
Abstract
Repeat expansion in C9orf72 causes amyotrophic lateral sclerosis and frontotemporal lobar degeneration. Expanded sense and antisense repeat RNA transcripts in C9orf72 are translated into five dipeptide-repeat proteins (DPRs) in an AUG-independent manner. We previously identified the heterogeneous ribonucleoprotein (hnRNP) A3 as an interactor of the sense repeat RNA that reduces its translation into DPRs. Furthermore, we found that hnRNPA3 is depleted from the nucleus and partially mislocalized to cytoplasmic poly-GA inclusions in C9orf72 patients, suggesting that poly-GA sequesters hnRNPA3 within the cytoplasm. We now demonstrate that hnRNPA3 also binds to the antisense repeat RNA. Both DPR production and deposition from sense and antisense RNA repeats are increased upon hnRNPA3 reduction. All DPRs induced DNA double strand breaks (DSB), which was further enhanced upon reduction of hnRNPA3. Poly-glycine–arginine and poly-proline-arginine increased foci formed by phosphorylated Ataxia Telangiectasia Mutated (pATM), a major sensor of DSBs, whereas poly-glycine–alanine (poly-GA) evoked a reduction of pATM foci. In dentate gyri of C9orf72 patients, lower nuclear hnRNPA3 levels were associated with increased DNA damage. Moreover, enhanced poly-GA deposition correlated with reduced pATM foci. Since cytoplasmic pATM deposits partially colocalized with poly-GA deposits, these results suggest that poly-GA, the most frequent DPR observed in C9orf72 patients, differentially causes DNA damage and that poly-GA selectively sequesters pATM in the cytoplasm inhibiting its recruitment to sites of DNA damage. Thus, mislocalization of nuclear hnRNPA3 caused by poly-GA leads to increased poly-GA production, which partially depletes pATM, and consequently enhances DSB.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
C9orf72 repeat expansion is the most common cause of autosomal dominant FTLD, FTLD/ALS, and ALS [14, 20, 50]. While unaffected people generally have less than 30 (G4C2)n repeats, mutation carriers have a few hundred or even thousands of repeats [50]. Sense and antisense repeat RNAs accumulate within intranuclear RNA foci [14]. Furthermore, sense and antisense transcripts are translated in all reading frames into dipeptide-repeat proteins (DPRs) in an AUG-independent manner [2, 43]. Accumulating evidence suggests that neurotoxicity occurs via various cellular pathways, such as RNA mis-splicing and reduced transcription of the C9orf72 gene [27, 29], nucleocytoplasmic transport dysfunction [8,9,10,, 28, 65, 66], nucleolar stress [23, 38, 60], and DNA damage [15,16,, 32, 58].
We previously identified the heterogeneous ribonucleoprotein (hnRNP) A3 as an interactor of the sense repeat RNA. We and others also found that hnRNPA3 is mislocalized from the nucleus to the cytoplasm specifically in hippocampal, cerebellar, and spinal motor neurons of C9orf72 patients [17, 41]. Moreover, mislocalized hnRNPA3 colocalizes with poly-glycine-alanine (poly-GA) deposits [42]. Reduction of nuclear hnRNPA3 increases (G4C2) repeat RNA foci. Furthermore, repeat RNA foci and DPRs may enhance nucleocytoplasmic transport dysfunction, reducing nuclear hnRNPA3 and thus initiating a vicious cycle [42]. Thus, reduction of nuclear hnRNPA3 may be specifically associated with C9orf72-dependent neurotoxicity, though the cellular mechanisms remain unclear. There is evidence that C9orf72-linked neurotoxicity is driven by genomic instability [58]. Moreover, impairment of DNA damage repair is implicated in several other neurodegenerative diseases, such as Alzheimer disease, Parkinson’s disease, and Huntington disease [6, 8, 9, 11, 21, 25, 33, 34, 36, 64]. DNA double strand breaks (DSBs) are the most severe type of DNA damage and frequently lead to cell death if not correctly repaired. Homologous recombination (HR) and non-homologous end joining (NHEJ) are the two major cellular mechanisms that repair DSBs in mammalian cells. HR fully restores the original sequence using the sister chromatid as a template, but is, therefore, limited to the late S to G2/M phase of the cell cycle [37, 61]. NHEJ is a rather error-prone repair mechanism that rejoins broken DNA ends without a template DNA and can, therefore, occur throughout the cell cycle [47]. Since neurons are arrested in the G0 phase and are non-proliferative, their main repair mechanism is limited to NHEJ. Due to their error-prone repair pathway, neurons are quite vulnerable to DNA damage. This may be particularly important for expanded C9orf72 repeats, as they form G-quadruplex structures and promote the formation of RNA:DNA hybrids (R-loops) [18, 23, 63], which are prone to DSBs. Walker et al. reported that expanded hexanucleotide repeats and poly-GA impair Ataxia Telangiectasia Mutated (ATM)-mediated DNA repair [58]. Moreover, reduced expression of hnRNPA3 itself may enhance DSBs [13]. Furthermore, many hnRNPs, which are genetically associated with FTD/ALS, such as hnRNPA1, A2B1, and FUS (hnRNPP2) are reported to be involved in DNA damage and repair [4, 12, 24, 44, 48, 55], and hnRNPA3 is a homolog of hnRNPA1 and A2B1 [9, 59]. We speculated that cytoplasmic mislocalization of hnRNPA3 may affect ATM-mediated DNA damage directly [13] via increased repeat RNA foci and DPR production. We now investigated the association of hnRNPA3 expression, RNA foci formation, DPR production, and DNA damage in cultured cells, including patient-derived human neurons and brains of C9orf72 carriers. Our findings suggest that the most frequent DPRs (poly-GA) observed in C9orf72 patients differentially cause DNA damage by selectively sequestering phosphorylated ATM (pATM) in the cytoplasm and inhibiting its recruitment to sites of DNA damage.
Materials and methods
DNA synthesis and plasmid construction for in vitro transcription
We synthesized the plasmid containing hexanucleotide repeats for in vitro transcription by using a previously reported protocol [41]. In brief, 124 base single-stranded DNA containing G4C2, C4G2, or A4C2 hexanucleotide repeats with restriction enzyme sites (NheI or HindIII) were synthesized (Suppl. Fig. 1a). 100 μM of complementary DNA strands were annealed in the presence of 10% GC-RICH solution (Roche) and GC-RICH PCR Reaction buffer (Roche) and cloned into pcDNA3.1(+) vector (Invitrogen). Plasmids containing 17 repeats of G4C2, C4G2, and A4C2 were obtained. The DNA sequence of all constructs was verified.
In vitro transcription of RNA probes
pcDNA3.1-(G4C2)17, pcDNA3.1-(C4G2)17, and pcDNA3.1-(A4C2)17 constructs were linearized with HindIII and used as templates for RNA synthesis (Suppl. Fig. 1a). In vitro RNA transcription was performed with T7 Ribomax Express Large Scale RNA Production System (Promega) supplemented with 40 U of RNase inhibitor (RiboLock, Thermo Scientific) as described by the manufacturer. To achieve equal levels of biotinylation between these probes, different concentrations of biotin-14-CTP (1 mM for G4C2 probe, 0.08 mM for C4G2 probe, and 1.2 mM for A4C2 probe) were added in each reaction. Following DNase treatment, biotinylated RNA products were purified with phenol/chloroform. For competition experiments non-biotinylated C4G2 repeat RNA was in vitro RNA transcribed using the MEGA script kit (Ambion) as described by the manufacturer. The expected lengths of repeat RNA probes and competitor were confirmed by formaldehyde gel electrophoresis (Suppl. Fig. 1b). Biotinylation efficacy of RNA probes was evaluated using the BrightStar BioDetect kit (Ambion) (Suppl. Fig. 1c).
Purification of hexanucleotide repeat-binding proteins
Purification of hexanucleotide repeat-binding proteins was performed following our previously described protocol [41]. In brief, a total of 0.6 mg of HeLa cell nuclear extract was diluted in 4 ml of protein-binding buffer (10% glycerol, 10 mM HEPES, 50 mM KCl, 1 U/ml RNase inhibitor, 0.15 lg/ml yeast tRNA (10109495001, Roche), 1 mM EDTA, 1 mM DTT and 0.5% Triton X100 in DEPC water). The diluted extract was then precleared with heparin-agarose (H6508, Sigma-Aldrich) and streptavidin-agarose (15942-050, Invitrogen). The precleared nuclear extracts were incubated with streptavidin μMACS-microbeads (MACS molecular) and 150 pmol of biotinylated repeat RNA in the presence of 50 mM KCl for 1 h. For competition experiments, nuclear extracts were incubated for 1 h with 7.5 nmol (50-fold excess) of non-biotinylated (C4G2)17 competitor RNA before addition of the biotinylated probe. The reaction mixture was loaded on a μMACS column and subsequently washed three times with protein-binding buffer. Hexanucleotide repeat binding proteins were sequentially eluted with increasing concentrations of NaCl. Each eluate was TCA precipitated and subjected to SDS-PAGE.
