
Abstract
Synaptic failure is an immediate cause of cognitive decline and memory dysfunction in Alzheimer’s disease. Dendritic spines are specialized structures on neuronal processes, on which excitatory synaptic contacts take place and the loss of dendritic spines directly correlates with the loss of synaptic function. Dendritic spines are readily accessible for both in vitro and in vivo experiments and have, therefore, been studied in great detail in Alzheimer’s disease mouse models. To date, a large number of different mechanisms have been proposed to cause dendritic spine dysfunction and loss in Alzheimer’s disease. For instance, amyloid beta fibrils, diffusible oligomers or the intracellular accumulation of amyloid beta have been found to alter the function and structure of dendritic spines by distinct mechanisms. Furthermore, tau hyperphosphorylation and microglia activation, which are thought to be consequences of amyloidosis in Alzheimer’s disease, may also contribute to spine loss. Lastly, genetic and therapeutic interventions employed to model the disease and elucidate its pathogenetic mechanisms in experimental animals may cause alterations of dendritic spines on their own. However, to date none of these mechanisms have been translated into successful therapeutic approaches for the human disease. Here, we critically review the most intensely studied mechanisms of spine loss in Alzheimer’s disease as well as the possible pitfalls inherent in the animal models of such a complex neurodegenerative disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Neuropathology of Alzheimer’s disease
In 1906, Alois Alzheimer examined the brain of a 54-year-old woman, who had died after a three-year course of severe cognitive impairment and memory loss. He noticed distinct histological alterations in the cortex, such as fibrillary tangles inside neurons and extracellular deposits of a substance unknown to him, which has later been identified as amyloid beta [4]. Quantification of these neuropathological alterations during autopsy is used today to assess whether an individual suffered from the disease now bearing Alzheimer’s name and how far the disease has progressed [26, 196]. These alterations are thought to be caused by an imbalance of amyloid beta production and its removal from the brain, causing the aggregation of characteristic fibrillar amyloid deposits. In turn, amyloid toxicity, which may be mediated by oligomeric intermediates and/or fibrillar amyloid beta, is thought to cause tau hyperphosphorylation and inflammatory changes as endogenous reactions to the presence of noxic stimuli. This pathogenic mechanism, which is essentially covered by the amyloid cascade hypothesis [80], is founded on numerous animal models which are genetically engineered to develop amyloid plaques. These animal models recapitulate some but not all the typical histologic alterations such as amyloidosis, synapse and neuron loss, tau hyperphosphorylation and inflammation. Another line of evidence is that humans with Down syndrome develop similar pathological changes as a result of the triplication of chromosome 21, on which the amyloid precursor protein (APP) is encoded [212]. Also, familial forms of Alzheimer’s disease are caused by mutations either in APP or in one of the two presenilin genes, which code for the enzymes processing APP to beta amyloid, ultimately leading to an overproduction of beta amyloid [175]. Lastly, one of the main risk factors of sporadic Alzheimer’s is homozygosity for the ε4 variant of the apolipoprotein E gene (ApoE4) [14], which causes reduced amyloid beta clearance [34]. While the amyloid cascade hypothesis is not without controversy [33], there is ample evidence that amyloid beta and hyperphosphorylated tau protein as well as the resultant inflammation may damage synaptic function.
Dendritic spines
Dendritic spine structure
Dendritic spines are the morphologic correlates of excitatory postsynapses. Morphologically, spines are specialized protrusions from a dendrite’s shaft, where neurons form synapses to receive and integrate information [69]. Typically, three different spine shapes are distinguished: Mushroom spines, which have a large head and a thin neck; stubby spines which have a large head but no discernible neck; and thin spines, which are slender, filopodia-like protrusions without a discernible head. A number of specialized synaptic proteins, including scaffolding proteins and ion channels, are clustered [136, 176] at dendritic spines. Spine size and morphology may reflect anatomical circumstances. For instance, longer spines may be observed in brain regions where target axons are located farther away from dendrites, such as in the reticular nucleus of the thalamus and in the gelatinous substance of the spinal cord dorsal horn [66]. Most importantly, however, dynamic alterations in spine morphology affect functional characteristics. For instance, increase in spine head size helps accommodate higher receptor numbers, while shortening and widening of spine necks decrease the electrical resistance of the spine neck, thereby leading to larger excitatory postsynaptic potentials [220].
