat diet. Acta Neuropathol 132:127–144. https://doi.org/10.1007/s00401-016-1550-4
Sacheck JM, Hyatt JP, Raffaello A, Jagoe RT, Roy RR, Edgerton VR et al (2007) Rapid disuse and denervation atrophy involve transcriptional changes similar to those of muscle wasting during systemic diseases. FASEB J 21:140–155. https://doi.org/10.1096/fj.06-6604com
Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E et al (2013) BMP signaling controls muscle mass. Nat Genet 45:1309–1318. https://doi.org/10.1038/ng.2772
Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S et al (2010) Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington's disease. Hum Mol Genet 19:65–78. https://doi.org/10.1093/hmg/ddp467
Scaramuzzino C, Casci I, Parodi S, Lievens PMJ, Polanco MJ, Milioto C et al (2015) Protein arginine methyltransferase 6 enhances polyglutamine-expanded androgen receptor function and toxicity in spinal and bulbar muscular atrophy. Neuron 85:88–100. https://doi.org/10.1016/j.neuron.2014.12.031
Schiaffino S, Dyar KA, Ciciliot S, Blaauw B, Sandri M (2013) Mechanisms regulating skeletal muscle growth and atrophy. FEBS J 280:4294–4314. https://doi.org/10.1111/febs.12253
Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A (1989) X-linked recessive bulbospinal neuronapathy. A clinicopathological study. Brain 112(Pt 1):209–232
Sofer A, Lei K, Johannessen CM, Ellisen LW (2005) Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol 25:5834–5845. https://doi.org/10.1128/MCB.25.14.5834-5845.2005
Soraru G, D'Ascenzo C, Polo A, Palmieri A, Baggio L, Vergani L et al (2008) Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci 264:100–105. https://doi.org/10.1016/j.jns.2007.08.012
Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H et al (2000) The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 97:6763–6768. https://doi.org/10.1073/pnas.100110097
Uversky VN (2015) Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders. Front Aging Neurosci 7:18. https://doi.org/10.3389/fnagi.2015.00018
Walcott JL, Merry DE (2002) Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J Biol Chem 277:50855–50859. https://doi.org/10.1074/jbc.M209466200
Wales S, Hashemi S, Blais A, McDermott JC (2014) Global MEF2 target gene analysis in cardiac and skeletal muscle reveals novel regulation of DUSP6 by p38MAPK-MEF2 signaling. Nucleic Acids Res 42:11349–11362. https://doi.org/10.1093/nar/gku813
Wang AM, Miyata Y, Klinedinst S, Peng H-MM, Chua JP, Komiyama T et al (2013) Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 9:112–118. https://doi.org/10.1038/nchembio.1140
Woronicz JD, Lina A, Calnan BJ, Szychowski S, Cheng L, Winoto A (1995) Regulation of the Nur77 orphan steroid receptor in activation-induced apoptosis. Mol Cell Biol 15:6364–6376. https://doi.org/10.1128/mcb.15.11.6364
Yamamoto H, Williams EG, Mouchiroud L, Canto C, Fan W, Downes M et al (2011) NCoR1 is a conserved physiological modulator of muscle mass and oxidative function. Cell 147:827–839. https://doi.org/10.1016/j.cell.2011.10.017
Yu YT, Breitbart RE, Smoot LB, Lee Y, Mahdavi V, Nadal-Ginard B (1992) Human myocyte-specific enhancer factor 2 comprises a group of tissue-restricted MADS box transcription factors. Genes Dev 6:1783–1798. https://doi.org/10.1101/gad.6.9.1783
Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM et al (2006) Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Investig 116:2663–2672. https://doi.org/10.1172/JCI28773
Yu Z, Dadgar N, Albertelli M, Scheller A, Albin RL, Robins DM et al (2006) Abnormalities of germ cell maturation and sertoli cell cytoskeleton in androgen receptor 113 CAG knock-in mice reveal toxic effects of the mutant protein. Am J Pathol 168:195–204. https://doi.org/10.2353/ajpath.2006.050619
Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F et al (1999) Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol 19:21–30. https://doi.org/10.1128/mcb.19.1.21
Acknowledgements
We thank Michael Greenberg for providing the MEF2-VP16 (CA-MEF2) and MEF2ΔDBD-VP16 (DBD-MEF2) plasmids.
Funding
This work was supported by the NIH (R01 NS055746 to APL; T32 GM007863 and T32 GM007315 to SRN), a University of Michigan Rackham Predoctoral Fellowship to SRN and the CHDI Foundation to GPB.
Author information
Authors and Affiliations
Contributions
SRN, MLL, ZY, CM, STJ, ECD, and KVP performed experiments. SRN, MLL, ZY, CM, STJ, KVP, MP, GPB and APL planned experiments and interpreted data. STJ, ECD, GS, GPB, and DMR provided critical reagents. SRN and APL wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors have declared that no conflict of interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Nath, S.R., Lieberman, M.L., Yu, Z. et al. MEF2 impairment underlies skeletal muscle atrophy in polyglutamine disease. Acta Neuropathol 140, 63–80 (2020). https://doi.org/10.1007/s00401-020-02156-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-020-02156-4