Cell culture
HeLa cells were cultured in DMEM containing 10% FCS and Penicillin/Streptomycin at 37 °C with 5%CO2.
Patient-derived fibroblasts
We included cell lines from 3 C9orf72 ALS patients and 3 control cases from our previous report [42]. All procedures were in accordance with the Helsinki convention and approved by the Ethical Committee of the University of Dresden (EK45022009; EK393122012). Patients were genotyped using EDTA blood in the clinical setting after given written consent according to German legislation independent of any scientific study by a diagnostic human genetic laboratory (CEGAT, Tübingen, Germany or Department of Human Genetics, University of Ulm, Germany) using diagnostic standards.
For further details of patients’ background, see Suppl. Table 1.
siRNA-mediated knockdown in fibroblasts and iPSC-derived neurons
The following siRNAs were obtained from Dharmacon: ON-TARGETplus human non-targeting siRNA D-001810-01, hnRNPA3 J-019347-08 ACAAUGAAGGAGGAAAUUU, hnRNPA3 J-019347-06 GGAGGGAACUUUGGAGGUG. 10nMol of each siRNA was reverse transfected using RNAiMax (Thermo Fisher Scientific) and OPTI-MEM. Media were exchanged after overnight incubation. Cells on coverslips were fixed on the next day (48 h after a transfection of siRNA containing solution).
Plasmid transfection
For plasmid transfection in HeLa cells, 0.5 μg/well (in the case of 24-well plate) of DNA was transfected with lipofectamine LTX with plus reagent (Thermo Fisher Scientific) in OPTI-MEM. Plasmid containing media were exchanged after 6 h-incubation. Cells were harvested 48 h after plasmid transfection.
Fluorescence in situ hybridization for antisense RNA foci
Fluorescence in situ hybridization (FISH) was performed as previously described [39, 42, 51] with slight modifications. 2% paraformaldehyde fixed and perforated cells on glass coverslips were rinsed twice with 2 × saline-sodium citrate buffer (SSC) and then incubated in prehybridization solution (40% formamide (Life Technologies, 15515-026)/2 × SSC, 2.5% bovine serum albumin (BSA)) at 57 °C for 30 min. Cells were then incubated with hybridization solution (40% formamide, 2 × SSC, 0.8 mg/ml tRNA (Roche), 0.8 mg/ml single strand salmon sperm DNA (Sigma-Aldrich, D7656), 0.16% BSA, 8% Dextran sulfate (Sigma-Aldrich), 1.6 mM Ribonucleoside vanadyl complex (New England Biolabs, S1402S), 5 mM EDTA, 10 ng/ml 5′ Cy3-labbeled 2′-O-methyl-(CCGGGG) × 4 probe (IDT)) at 57 °C. The following day, cells were sequentially washed in 40% formamide/0.5 × SSC for 3 times 30 min each at 57 °C and then with 0.5 × SSC 3 times 10 min each at room temperature. After a brief rinse with PBS, nuclei were counterstained with 0.5 μg/ml of DAPI for 20 min and then washed with PBS for 3 times 3 min each. Glass coverslips were mounted using Prolong Gold antifade (Life Technologies) and analyzed with LSM710 confocal microscopy with ZEN2011 software (Zeiss). For FISH of fibroblasts, after incubation with hybridization solution and washing, blocking with 5% FCS (30 min) and nucleolin (NCL) staining (4 °C overnight) was performed. Nuclei were counterstained with DAPI and glass coverslips were mounted on the following day.
Differentiation of human neural precursor cells (NPCs) to spinal motor neurons (MNs)
The generation of all iPSCs was recently published [22, 26, 52]. The generation of human NPCs and MNs was accomplished following the protocol from Naumann et al. [44]. In brief, colonies of iPSCs were collected and stem cell medium, containing 10 µM SB-431542, 1 µM Dorsomorphin, 3 µM CHIR 99021 and 0.5 µM pumorphamine (PMA), was added. After 2 days hESC medium was replaced with basal media (DMEM-F12/Neurobasal 50:50 with 1:200 N2 Supplement, 1:100 B27 lacking Vitamin A and 1% penicillin/streptomycin/glutamine). On day 4 150 µM ascorbic acid was added, while Dorsomorphin and SB-431542 were withdrawn. 2 Days later the EBs were mechanically separated and replated on Matrigel coated dishes. For this purpose, Matrigel was diluted (1:100) in DMEM-F12 and kept on the dishes over night at room temperature. Possessing a ventralized and caudalized character the arising so called small molecule NPCs (smNPC) formed homogenous colonies during the course of further cultivation. It was necessary to split them at a ratio of 1:10–1:20 once a week using Accutase for 10 min at 37 °C.
To identify Hb9-positive neurons, we transduced the Hb9::GFP promoter into the neural progenitor cells using a lentivirus system [30, 45], and the resulting neural progenitors were replated on Poly-L-Ornithine / laminin coated dish and cultured in basal media supplemented with 1 µM PMA. After 2 days 1 µM retinoic acid (RA) was added. On day 9 another split was performed to seed them on a desired cell culture system. Furthermore, the medium was modified to induce neural maturation. For this purpose, the developing neurons were treated with N2B27 containing 10 ng/µl BDNF, 500 µM dbcAMP and 10 ng/µl GDNF. Finally, motor neuron differentiation was confirmed by immunocytochemistry.
Plasmids
The (G4C2)80 and (C4G2)30 expression vectors are based on a previously published cDNA construct [42]. The modified vector expresses (C4G2)30 under the control of the CMV promoter including 620 bp of the 5′ flanking region of the human C9orf72 C4G2 repeat which was PCR subcloned from patient fibroblast derived genomic DNA. This patient has a GGGCCCGCCCCC insertion just before the beginning of the CCCCGG repeat, where frequent sequence variations have been observed [40, 46, 57]. The 5′ flanking region contains multiple stop codons in each reading frame.
Synthetic DPR constructs containing an ATG start codon for transient expressions in HeLa cells were constructed as previously described [7] with slight modifications. Codon optimized DNA sequences encoding 12 repeats of each DPR, were designed. Restriction sites for XhoI-FokI and BbsI-XbaI were placed at the beginning and end of the DNA oligonucleotide. The synthesized DNA oligonucleotide was subcloned into the pEGFP-C1 vector (Clontech) using XhoI and XbaI sites. The codon optimized 11 repeat coding fragments were isolated through double digestion with FokI and BbsI, and then ligated into BbsI site of the parental 12 repeats vector to double the repeat-length. This was repeated until 175–177 repeats were achieved (12 + 11 = 23 repeats, 23 + 22 = 45 repeats, 45 + 44 = 89 repeats, 89 + 86 = 175 repeats (poly-GA), 89 + 88 = 177 repeats (poly-glycine–arginine: poly-GR), 89 + 87 = 176 repeats (poly-proline-arginine: poly-PR). The repeat number differences at the last step are due to clonal instability). The following oligonucleotides were used.
XhoI-FokI-GA12-BbsI-XbaI: 5′-CTCGAGGGATGTTGAATTCTGGTGCTGGCGCGGGAGCAGGCGCTGGTGCTGGTGCAGGAGCGGGTGCGGGAGCTGGTGCCGGCGCAGGTGCTGTCTTCGGATCCTAGTCTAGA-3′.
XhoI-FokI-GR12-BbsI-XbaI: 5′-CTCGAGGGATGTTGAATTCTGGTCGTGGACGTGGACGAGGTCGAGGTCGAGGTCGTGGACGTGGTCGAGGTCGAGGTCGTGGACGTGGTCGTGTCTTCGGATCCTAGTCTAGA-3′.
XhoI-FokI-PR12-BbsI-XbaI: 5′-CTCGAGGGATGTTGAATTCTCCGCGACCTCGACCGCGGCCACGCCCACGCCCTCGGCCCAGACCACGTCCTAGGCCCAGACCCAGACCGCGAGTCTTCGGATCCTAGTCTAGA-3′.
Synthetic DPR constructs containing an ATG start codon for lentiviral expression (synapsin promoter) and transient transfection (EF1 promoter) were described before [9].