Role of dendritic spines in synaptic plasticity
Synaptic plasticity is in part mediated by altering the number of synaptic AMPA receptors through fast trafficking mechanisms [121]. However, these functional alterations are accompanied by morphological adaptations of dendritic spines, such as changes both in the number and shape of spines, which are termed structural plasticity [69] and are the focus of this review (Fig. 1). Such alterations have been observed to occur within minutes [108], yet they may also endure over longer time spans [218]. For instance, learning of motor tasks is associated with an increased spine formation [217] and a fraction of these newly formed spines may persist permanently [218]. Conversely, keeping animals in an enriched environment, which broadly stimulates motor, sensory and cognitive systems, increases the turnover, i.e., both the formation as well as the elimination of dendritic spines and this turnover. The net effect of these changes is an increased density of spines [93]. A key mediator for this effect is brain-derived neurotrophic factor (BDNF) [64], which acts via two receptors, NTRK2 (also known as TRKB) and NGFR (also known as p75NGF). A central regulator for structural plasticity is the enzyme glycogen synthase kinase 3β (GSK3β), which is a target of many psychotropic drugs [8]: Long-term potentiation, which is a functional correlate of synaptic plasticity, leads to inhibition of GSK3β [56, 146], which in turn increases structural plasticity by destabilization and increased turnover of dendritic spines [140]. Furthermore, a host of cytoskeletal proteins [178], as well as local protein translation [197], is required for the proper maintenance and turnover of dendritic spines.

Dendritic spines are remodeled in enriched environment. a Chronic in vivo imaging of the same apical dendrites from layer V pyramidal neurons in the somatosensory cortex over 43 days when GFP-M mice were housed in standard conditions (SC) or enriched environment (EE). Blue and green arrowheads point to preexisting spines which are shown in the first image and new-gained spines that emerged over two consecutive imaging sessions, respectively. Empty arrowheads indicate the lost preexisting spines (blue) and new-gained spines (green). The images show high-contrast representations of maximum-intensity projected multiphoton images. b Graphical representations of the relative spine density. c, d The fate of preexisting spines in the first imaging time point and new-gained spines at the second imaging time point. e Apical dendrites (black) from layer V pyramidal neurons housed in SC or EE and 3D reconstructions (gray) generated in Imaris. The images show high-contrast representations of maximum-intensity projected confocal images. Mushroom, thin and stubby spines are colored in green, blue and red. f Plot of mushroom, thin and stubby spine fractions when mice were exposed to SC or EE. g Cumulative distributions of spine length and head volume when mice were exposed to SC or EE. *p < 0.05, **p < 0.01 (Two-way ANOVA in b–d, Student’s t test in f and Komogornov–Smirnov test in g)
Dendritic spine pathology
Disturbance of the physiologic spine homeostasis underlies a number of neuropsychiatric disorders [147]. The most prominent example is loss of dendritic spines, which is encountered in most neurodegenerative disorders. Pathological spine loss can be caused by altering presynaptic input due to neuron-autonomous or extra-neuronal factors (Fig. 2). Synaptic factors for pathological spine loss may be deafferentation, which leads to a loss of complete dendrites [47, 90, 124], or sensory deprivation, which causes more complex changes: a retinal lesion, for instance, causes a complete replacement of spines in the deafferented cortex [95]. Pathological activation of NMDA receptors during excitotoxicity [78, 82] or disruption of dendritic transport both lead to spine loss and can be considered neuron-autonomous causes. Similarly, disruption of local protein synthesis at the spine may alter spine densities and morphology [199]. Examples for extraneuronally caused spine loss are trauma or inflammation, which in turn act through multiple mechanisms. Trauma causes initial spine loss mediated by calcineurin, followed by an overgrowth of spines [31]. Inflammation causes secretion of interleukin 1β, which antagonizes the action of BDNF, thereby leading to spine loss [201]. Tumor necrosis factor α (TNFα) from activated microglia leads to phosphorylation and upregulation of AMPA receptors, which in turn causes excitotoxicity [65, 109], thereby leading to spine loss [36]. Lastly, alterations in the composition of the extracellular matrix are associated with synapse loss [132].