Crispr/Cas9 genome editing
Knockout of hnRNPA3 was performed in HeLa cells using the Crispr/Cas9 [49]. Guide RNA sequences targeting hnRNPA3 genetic loci were designed using the gRNA tool from the Zhang laboratory [https://crispr.mit.edu (Suppl. Fig. 2a)]. Duplexed sgRNA oligos were digested and ligated into pSpCas9(BB)-2A-Puro (PX459) to generate hnRNPA3 Puro plasmid. CRISPR knock-out cells were generated by transfection with hnRNPA3 Puro plasmid followed by selection with 1 µg/ml puromycin. Individual clones were generated by plating cells at low density and isolating individual colonies. hnRNPA3 knockout was confirmed by DNA sequencing (Suppl. Fig. 2b) and western blotting (Fig. 2a). For sequencing of hnRNPA3, genomic loci were amplified by PCR using the following primers:
Forward: 5′-GTATGTCAGCCGCGTTTT-3′, Reverse: 5′- CGGCGGATCAATGTCAAT-3′.
Western blotting
To detect the protein expression (except for pATM), samples were separated on 12% Tris-glycine gel and transferred on PVDF membranes. After blocking for 1 h with 0.2% I-Block (Applied Biosystems) in TBST (TBS with 0.5% Tween20), membranes were incubated with indicated antibody overnight. The antibody signal was detected with HRP-conjugated secondary antibodies (Promega) using the ECL reagents (GE healthcare) and exposed to X-ray films (SuperRX, Fujifilm).
For detection of pATM, samples were separated on NuPAGE 3–8% Tris-Acetate protein gel (Invitrogen) and transferred on PVDF membrane with 25 V for 15 h. After blocking for 1 h with 5% PhosphoBLOCKER (Cell Biolabs, AKR-104) in TBST, membranes were incubated with indicated antibody for 2 h at room temperature. All subsequent steps were performed as described above.
Co-immunoprecipitation (co-IP)
HeLa cells were lysed in co-IP buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.4, 0.5% NP-40, 0.5% Na deoxycholate, 5 mM EDTA) supplemented with EDTA-free Protease Inhibitor Cocktail (Roche, 04 693 132 001) and PhosSTOP (Roche, 04 906 845 001), then incubated for 30 min on ice. Lysates were centrifuged for 5 min at 5000 rpm, and the supernatant was collected. For co-IP, the Dynabeads Protein G immunoprecipitation kit (Invitrogen, 10007D) was used. The bead-antibody-antigen complex was isolated using a magnetic tool (Invitrogen, 12321D) and resuspended in elution buffer and NuPAGE LDS sample buffer (Invitrogen, NP0007) supplemented with DTT. The complex was heated for 10 min at 70 °C and captured proteins were separated from bead-antibody complexes using a magnetic tool. Isolated antigens were analyzed by western blotting.
Cell fractionation
Cell fractionation was performed as previously described [5]. Briefly, cells were washed twice in ice-cold PBS. Cells were harvested in 400 μl buffer 1 (50 mM Tris (pH 7.9), 10 mM KCl, 1 mM EDTA, 0.05% NP-40, 10% glycerol and protease/phosphatase inhibitors) and centrifuged at 6,000 r.p.m. for 3 min at 4 °C. The supernatants were used for the analysis of cytoplasmic proteins. The nuclear pellet was lysed with 150 μl buffer 2 (20 mM HEPES (pH 7.9), 400 mM NaCl, 10 mM KCl, 1% NP-40, 20% glycerol, 1 mM EDTA and protease/phosphatase inhibitors) for 20 min at 4 °C, then centrifuged at 14,000 r.p.m. for 10 min. The supernatants were used for the analysis of nuclear proteins.
Immunofluorescent staining for HeLa cells and neurons
Cells on glass coverslips were washed with PBS and fixed with 4%PFA for 15 min on ice and permeabilized with 0.2% Triton-X100 in PBS for 5 min at room temperature. After that cover slips were washed with PBS 3 times and blocked with 5%FCS in PBS. Primary antibody incubation was performed overnight at 4 °C, and incubation of secondary antibody conjugated with Alexa Fluor 555 or Alexa Fluor 488 (anti-rabbit, anti-mouse, and anti-rat) was performed for 1 h at room temperature. After a brief rinse with PBS, nuclei were counterstained with 0.5 μg/ml of DAPI for 20 min and then washed with PBS for 3 times 5 min. Finally, coverslips were mounted using Prolong Gold antifade (Life Technologies).
Quantitative immunofluorescence in HeLa cells and neurons
Three to ten fluorescent images of HeLa cells and cultured neurons were obtained from each glass coverslip using LSM710 (Zeiss) microscope with a 63 × oil immersion objective. Raw data tif files of each channel were exported from czi files using Zen2011 software. Quantification was performed using ImageJ software. The numbers of γH2AX foci positive HeLa cells and iPSC-derived neurons were determined with cell counter plugin of Image J.
Human brain samples
All cases were provided by the Neurobiobank Munich, Ludwig-Maximilians-University (LMU) Munich, Germany and the ALS Tissue Bank Kantonspital St. Gallen, Switzerland and collected and distributed according to the guidelines of the local ethics committees. Autopsies were performed on basis of informed consent. For further details, see Table 1.
Immunofluorescent staining for human brain sections
Immunohistochemical staining was performed on 5 μm-thick paraffin sections. Following antigen retrieval by microwaving for 30 min in citrate buffer (10 mM citric acid, 0.05% Tween20, pH 6.2) for co-staining of hnRNPA3 and poly-GA, or citrate-EDTA buffer (10 mM citric acid, 2 mM EDTA, 0.05% Tween20, pH 6.2) for all other stainings and settling the sections at room temperature for 20 min, sections were rinsed with PBS-Tween20 for 2 min, 2 times. After blocking with 2% FCS in PBS for 30 min, sections were incubated in primary antibody overnight at 4 °C. Sections were then rinsed with PBS for 5 min, 3 times, and then incubated in secondary antibody solution for 1 h at room temperature. After rinsing with PBS 2 times, 2.5 min each, nuclei were counterstained with 0.5 μg/ml of DAPI for 20 min and sections were then washed with PBS for 2 times 2.5 min each. To reduce autofluorescence, slides were incubated with 0.3% Sudan Black in 70% ethanol for 1 min. After a rinse with tap water, sections were mounted with using Prolong Gold antifade (Life Technologies).
Quantitative immunofluorescence in human brain sections
To quantify nuclear hnRNPA3 signal intensity in granular cells of human dentate gyri, we followed the protocol that we previously reported [42]. Fluorescent images of three different areas of hippocampal dentate gyrus granular cell layer at the level of lateral geniculate body were obtained from each case as indicated in Suppl. Fig. 3 using a LSM710 (Zeiss) microscope with a 63 × oil immersion objective. One image was shot for each area. Raw data tif files of each channel were exported from czi files using Zen2011 software. Quantification was performed using ImageJ software. Binary images from DAPI staining were used for the nuclear regions. Vascular structures were manually excluded from the analysis. Granular cell numbers of each image were counted. Nuclear hnRNPA3 was defined as hnRNPA3 signal overlapping with DAPI staining. The hnRNPA3 signal from structures outside of cell bodies was regarded as background. After subtraction of background, nuclear hnRNPA3 intensities of 242–636 granular layer cells/case were quantified in 16 C9orf72 cases. Median nuclear hnRNPA3 intensities (arbitrary unit) of these cells per micrograph were defined as the nuclear hnRNPA3 level of each image. An average of the nuclear hnRNPA3 levels of 3 micrographs from a C9orf72 case was regarded as the nuclear hnRNPA3 level of the case. The numbers of γH2AX foci positive cells, poly-GA aggregates positive cells, and pATM foci positive cells were determined by cell counter, the plugin of ImageJ software.
Filter trap analysis in HeLa cells
Cells cultured in 6-well plates were lysed in 600 μl lysis buffer (25 mM HEPES pH 7.6, 150 mM NaCl, 3% SDS, 0.5% sodium deoxycholate, 1% Triton X-100) supplemented with protease inhibitor cocktail (Sigma-Aldrich) for 10 min and passed through 27G needle for 10 times. The lysates were further diluted to 2 μg or 0.5 μg protein per 200 μl with lysis buffer. 100 μl of each sample were filtered through a nitrocellulose membrane (0.45 μm pore). The membrane was subsequently washed once with TBST, and then blocked in I-Block/TBS/0.5%TritonX-100. Levels of each DPR were analyzed with antibodies against each three different tag (FLAG, myc, HA). Quantified signals from 2 or 3 independent filter trap analyses probed with 3 different tag antibodies (total 6 or 8 membranes) are shown as fold expression.
Antibodies
The following antibodies were used for Western blotting (WB), immunoprecipitation (IP), and immunofluorescence (IF): anti-hnRNP A1 antibody, clone 4B10 (Sigma-Aldrich, 05-1521) WB 1/8,000, anti-hnRNP A2/B1 antibody, clone DP3B3 (Sigma-Aldrich, R4653) WB 1/10,000, anti-hnRNP A3 antibody, ab1 (Sigma-Aldrich, AV41195, lot No. QC10071) WB 1/2,000 and 1/150 (human brain sections), anti-DYKDDDDK(FLAG) Tag antibody (Cell Signaling #2368S) WB 1/1,000, anti-myc clone 9E10 (Santa Cruz, sc-40) WB 1/1,000, anti-HA Tag antibody, clone 3F10 (Roche, 12158167001) WB 1/1,000, anti-GFP antibody, clone N86/8 (Neuromab) WB 1/3,000, anti-GFP antibody (abcam, ab6556) IF 1/2,000, anti-GFP antibody (Novus Biologicals, NB100-1770) IF 1/200, anti-β-actin antibody (Sigma-Aldrich, A5316) WB 1/2,000, anti-tubulin βIII antibody, clone 2G10 (Millipore, 05-559) WB 1/5,000, IF 1/200, anti-NCL antibody (Abcam, ab136649) IF 1/1,000, anti-phospho-Histone H2A.X (Ser139) antibody, clone JBW301 (Merck Millipore, 05-636) IF 1/250 (for HeLa cells and human brain sections), anti-ATM (phospho S1981) antibody (abcam, ab81292) IP 1/30, WB 1/2,500, anti-ATM (phospho S1981) antibody (abcam, ab36810) IF 1/250, anti-FUS antibody (Bethyl, A300-294A) IF 1/1,500, anti-HuR antibody (SantaCruz, sc-5261) IF 1/100, anti-α-tubulin (Sigma-Aldrich, T5168) WB 1/4,000, anti-Lamin A/C antibody (Cell Signaling, #2032) WB 1/1,000 and anti-GA antibody (Mackenzie et al. [35]) IF 1/500.
Quantitative reverse transcription PCR (qPCR)
We performed qPCR following the protocol from our previous report [42]. Total RNA was prepared using the RNeasy and Qiashredder kit (Qiagen). RNA preparations were treated with Turbo DNA-free kit (Thermo Fisher Scientific) to minimize residual DNA contamination. Two micrograms of RNA were used for reverse transcription with M-MLV Reverse Transcriptase (Promega) using oligo-(dT) 12–18 primer (Invitrogen). qPCR was performed using the 7500 Fast Real-Time PCR System (Applied Biosystems) with TaqMan technology. Primers and probes were designed (IDT) for the detection of a region flanking the TAG 3′ of the G4C2 repeat of repeat constructs (repeat TAG primer). Sense repeat, primer 1: TCT CAA ACT GGG ATG CGT AC, primer 2: GTA GTC AAG CGT AGT CTG GG, probe: 5′-/56-FAM/TG CAG ATA T/Zen/C CAG CAC AGT GGC G/3IABkFQ/-3′. Antisense repeat, primer 1: CAA ACC CGG GTA CCC ATA C, primer 2: CGG GCC CTC TAG ACT ACT T, probe: 5′-/56-FAM/ACG TCC CAG/Zen/ACT ACG CTT GAC TAC A/3IABkFQ/-3′. A primer/probe set for Human GAPDH, 4326317E (Applied Biosystems) was used as endogenous control. Each sample was paired with no reverse transcription controls showing < 1/210 (ΔCT > 10) signal when compared to reverse transcribed samples, thus excluding contamination of plasmid DNA-derived signal. Signals from repeat construct derived cDNAs were normalized to GAPDH cDNA according to the ΔΔCT method.
Statistics
Statistical analysis was performed using IBM SPSS Statistics 23 software. All statistical results are shown in Suppl. Table 2.
Results
hnRNPA3 binds to the C9orf72 antisense repeat RNA
We previously reported that hnRNPA3 binds to the sense repeat RNA and that reduction of hnRNPA3 increases sense repeat RNA foci [41, 42]. We now investigated if hnRNPA3 also binds to the antisense repeat RNA and if reduction of hnRNPA3 increases the number of antisense repeat RNA foci. To verify binding of hnRNPA3 to the antisense repeat RNA, we performed RNA pull down assays using in vitro-transcribed biotinylated RNAs containing either 17 GGGGCC (G4C2; sense), 17 CCCCGG (C4G2; antisense), or 17 AAAACC (A4C2; control) repeats (Suppl. Fig. 1a). Length and amount of the in vitro-transcribed RNA was confirmed by RNA electrophoresis (Suppl. Fig. 1b) and dot blot assays (Suppl. Fig. 1c). Biotinylated in vitro-transcribed RNA probes were incubated with nuclear extracts from HeLa cells in the absence or presence of a 50-fold excess of nonbiotinylated competitor RNA containing the C4G2 repeat. Bound proteins were eluted using increasing salt concentrations and hnRNPA3 was detected in the eluate by western blotting using an anti-hnRNPA3 polyclonal antibody (Fig. 1a). This elution revealed specific binding of hnRNPA3 not only to the sense but also to the antisense repeat RNA, which was blocked by excess amounts of the non-biotinylated competitor RNA (Fig. 1a). Similarly, the homologous hnRNPA1 [9] also bound specifically to the antisense repeat RNA, and again binding could be significantly reduced by excess amounts of the non-biotinylated competitor RNA (Fig. 1b). Thus, hnRNPA3 interacts with both the sense and the antisense repeat RNA.

hnRNPA3 and hnRNPA1 bind to sense and antisense repeat RNAs. Nuclear extracts of HeLa cells were incubated with the indicated biotinylated RNA probes with (+) or without (−) 50-fold excess of non-biotinylated RNA competitor and pulled down by magnet beads. a Western blotting with an anti-hnRNPA3 antibody reveals that hnRNPA3 selectively binds to sense repeats (repeat probe: S) and antisense repeats (repeat probe: AS), which is inhibited by an excess of the non-biotinylated competitor RNA. b Western blotting with an anti-hnRNPA1 antibody reveals that hnRNPA1 binds to sense repeats (repeat probe: S) and antisense repeats (repeat probe: AS), which is inhibited by an excess of the non-biotinylated competitor RNA. C: (A4C2)17-control repeat, S: (G4C2)17-sense repeat, AS: (C4G2)17-antisense repeat
Reduction of hnRNPA3 leads to an increase of antisense RNA foci
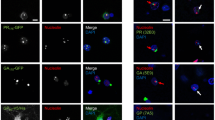
To investigate if reduction of hnRNPA3 leads to increased antisense RNA foci as observed before for sense RNA foci [42], we performed fluorescent in situ hybridization (FISH) using three independent fibroblast lines derived from patients with C9orf72 repeat expansions [42]. Upon siRNA-mediated hnRNPA3 knockdown (Fig. 2a), the number of foci per cell as well as foci positive cell increased compared to control siRNA or mock-treated cells (Fig. 2b–d). Furthermore, in iPSC-derived human neurons generated from fibroblasts derived from C9-case 1 and 2, siRNA-mediated knockdown of hnRNPA3 (Fig. 2e) also led to a significant increase of nuclear antisense RNA foci as well RNA foci positive cell (Fig. 2f–h). Thus, reduced nuclear hnRNPA3 leads to increased formation of sense and antisense repeat RNA foci.