Putative pathophysiological mechanisms for dendritic spine loss. 1 Fibrillar amyloid plaques cause spine loss in their immediate vicinity. 2 Spine loss at dystrophic dendrites. 3 Secondary spine loss due to presynaptic failure. 4 Amyloid oligomers engaging synaptic targets. 5 Amyloid uptake. 6 Axonal amyloid beta. 7 Hyperphosphorylated tau protein. 8 Deafferentation. 9 NMDA receptor-mediated excitotoxicity. 10 Spine pruning by microglia; Microglia activation leading to release of inflammatory mediators. 11 Immune activation, reactive oxygen species and activation of pro-apoptotic pathways
On the other hand, not only spine loss, but also an increased stability or density of spines may be consequences of pathological mechanisms. Patients suffering from fragile X syndrome, which causes severe mental retardation, have elevated spine numbers [87]. Similarly, some neuropsychiatric diseases are accompanied with elevated spine densities in specific brain regions [147]. Thus, high spine densities per se are not necessarily desirable, either.
Evidence for synapse loss in Alzheimer’s disease
A landmark study in the 1990s analyzed biopsies from individuals with clinically manifest Alzheimer’s disease using electron microscopy and correlated synapse numbers with results from the Mini-Mental Status examination, which is a measure for cognitive function: Patients with Alzheimer’s disease showed a significant loss of synapses compared with cognitively normal controls, and their cognitive capabilities correlated with synapse density [53, 195]. Follow-up studies on post-mortem tissue were able to analyze brain regions which are not amenable to biopsy. These showed, for instance, that individuals with early forms of Alzheimer’s disease had significantly fewer synapses in the inferior temporal gyrus, which plays an important role in verbal fluency [171], in the CA1 region [169], in the dentate gyrus [170] and in the posterior cingulate gyrus, which is a cortical region affected early during the onset of Alzheimer’s disease [168]. Immunohistochemical analyses showed loss of the presynaptic marker synaptophysin [123]. Moreover, a recent postmortem study using intracellular injections of Lucifer yellow in the brains of 5 Alzheimer’s disease patients revealed that intraneuronal tau aggregates are associated with a progressive alteration of dendritic spines [127].
Further evidence for the loss of synaptic function comes from in vivo PET imaging studies. These use radionuclide-labeled agonists for specific neurotransmitter receptors to measure the abundance of these receptors in various brain regions. One such study showed loss of α4β2 nicotinic acetylcholine receptors in the medial frontal cortex and nucleus basalis magnocellularis, which suggests loss of cholinergic synapses. This loss of α4β2 receptors correlated with increasing amyloid levels and with a loss of specific cognitive functions [141]. CB1 cannabinoid receptors, in contrast, were not altered [2] and 5HT4 serotonin receptors were increased with increased amyloid deposition [96, 118]. 5HT1 serotonin receptors were lost in late stages of Alzheimer’s disease [129]. These changes at the synaptic level have prominent effects on a more global scale, leading to paradoxical hyperexcitability and disruption of large-scale networks [76, 182, 183], which in turn are thought to be functional correlates of clinically apparent symptoms like impaired memory and cognition.
Pathogenesis of dendritic spine loss in Alzheimer’s disease
Accumulation of amyloid beta is thought to be the initial causative factor leading to progressive synaptic injury [80]. However, secondary neuropathological alterations such as tau hyperphosphorylation or inflammation and consecutive dendritic and axonal dysfunction may cause synaptic damage on their own or exacerbate damage caused by amyloid beta.