Reduction of hnRNPA3 increases RNA foci number. Knockdown of hnRNPA3 increases antisense RNA foci in fibroblasts derived from C9orf72 mutation carriers. a Western blotting with anti-hnRNPA3, hnRNPA1, and hnRNPA2B1 antibodies confirms selective and efficient siRNA-mediated knockdown of hnRNPA3. β-actin serves as a loading control. b Fluorescent in situ hybridization (FISH) of antisense repeat RNA foci in fibroblasts from 3 cases with a C9orf72 repeat extension (C9) and 1 control case (Ct). Arrows indicate Cy3-positive antisense RNA foci in the nucleus. c, d Quantification of RNA foci number and positivity upon siRNA-mediated knockdown of hnRNPA3. N = 126–234 cells from 3 biological replicates in each line. e Western blotting of cell lysates from iPSC-derived human neurons. Western blotting with antibodies to β-actin and βIII-tubulin confirmed selective and efficient knockdown of hnRNPA3. f FISH of antisense RNA foci in neurons from control cases (Ct) and C9orf72 carriers (C9). siCt: control siRNA, siA3: hnRNPA3-targeted siRNA. g, h Knockdown of hnRNPA3 increases antisense RNA foci number and positivity in iPSC-derived neurons from C9orf72 mutation carriers. N = 72–102 cells from 3 biological replicates in each line. Data of 6 images in each line were used for analysis. NT non-treatment, siCt control siRNA, siA3 hnRNPA3-targeted siRNA, NCL nucleolin. All graphs are shown as mean ± SEM. *p < 0.05; two tailed paired t test. Scale bar 10 μm
hnRNPA3 reduction increases DPR generation from the antisense repeat RNA
Since knockdown of hnRNPA3 increased not only sense RNA foci [42] but also antisense RNA foci (Fig. 2), we thought that the antisense-derived DPRs poly-PA/PR/GP may also increase concomitantly. To prove this, we first generated hnRNPA3 knockout (A3KO) HeLa cells using Crispr/Cas9 gene editing (Suppl. Fig. 2a, b). We verified the introduction of a frame shift followed by a premature stop codon in selected cell clones (Suppl. Fig. 2b) and then confirmed absence of hnRNPA3 expression by western blotting (Fig. 3a). Clone Y-14 was selected and transfected with sense and antisense repeat constructs that express fluorescent tags in all three reading frames of both repeat encoding DNA strands (Fig. 3b). First, we confirmed by qPCR that repeat RNAs from both strands were increased in the absence of hnRNPA3 (Fig. 3c). Re-expression of hnRNPA3 reduced the increase of repeat RNAs observed in the hnRNPA3 knockout (Fig. 3c). We then used filter trap analyses [31] to quantify DPR production. This revealed that all DPRs made from sense and antisense repeat RNA were significantly increased in the absence of hnRNPA3 (Fig. 3d). Thus, reduction of hnRNPA3 increases sense and antisense repeat RNA foci as well as all DPRs translated from sense and antisense repeat RNA. In contrast, overexpression of hnRNPA3 decreased repeat RNAs (Suppl. Fig. 4a) and DPRs (Suppl. Fig. 4b). These findings indicate that hnRNPA3 is strongly associated with the degradation of repeat RNA and subsequent expression of DPRs.

Repeat RNAs and DPRs are increased upon hnRNPA3 knockout. a Western blotting confirms selective knockout of hnRNPA3 in cell lines Y-9 and Y-14. Line Y-14 was selected for all following analyses. b Schematic representation of cDNA constructs generated to express sense and antisense DPRs fused to individual tags. All graphs are shown as mean ± SEM. *p < 0.05, **p < 0.01; two tailed paired t test. c Quantification of sense and antisense repeat RNA upon hnRNPA3 knockout and rescue by exogenous expression of hnRNPA3. N = 3 biological replicates. NT non-treatment, EV empty vector, S sense repeat RNA, AS antisense repeat RNA, mCh mCherry, A3 hnRNPA3, WT HeLa wild type cells, A3KO HeLa cells with knockout of hnRNPA3. d Filter trap assay demonstrating the increase of all DPRs produced from sense and antisense repeat RNA upon hnRNPA3 knockout. N = 4 biological replicates. All graphs are shown as mean ± SEM. *p < 0.05, **p < 0.01; two tailed paired t-test
hnRNPA3 reduction increases DNA damage in DPR-expressing HeLa cells and C9orf72 patient-derived neurons
Since reduced hnRNPA3 enhances DSB formation [13], repeat RNA levels, and DPR expression (Figs. 2, 3), we investigated the link between hnRNPA3, DPR expression, and DNA damage/repair. To do so, we determined DPR-dependent DNA damage by quantifying γH2AX and pATM foci in the presence and absence of hnRNPA3. Expression of poly-GA, poly-GR, and poly-PR alone increased γH2AX foci formation in HeLa cells (Fig. 4a, b: p < 0.05 or < 0.01, see Suppl. Table 2), which was even more pronounced in the absence of hnRNPA3 (Fig. 4a, b: p < 0.05 or < 0.01, see Suppl. Table 2).

hnRNPA3 knockout enhances DNA damage in DPR expressing HeLa cells. a Immunocytochemical detection of γH2AX foci in GFP-tagged DPR-expressing HeLa WT and A3KO HeLa cells. b Fold change of γH2AX foci in HeLa WT and A3KO cells expressing the indicated DPRs relative to HeLa WT cells with GFP expression. N = 47–127 cells from 2 biological replicates. c Immunocytochemical detection of pATM foci in GFP-tagged DPR-expressing HeLa WT and A3KO HeLa cells. d Fold change of pATM foci in HeLa WT and A3KO cells expressing the indicated DPRs relative to HeLa WT cells with GFP expression. N = 71–263 cells from 2 biological replicates. GFP EGFP transfected, GA EGFP-tagged poly-GA 175 repeats transfected, GR EGFP-tagged poly-GR 177 repeats transfected, PR: EGFP-tagged poly-PR 176 repeats transfected. All graphs are shown as mean ± SEM. *p < 0.05, **p < 0.01; one-way ANOVA and Tukey’s post-hoc test. Scale bar 10 μm
Previously, it was reported that cells expressing expanded G4C2 repeat RNA or poly-GA showed decreased pATM foci in nuclei [58]; however, it is unclear if poly-GR and PR affect pATM. Therefore, we investigated the pATM foci number in cells expressing GFP or GFP-tagged DPR. Interestingly, only poly-GR and poly-PR, but not poly-GA, increased the number of pATM foci in the presence of hnRNPA3 (Fig. 4c, d: p < 0.05 or < 0.01, see Suppl. Table 2). Furthermore, in the absence of hnRNPA3, the number of pATM foci were further increased only by poly-GR and poly-PR but not by poly-GA (Fig. 4 c, d: p < 0.05 or < 0.01, see Suppl. Table 2). Surprisingly, even pATM foci formation evoked by GFP expression exceeded the amount of pATM foci evoked by poly-GA (Fig. 4d: p < 0.05, see Suppl. Table 2). These findings may suggest that poly-GA inhibits the recruitment of ATM to the site of DNA damage and thus enhances DNA damage by a mechanism different from that of arginine-containing DPRs.
We next asked if increased DNA damage after reduction of hnRNPA3 is also observed in human C9orf72 patient-derived neurons. siRNA-mediated knockdown of hnRNPA3 significantly increased the number of γH2AX foci in human patient-derived neurons (Fig. 5a, b). Similarly, pATM positive foci (Fig. 5c, d) were also increased upon knockdown of hnRNPA3 in patient-derived human neurons.

Reduction of hnRNPA3 enhances DNA damage in human neurons. a Immunocytochemical detection of γH2AX foci upon siRNA-mediated knockdown of hnRNPA3 in nuclei of motor neurons derived from iPS cells with or without C9orf72 repeat expansion. b Quantification of the number of γH2AX signals observed in a. N = 721–1558 cells from 2 biological replicates in each line. c Immunocytochemical detection of pATM foci upon siRNA-mediated knockdown of hnRNPA3 in the nuclei of neurons derived from iPSCs with or without C9orf72 repeat expansion. d Quantification of the number of pATM signals observed in c. N = 46–59 cells from 2 biological replicates. Ct control case, C9C9orf72 mutation carriers, siCt control siRNA, siA3 hnRNPA3-targeted siRNA. All graphs are shown as mean ± SEM. *p < 0.05, **p < 0.01; one-way ANOVA and Tukey’s post-hoc test. Scale bar 10 μm
Low hnRNPA3 expression in hippocampal dentate gyri of C9orf72 patients is associated with enhanced DNA damage
To investigate whether increased DNA damage also occurs in human brains and is dependent on hnRNPA3 expression levels, we measured the correlation between the presence of γH2AX foci, pATM deposits, and poly-GA inclusions with nuclear hnRNPA3 expression in granular cells of the hippocampal dentate gyrus derived from patients with C9orf72 mutations by immunohistochemistry (Fig. 6a, b, Suppl. Table 3). Indeed, an increased number of γH2AX foci positive cells were found with lowered nuclear signal intensity of hnRNPA3 (Fig. 6a, b; Pearson’s r: − 0.520, p < 0.05, Suppl. Table 3). Furthermore, the number of granular cells with poly-GA inclusions was negatively correlated with the number of granular cells with pATM foci (Fig. 6c, d. Pearson’s r: − 0.512, p < 0.05, Suppl. Table 3).