Amyloid beta
Amyloid beta is one of a multitude of enzymatic cleavage products of APP [135] and its secretion into the extracellular space is increased with neuronal activity [45, 94] through activation of extrasynaptic NMDA receptors [24]. To be more precise, however, amyloid beta does not refer to a singular chemical substance, but to several, depending on the exact cleavage sites and post-translational modifications including oxidation, phosphorylation, nitration, racemization, isomerization, pyroglutamylation, and glycosylation [103]. For instance, the 42 amino acid version (Aβ1–42) has a stronger propensity to aggregate than the 40 amino acid version (Aβ1–40). Modifications at the N-terminus further alter the protein’s biophysical properties. Of particular interest, pyroglutamate amyloid beta (Aβp3–42), which has a cyclized glutamate residue at the N-terminus, has an even stronger propensity to aggregate [172] and seems to be specific for fibrillar plaques [54]. Amyloid beta can be detected in the extracellular as well as in the intracellular compartment, in oligomeric as well as fibrillar states. All these forms have been implicated in synaptic damage, which will be discussed in the following subsections.
Amyloid beta plaques
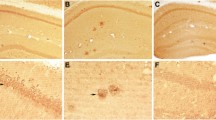
Amyloid plaques are the characteristic extracellular deposits of amyloid beta. Histologically, plaques may either appear either as diffuse plaques, which do not contain fibrils, and are detectable only in immunohistochemical stains using antibodies directed against APP epitopes. Alternatively, they may appear as cored plaques, which are composed of a fibrillar core and may be surrounded by a diffuse, non-fibrillar halo. The fibrillar core is detectable in H&E sections and can be stained with dyes specific for fibrillar aggregates such as Congo red or thioflavin S. Radiolabeled derivatives of these dyes, such as Pittsburgh compound B or florbetaben, are used as PET tracers to detect fibrillar amyloid beta in clinical settings, while fluorescent derivatives such as methoxy-X04 are used in animal studies for in vivo microscopy. As all of these compounds exclusively detect fibrillar protein aggregates, the majority of clinical and experimental in vivo studies have focused on fibrillar amyloid deposits. Indeed, there is ample evidence that fibrillar amyloid beta causes synaptic damage. In human cases, fibrillar amyloid plaques are typically surrounded by dystrophic neurites, which give rise to the so-called neuritic plaque appearance in silver stains or immunohistochemical stains against hyperphosphorylated tau protein using AT8 antibodies. In the human disease, dense neuritic AT8-staining also occurs distant to plaques in the form of neuropil threads, and the location and extent of this staining are the decisive measures to obtain the neuropathological staging according to the Braak and Braak criteria [26], which correlate best with the cognitive status. Some, but not all, mouse models of amyloidosis exhibit similar neuritic plaques, which are, like in the human disease, detectable using AT8 antibody staining or silver impregnation [39, 43, 154, 157, 184]. In contrast to the human disease, however, neuritic pathology in mouse models of amyloidosis is always limited to the immediate vicinity of fibrillar plaques (Fig. 3), while neuropil threads distant to plaques have not been observed. This might explain why in several animal models of Alzheimer’s disease which overexpress mutant human APP and/or presenilin alterations in spine density of layer 3 and 5 pyramidal neurons are apparent only in close vicinity of plaques (Fig. 4) [17, 97, 99, 214]. We found that spine loss occurred with a delay of at least 4 weeks after plaques had formed in one APP/PS1 mouse model [17]. However, the mechanisms leading to spine loss may differ between mouse models even if they share similar transgenes [228]. Nevertheless, in some mouse models, spine loss apparently independent of plaques was observed [19, 104]. In the triple transgenic mouse model co-expressing mutant APP, PS1 and tau, which we had analyzed, this spine loss occurred only at dystrophic dendrites with intracellular accumulation of both soluble amyloid beta and hyperphosphorylated tau protein [19]. Since substantial axonal damage occurs at amyloid plaques, secondary spine loss as a consequence of presynaptic failure [1] in those regions where damaged axons project to is very likely. Moreover, chronically altered synaptic input may affect the overall dendritic complexity and length in aged APP/PS1 mice while the dendritic spine density remains unaltered [179]. Both scenarios would explain a slight decrease in overall synapse density in some mouse models of amyloidosis in the absence of a significant reduction of the spine density of layer 3 and 5 neurons. Furthermore, functional alterations of neurons near plaques also point to a role of fibrillar amyloid beta in the damage of synaptic function, either direct or indirect [30]. Functional links between neuronal and synaptic dysfunction may be the disturbance of intracellular calcium dynamics [38, 100] or mitochondrial integrity [216]. Furthermore, perisomatic GABAergic terminals are lost close to plaques [70], which may also contribute to hyperexcitability and spine loss.