Elevated DNA double strand breaks are associated with reduced hnRNPA3 and pATM foci in the dentate gyrus of C9orf72 patients. a Immunohistochemical detection of hnRNPA3 (A3) and γH2AX foci, a marker for double strand breaks, in granular cells of hippocampal dentate gyri of a control (Ct) case and C9orf72 carriers (C9) with high (case 16) and low (case 3) nuclear hnRNPA3 expression. Scale bar 25 μm (low magnification figures)/10 μm (high magnification figures). b Scatter plot of nuclear hnRNPA3 signal intensities and percentages of nuclei with γH2AX foci of dentate gyrus granular cells in C9orf72 carriers. Nuclear A3 intensity negatively correlates with γH2AX foci. Pearson’s r: − 0.520, p < 0.05. N = 16 cases, total number of cells counted: 242–636 cells in each case. c Immunohistochemical detection of poly-GA and pATM in granular cells of hippocampal dentate gyri of a control (Ct) case and C9orf72 mutation carriers (C9). Poly-GA positive cells tend not to have nuclear pATM foci. Scale bar 25 μm (low magnification figures)/10 μm (high magnification figures). d Scatter plot of percentages of dentate gyrus granular cells with poly-GA inclusions and percentages of granular cells with pATM foci in C9orf72 carriers. Poly-GA positivity negatively correlates with pATM foci. Pearson’s r: − 0.512, p < 0.05. N = 16 cases, total counted cell number: 185–407 cells in each case
To rule out exacerbated RNA degradation in cases with low hnRNPA3 signals, we investigated the independent RNA binding proteins FUS and HuR (Suppl. Fig. 5). Nuclear signal intensities of both RNA proteins showed no significant correlation with the nuclear signal intensity of hnRNPA3 (Suppl. Fig. 5b, e). Moreover, nuclear signal intensities of FUS and HuR did also not correlate with γH2AX foci positivity (Suppl. Fig. 5c, f).
Poly-GA sequesters pATM
Notably, all C9 cases contained cytoplasmic pATM aggregates partially colocalizing with poly-GA inclusions (Fig. 7a, Suppl. Fig. 6a). On the other hand, control cases only rarely contained cytoplasmic pATM deposits, which were additionally much smaller than those in C9 cases (Fig. 7a, b, Suppl. Fig. 6b). Interestingly, hnRNPA3 aggregates also colocalize with poly-GA inclusions (Fig. 8), which is consistent with our previous study [42]. Therefore, we hypothesized that poly-GA aggregation sequesters pATM in cytoplasmic aggregates. To confirm co-aggregation of poly-GA and pATM, we performed co-immunoprecipitations in GFP-GA transfected HeLa cells (Fig. 9a). Strikingly, the anti-pATM antibody selectively co-immunoprecipitated aggregated poly-GA, which accumulated in the stacker of the gel during gel electrophoresis (Fig. 9a). In contrast, non-aggregated poly-GA was not precipitated (Fig. 9a). Poly-GR and poly-PR were not co-precipitated by the anti-pATM antibody even when using cell lysates with higher amounts of both DPRs for the co-immunoprecipitation assays (Fig. 9b; compare to Fig. 9a). Furthermore, the anti-pATM antibody did not cross-react with aggregated poly-GA (Suppl. Fig. 7) demonstrating the specificity of the co-immunoprecipitation in Fig. 9a. To further confirm that poly-GA aggregates sequester pATM in the cytoplasm, where most GA deposits occur [43], we performed co-immunoprecipitations upon from nuclear and cytoplasmic fractions (Suppl. Fig. 8a). Aggregated poly-GA selectively co-precipitated with pATM in the cytoplasmic fraction (Suppl. Fig. 8b), while aggregated poly-GA was not detected in the nuclear fraction and consequently no co-immunoprecipitation was observed (Suppl. Fig. 8c). Poly-GR and poly-PR did not co-precipitate with pATM in the nuclear fractions or the cytoplasmic fractions (Suppl. Fig. 8b–d). Thus, we conclude that poly-GA deposits selectively sequester pATM within the cytoplasm.

Poly-GA aggregates sequester pATM in cytoplasmic aggregates. a Poly-GA aggregates partially colocalize with pATM aggregates in granular cells of hippocampal dentate gyri of C9orf72 mutation carriers. Scale bar 10 μm. b Percentages of dentate gyrus granular cells with cytoplasmic pATM aggregates (diameter > 1 μm) in control cases (N = 9) and C9orf72 mutation carriers (N = 16). p < 0.05; two tailed paired t test. Ct control cases, C9C9orf72 mutation carriers

Poly-GA aggregates colocalize with cytoplasmic hnRNPA3 deposits. Immunohistochemical detection of poly-GA (GA) and hnRNPA3 (A3) in granular cells of hippocampal dentate gyri of C9orf72 carriers (C9). Poly-GA aggregates partially colocalize with cytoplasmic hnRNPA3 deposits in granular cells of hippocampal dentate gyri of C9orf72 mutation carriers. Scale bar 25 μm (low magnification figures)/10 μm (high magnification figures)

Poly-GA co-immunoprecipitation with pATM. a Aggregated poly-GA selectively co-immunoprecipitates with pATM. b To increase the sensitivity of DPR detection, cell lysates with 10 × higher protein concentrations as in a were used for co-immunoprecipitation. Although aggregated poly-GR and poly-PR are readily detected, only aggregated poly-GA co-immunoprecipitates with pATM. FT flow through, NT non-transfected, GFP EGFP transfected, GFP-GA/GR/PR EGFP-tagged poly-GA/GR/PR 89 repeats transfected, IgG-HC IgG-heavy chain, IgG-LC IgG-light chain
Discussion
Our study shows the functional and pathological consequences of loss of hnRNPA3 observed in C9orf72-associated neurodegeneration. First, hnRNPA3 not only binds to sense RNA repeats but also to antisense RNA repeats (Fig. 1). Second, lowering hnRNPA3 increases sense and antisense repeat RNAs, leading to enhanced RNA foci production as well as increased DPR generation (Figs. 2, 3). Third, reduced hnRNPA3 enhances DSB formation, which is induced by DPR (Fig. 4) and likely also by RNA foci (Fig. 5). Finally, poly-GA aggregates but no other DPRs sequester pATM within the cytoplasm and impede the recruitment of pATM to sites of DNA damage (Figs. 6, 7, 9, Suppl. Figs. 6, 8). Thus, the loss of hnRNPA3 expression likely potentiates DNA damage in C9orf72 patients’ brains.
In RNA pull-down assays we demonstrate that antisense repeat RNAs bind to hnRNPA3 similarly to sense repeat RNAs (Fig. 1a). Likewise, hnRNPA1 binds to both repeat transcripts (Fig. 1b). hnRNPA1 and A3 both share very similar RNA recognition motifs (RRM) [1, 59]. However, despite similar binding efficiencies, reduced hnRNPA3 increases poly-GA expression more than reduced hnRNPA1 [42].
Reduced nuclear hnRNPA3 expression positivity correlates with higher γH2AX foci in dentate gyrus granular cells of patients with C9orf72 mutations (Fig. 6a, b). This is in line with our cell culture models using HeLa cells and iPSC-derived human neurons (Figs. 4, 5). These findings indicate that reduction of hnRNPA3 further promotes DSB via increased RNA foci and DPR expression. This is also consistent with the finding that reduction of hnRNPA3 itself independently promotes DSB [13]. Indeed, depletion of hnRNPA3 increased γH2AX and pATM foci even in the cells with GFP only expression (Fig. 4). Thus, reduction of hnRNPA3 alone may enhance DSB, which is further increased by RNA foci and DPR expression.