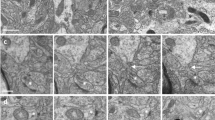
Neuritic pathology in a mouse model of amyloidosis. Electron micrograph of neuropil in the vicinity of a fibrillar plaque (not shown) of an APPPS1 mouse. Dystrophic axons (labeled “Axon”) appear enlarged and filled with electron-dense membranous material. Several synaptic densities are marked with arrows. Note that most synapses originate from unaltered synaptic boutons, while only a single synapse originates from the dystrophic axon (double arrow, far right), which may reflects spine loss as a result of presynaptic failure

Spine loss is observed on dendrites that are close to amyloid plaques. a GFP-labeled dendrites (black) and methoxy-X04-labeled amyloid plaques (blue) from APPPS1xGFP-M mice. The images show high-contrast representations of maximum-intensity projected multiphoton images. Black arrowheads point to dendrites that are located close (<30 µm) to or far from (>100 µm) plaques, which are shown enlarged in b. b Maximum intensity projected apical dendrites that are located close (<30 µm) to or far from (>100 µm) amyloid plaques. The same dendrites were repeatedly imaged 1 week apart. Red arrowheads point at spines eliminated over 1 week and green arrowheads point at newly formed spines. c Spine densities of apical dendrites on different localizations (>100 or <30 µm from plaques). Spines that were new-gained d or lost e over 1 week on dendrites (>100 or <30 µm from amyloid plaques). *p < 0.05, **p < 0.01 (Student’s t test)
Amyloid beta oligomers
Amyloid beta readily aggregates into soluble oligomers, which are still diffusible, in contrast to insoluble fibrils. As oligomers may exert effects distant to the place of their generation, they offer a convenient explanation for one of the major unsolved elements in the amyloid cascade hypothesis: the spatial separation of initial amyloid deposition and tau pathology in the brain. In the early stages of Alzheimer’s disease, amyloid beta accumulates preferentially in neocortical regions [196], while tau pathology typically starts in the brainstem and the transentorhinal region [26, 27]. Furthermore, the early stages of tau pathology often occur in individuals who do not show extracellular fibrillar amyloid deposits [27]. Similar to amyloid beta itself, amyloid oligomers are a highly heterogeneous group of chemicals, which are often classified according to their structural properties. The following variants are commonly distinguished [73, 77]: Protofibrils, which are beta-sheet containing intermediates of synthetic amyloid beta fibrillization; Annular assemblies and globulomers, which are both synthetic products; Amyloid beta-derived diffusible ligands, which are small, diffusible synthetic products; Aβ*56, which are endogenous products, found in APP transgenic mice and correspond to 12-mers; Secreted amyloid beta dimers and trimers, which are produced by cultured cells and are resistant to proteolytic degradation. It is important to note, however, that this heterogeneity not only reflects biological variation, but also technical variation in the methods to produce synthetic oligomers or to isolate them from biological tissues [12].