Nuclear pATM foci as well as γH2AX foci are increased upon reduction of hnRNPA3 in cells expressing poly-GR and poly-PR (Fig. 4). In contrast, poly-GA decreased nuclear pATM foci, and, consequently, poly-GA deposition negatively correlated with nuclear pATM foci (Figs. 4c, d, 6c, d). Interestingly, cytoplasmic pATM aggregates were found in the dentate gyri of C9orf72 patients but only rarely in control cases (Fig. 7a, b). Co-IPs revealed that pATM bound selectively to aggregated poly-GA but not to soluble poly-GA (Fig. 9, Suppl. Fig. 8), which is in line with the partial co-localization of poly-GA deposits with pATM (Fig. 7a). These findings, therefore, suggest that pATM is selectively sequestered by poly-GA deposits and that sequestration impairs DSB repair in C9orf72 cases. Interestingly, it was reported that mutant huntingtin also sequesters pATM in the cytoplasm and impairs the recruitment of pATM to sites of irradiation-induced DSB [16]. Therefore, we speculate that pATM sequestration might be a common feature in repeat expansion disorders. However, alternative pathways such as defective nucleo-cytoplasmic transport may also impair pATM recruitment. ATM exists as a dimer in the cytoplasm [3, 56]. In response to DSB, ATM is oxidized, monomerized, and auto-phosphorylated. Subsequently, pATM binds to importin and shuttles into the nucleus [56]. DPR and C9orf72 repeat RNA are thought to impair nucleocytoplasmic transport [19, 28, 65, 66]. Therefore, nucleocytoplasmic transport dysfunction may additionally impair the recruitment of pATM to the nucleus. Walker et al. reported that repeat RNA increased γH2AX foci, and decreased pATM foci in the nucleus [58]. They concluded that activation of ATM was impaired by DPRs and repeat RNA. We now demonstrate that DPRs may affect ATM activation via differential individual mechanisms, i.e., poly-GA, but not poly-GR or poly-PR, sequesters ATM in cytoplasmic deposits. We have not further investigated how poly-GR and poly-PR enhance DSB without sequestration of pATM; however, there are some reports that show an association of poly-GR and poly-PR with DNA damage repair. Lopez-Gonzalez et al. reported that poly-GR binds to mitochondrial ribosomal proteins and leads to mitochondrial dysfunction, oxidative stress, and DNA damage [32]. Others reported that poly-GR and poly-PR change the intranuclear distribution of NPM1, which is protective against DNA damage [15, 54, 62]. Thus, each neurotoxic DPR may affect DNA damage in an individual way. On the other hand, the prevalence of poly-GR and poly-PR depositions is much lower than poly-GA deposits in human brains [51]. Poly-GA may, therefore, be the most relevant DPR in the pathological cascade of DNA damage in human brains.
Finally, we have not fully defined how H2AX is phosphorylated in the absence of pATM recruitment. After pATM is recruited to sites of DSB, pATM normally phosphorylates H2AX and converts it to γH2AX [10]. Thus, it is rather surprising that poly-GA increases γH2AX while decreasing pATM. We speculate that under these conditions γH2AX is phosphorylated by the poly-GA induced DNA-PK, which is a known H2AX kinase [53]. Therefore, DNA-PK may be able to phosphorylate H2AX at the sites of DSB, though precipitation of pATM into poly-GA aggregates may nevertheless cause a loss of function.
Taken together, we conclude that reduced hnRNPA3 enhances DNA damage (1) by increasing (G4C2)n and (C4G2)n expressions and foci formation (2) by increasing DPR expression, and (3) by impairment of ATM-mediated DNA damage repair through the sequestration of pATM by poly-GA (Fig. 10).

Schematic model demonstrating the cellular consequences of reduced hnRNPA3
References
Akindahunsi AA, Bandiera A, Manzini G (2005) Vertebrate 2xRBD hnRNP proteins: a comparative analysis of genome, mRNA and protein sequences. Comput Biol Chem 29:13–23. https://doi.org/10.1016/j.compbiolchem.2004.11.002
Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77:639–646. https://doi.org/10.1016/j.neuron.2013.02.004
Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–506. https://doi.org/10.1038/nature01368
Barker S, Weinfeld M, Zheng J, Li L, Murray D (2005) Identification of mammalian proteins cross-linked to DNA by ionizing radiation. J Biol Chem 280:33826–33838. https://doi.org/10.1074/jbc.M502477200
Barroso-Gonzalez J, Auclair S, Luan S, Thomas L, Atkins KM, Aslan JE et al (2016) PACS-2 mediates the ATM and NF-kappaB-dependent induction of anti-apoptotic Bcl-xL in response to DNA damage. Cell Death Differ 23:1448–1457. https://doi.org/10.1038/cdd.2016.23
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH et al (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. https://doi.org/10.1038/ng1769
Bennion Callister J, Ryan S, Sim J, Rollinson S, Pickering-Brown SM (2016) Modelling C9orf72 dipeptide repeat proteins of a physiologically relevant size. Hum Mol Genet 25:5069–5082. https://doi.org/10.1093/hmg/ddw327
Boeynaems S, Gitler AD (2018) Pour Some Sugar on TDP(-43). Mol Cell 71:649–651. https://doi.org/10.1016/j.molcel.2018.08.032
Bradley-Whitman MA, Timmons MD, Beckett TL, Murphy MP, Lynn BC, Lovell MA (2014) Nucleic acid oxidation: an early feature of Alzheimer's disease. J Neurochem 128:294–304. https://doi.org/10.1111/jnc.12444
Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ (2001) ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467. https://doi.org/10.1074/jbc.C100466200
Camins A, Pizarro JG, Alvira D, Gutierrez-Cuesta J, de la Torre AV, Folch J et al (2010) Activation of ataxia telangiectasia muted under experimental models and human Parkinson's disease. Cell Mol Life Sci 67:3865–3882. https://doi.org/10.1007/s00018-010-0408-5
Cloutier A, Shkreta L, Toutant J, Durand M, Thibault P, Chabot B (2018) hnRNP A1/A2 and Sam68 collaborate with SRSF10 to control the alternative splicing response to oxaliplatin-mediated DNA damage. Sci Rep 8:2206. https://doi.org/10.1038/s41598-018-20360-x
Comegna M, Succoio M, Napolitano M, Vitale M, D'Ambrosio C, Scaloni A et al (2014) Identification of miR-494 direct targets involved in senescence of human diploid fibroblasts. FASEB J 28:3720–3733. https://doi.org/10.1096/fj.13-239129
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72:245–256. https://doi.org/10.1016/j.neuron.2011.09.011
Farg MA, Konopka A, Ying Soo K, Ito D, Atkin JD (2017) The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet 26:2882–2896. https://doi.org/10.1093/hmg/ddx170
Ferlazzo ML, Sonzogni L, Granzotto A, Bodgi L, Lartin O, Devic C et al (2014) Mutations of the Huntington's disease protein impact on the ATM-dependent signaling and repair pathways of the radiation-induced DNA double-strand breaks: corrective effect of statins and bisphosphonates. Mol Neurobiol 49:1200–1211. https://doi.org/10.1007/s12035-013-8591-7
Fifita JA, Zhang KY, Galper J, Williams KL, McCann EP, Hogan AL et al (2017) Genetic and pathological assessment of hnRNPA1, hnRNPA2/B1, and hnRNPA3 in familial and sporadic amyotrophic lateral sclerosis. Neurodegener Dis 17:304–312. https://doi.org/10.1159/000481258
Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G et al (2012) C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci Rep 2:1016. https://doi.org/10.1038/srep01016
Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525:129–133. https://doi.org/10.1038/nature14974
Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G et al (2012) A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11:54–65. https://doi.org/10.1016/s1474-4422(11)70261-7
Giuliano P, De Cristofaro T, Affaitati A, Pizzulo GM, Feliciello A, Criscuolo C et al (2003) DNA damage induced by polyglutamine-expanded proteins. Hum Mol Genet 12:2301–2309. https://doi.org/10.1093/hmg/ddg242
Glass H, Pal A, Reinhardt P, Sterneckert J, Wegner F, Storch A et al (2018) Defective mitochondrial and lysosomal trafficking in chorea-acanthocytosis is independent of Src-kinase signaling. Mol Cell Neurosci 92:137–148. https://doi.org/10.1016/j.mcn.2018.08.002
Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507:195–200. https://doi.org/10.1038/nature13124
Haley B, Paunesku T, Protic M, Woloschak GE (2009) Response of heterogeneous ribonuclear proteins (hnRNP) to ionising radiation and their involvement in DNA damage repair. Int J Radiat Biol 85:643–655. https://doi.org/10.1080/09553000903009548
Horton TM, Graham BH, Corral-Debrinski M, Shoffner JM, Kaufman AE, Beal MF et al (1995) Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington's disease patients. Neurology 45:1879–1883
Japtok J, Lojewski X, Naumann M, Klingenstein M, Reinhardt P, Sterneckert J et al (2015) Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis 82:420–429. https://doi.org/10.1016/j.nbd.2015.07.017
Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB et al (2016) Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Hum Mol Genet. https://doi.org/10.1093/hmg/ddw052
Khosravi B, Hartmann H, May S, Mohl C, Ederle H, Michaelsen M et al (2017) Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD. Hum Mol Genet 26:790–800. https://doi.org/10.1093/hmg/ddw432
Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T et al (2014) Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345:1139–1145. https://doi.org/10.1126/science.1254917
Lee SK, Jurata LW, Funahashi J, Ruiz EC, Pfaff SL (2004) Analysis of embryonic motoneuron gene regulation: derepression of general activators function in concert with enhancer factors. Development 131:3295–3306. https://doi.org/10.1242/dev.01179
Li H, Wyman T, Yu ZX, Li SH, Li XJ (2003) Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet 12:2021–2030. https://doi.org/10.1093/hmg/ddg218
Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D et al (2016) Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92:383–391. https://doi.org/10.1016/j.neuron.2016.09.015
Loughlin FE, Lukavsky PJ, Kazeeva T, Reber S, Hock EM, Colombo M et al (2019) The solution structure of FUS bound to RNA reveals a bipartite mode of RNA recognition with both sequence and shape specificity. Mol Cell 73(490–504):e496. https://doi.org/10.1016/j.molcel.2018.11.012
Lovell MA, Markesbery WR (2007) Oxidative DNA damage in mild cognitive impairment and late-stage Alzheimer's disease. Nucleic Acids Res 35:7497–7504. https://doi.org/10.1093/nar/gkm821
Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta Neuropathol 126:859–879. https://doi.org/10.1007/s00401-013-1181-y
McGurk L, Gomes E, Guo L, Mojsilovic-Petrovic J, Tran V, Kalb RG et al (2018) Poly(ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell 71(703–717):e709. https://doi.org/10.1016/j.molcel.2018.07.002
Merlo D, Mollinari C, Racaniello M, Garaci E, Cardinale A (2016) DNA double strand breaks: a common theme in neurodegenerative diseases. Curr Alzheimer Res 13:1208–1218. https://doi.org/10.2174/15672050136661604011
Mizielinska S, Ridler CE, Balendra R, Thoeng A, Woodling NS, Grasser FA et al (2017) Bidirectional nucleolar dysfunction in C9orf72 frontotemporal lobar degeneration. Acta Neuropathol Commun 5:29. https://doi.org/10.1186/s40478-017-0432-x
Mizielinska SL, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 126:845–857. https://doi.org/10.1007/s00401-013-1200-z
Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K et al (2013) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126:881–893. https://doi.org/10.1007/s00401-013-1189-3
Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H et al (2013) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125:413–423. https://doi.org/10.1007/s00401-013-1088-7
Mori K, Nihei Y, Arzberger T, Zhou Q, Mackenzie IR, Hermann A et al (2016) Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide-repeat protein deposition. EMBO Rep 17:1314–1325. https://doi.org/10.15252/embr.201541724
Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E et al (2013) The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339:1335–1338. https://doi.org/10.1126/science.1232927
Naumann M, Pal A, Goswami A, Lojewski X, Japtok J, Vehlow A et al (2018) Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 9:335. https://doi.org/10.1038/s41467-017-02299-1
Nihei Y, Ito D, Okada Y, Akamatsu W, Yagi T, Yoshizaki T et al (2013) Enhanced aggregation of androgen receptor in induced pluripotent stem cell-derived neurons from spinal and bulbar muscular atrophy. J Biol Chem 288:8043–8052. https://doi.org/10.1074/jbc.M112.408211
Nordin A, Akimoto C, Wuolikainen A, Alstermark H, Forsberg K, Baumann P et al (2017) Sequence variations in C9orf72 downstream of the hexanucleotide repeat region and its effect on repeat-primed PCR interpretation: a large multinational screening study. Amyotroph Lateral Scler Frontotemporal Degener 18:256–264. https://doi.org/10.1080/21678421.2016.1262423
O'Driscoll M, Jeggo PA (2006) The role of double-strand break repair - insights from human genetics. Nat Rev Genet 7:45–54. https://doi.org/10.1038/nrg1746
Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY et al (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162:1066–1077. https://doi.org/10.1016/j.cell.2015.07.047
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. https://doi.org/10.1038/nprot.2013.143
Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268. https://doi.org/10.1016/j.neuron.2011.09.010
Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C et al (2015) Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta Neuropathol 130:537–555. https://doi.org/10.1007/s00401-015-1450-z
Sivadasan R, Hornburg D, Drepper C, Frank N, Jablonka S, Hansel A et al (2016) C9ORF72 interaction with cofilin modulates actin dynamics in motor neurons. Nat Neurosci. https://doi.org/10.1038/nn.4407
Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA (2004) ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res 64:2390–2396
Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X et al (2015) Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum Mol Genet 24:2426–2441. https://doi.org/10.1093/hmg/ddv005
Tchurikov NA, Kretova OV, Fedoseeva DM, Sosin DV, Grachev SA, Serebraykova MV et al (2013) DNA double-strand breaks coupled with PARP1 and HNRNPA2B1 binding sites flank coordinately expressed domains in human chromosomes. PLoS Genet 9:e1003429. https://doi.org/10.1371/journal.pgen.1003429
The COPERNIC project investigators, Granzotto A, Benadjaoud MA, Vogin G, Devic C, Ferlazzo ML et al (2016) Influence of nucleoshuttling of the ATM protein in the healthy tissues response to radiation therapy: toward a molecular classification of human radiosensitivity. Int J Radiat Oncol Biol Phys 94:450–460. https://doi.org/10.1016/j.ijrobp.2015.11.013
van der Zee J, Gijselinck I, Dillen L, Van Langenhove T, Theuns J, Engelborghs S et al (2013) A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum Mutat 34:363–373. https://doi.org/10.1002/humu.22244
Walker C, Herranz-Martin S, Karyka E, Liao C, Lewis K, Elsayed W et al (2017) C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci. https://doi.org/10.1038/nn.4604
Wei Y (2014) Characterization of hnRNP A3 isoforms in the A2RE-proteome. School of Chemistry and Molecular Biosciences, The University of Queensland, City
Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y et al (2014) Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84:1213–1225. https://doi.org/10.1016/j.neuron.2014.12.010
West SC (2003) Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 4:435–445. https://doi.org/10.1038/nrm1127
White MR, Mitrea DM, Zhang P, Stanley CB, Cassidy DE, Nourse A et al (2019) C9orf72 Poly(PR) dipeptide repeats disturb biomolecular phase separation and disrupt nucleolar function. Mol Cell. https://doi.org/10.1016/j.molcel.2019.03.019
Zamiri B, Reddy K, Macgregor RB Jr, Pearson CE (2014) TMPyP4 porphyrin distorts RNA G-quadruplex structures of the disease-associated r(GGGGCC)n repeat of the C9orf72 gene and blocks interaction of RNA-binding proteins. J Biol Chem 289:4653–4659. https://doi.org/10.1074/jbc.C113.502336
Zhang J, Perry G, Smith MA, Robertson D, Olson SJ, Graham DG et al (1999) Parkinson's disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154:1423–1429. https://doi.org/10.1016/s0002-9440(10)65396-5
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525:56–61. https://doi.org/10.1038/nature14973
Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF et al (2016) C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nat Neurosci 19:668–677. https://doi.org/10.1038/nn.4272
Acknowledgements
This work was supported by the NOMIS foundation (to C.H., D.E. and A.H.), the Munich Cluster of Systems Neurology (SyNergy) (to C.H and D.E.) and the Helmholtz Virtual Institute “RNA dysmetabolism in ALS and FTD (VH-VI-510)” (to C.H. and A.H). KM is supported by grants from JSPS KAKENHI (JP16H05379, JP17H05091, JP18K19515), AMED (JP18ek0109316), Mochida Memorial Foundation, Takeda Science Foundation and SENSHIN Medical Research Foundation, AH is supported by the Hermann und Lilly Schilling-Stiftung für medizinische Forschung im Stifterverband. DE was supported by the European Community's Health Seventh Framework Programme under grant agreement 617198 [DPR-MODELS]. AS and MW were supported by the Swiss ALS Foundation. We were grateful to Daisuke Ito (Keio University School of Medicine, Department of Neurology) for providing Lenti-Hb9-GFP plasmid, Xenia Lojewski and Hannes Glass for the help of the generation of iPSCs, and Jane Hettinger for carefully proofreading the manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
CH collaborates with Denali Therapeutics on a topic not related to C9orf72, participated on one advisory board meeting of Biogen, and received a speaker honorarium from Novartis and Roche. All other authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants or participants’ families included in this study.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Nihei, Y., Mori, K., Werner, G. et al. Poly-glycine–alanine exacerbates C9orf72 repeat expansion-mediated DNA damage via sequestration of phosphorylated ATM and loss of nuclear hnRNPA3. Acta Neuropathol 139, 99–118 (2020). https://doi.org/10.1007/s00401-019-02082-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-019-02082-0