Nevertheless, there are several studies which point to functional relevance of oligomers in the human disease. For instance, in subjects with plaque pathology, the concentrations of amyloid oligomers were indicative of whether the subject had suffered from dementia [63] and synapse loss correlated with oligomer levels [98]. Dimers, trimers and Aβ*56 have been found in human subjects, with different associations to aging and Alzheimer’s disease [111]. Among these, Aβ*56 levels seem to correlate with synapse loss and the presence of tau oligomers [35, 111]. One animal study showed that removal of oligomers by genetically switching off their production led to cognitive improvement [68]. In another study, we showed that immunotherapy directed against globulomers abolished synapse loss distant from plaques, while in proximity to plaques synapse loss was attenuated only by a small degree [59], suggesting that plaque-associated pathology is not primarily mediated by oligomers. To assess the mechanism by which amyloid oligomers are synaptotoxic, oligomers are usually generated in vitro and then tested for their specific effects. However, a large variety of oligomers may be generated, depending on the exact experimental protocol used [89], and no single form of oligomers is accepted as the major contributor in Alzheimer’s disease [12]. Therefore, oligomers have been found to exert a wide variety of harmful effects on synapses [11]. Indeed, amyloid oligomers have been shown to bind preferentially to synapses [223]. There, they interact with a wide range of synaptic targets, such as prion protein [205], EphB2, RAGE, α7 nicotinic acetylcholine receptors, glutamate transporters [105], the Wnt receptor frizzled [120], insulin receptors, presynaptic P/Q channels, the neutrophin receptor p75 [150], mGluR5 [155, 204], β2 Adrenergic receptors [210], or calcineurin [214]. They may disrupt the neuritic cytoskeleton [223] and exacerbate neuronal activity-dependent DNA damage [165]. Finally, oligomers may also form ion-permeable pores in cell membranes [106], causing unregulated calcium entry and thereby leading to synaptic toxicity. It should be noted that most studies showing effects on the binding of synthetic amyloid oligomers to certain synaptic and extrasynaptic receptors lack control studies that guarantee specificity for the effects. Often equal concentrations’ monomers are used as controls, yet amyloid beta has a propensity to spontaneously aggregate to oligomers under various conditions in vitro [16, 83, 209]. Also scrambled peptides are used as controls. These are inappropriate controls for the specificity of an oligomeric protein complex, as scrambled peptides may lose their propensity to aggregate. So other either naturally occurring oligomeric proteins, such as tau, α-synuclein, gp120, PrP106–126 or synthetic oligomeric protein preparations [156], would be more appropriate to prove that the proposed effects of amyloid beta oligomers are truly specific for amyloid beta oligomers rather than oligomeric aggregates in general. Another commonly used control are fibrillar amyloid beta preparations which are observed to be less or not toxic to syanpses. The problem here is that the concentration of protein complexes injected into the brain is impossible to estimate [11, 12]. Thus, because of the complex biochemical behavior of amyloid oligomers, a single control peptide may not suffice to cover all possible unspecific effects. Furthermore, our in vivo studies show convincing evidence that fibrillar amyloid beta is highly toxic to synapses [17, 19, 59]. Amyloid deposition and evidence of fibrillar amyloid are time-dependent processes, not only in humans but also in animal models of Alzheimer’s disease. Although the exact age at which fibrillar deposits can be detected varies between models, most show an initial phase where no or only very few plaques are present. Several studies showed spine loss or synaptic dysfunction in these models before the appearance of plaques, which was generally interpreted as effects being mediated by soluble amyloid beta [29, 32]. Similarly, mouse models with the Osaka mutation, which generates oligomeric, but not fibrillar amyloid, develop synapse loss [200]. Additionally, if crossed with human tau expressing mice, these mice also develop tau pathology [206].
It is important to note, however, that the absence of plaques does not necessarily attribute causation to soluble amyloid beta, as intraneuronally accumulated amyloid beta or the genetic manipulations to obtain animal models may affect dendritic spines on their own (see below).
Intraneuronal amyloid beta
Intraneuronal accumulation of amyloid beta has been observed in Down syndrome as well as in the early stages of Alzheimer’s disease [74, 125, 133]. However, in older individuals with Down syndrome and in late stages of Alzheimer’s disease, when abundant plaques are present in the brain, intraneuronal accumulation of amyloid beta is less evident [74, 133], although still present [138, 192]. While most studies suggested that intraneuronal amyloid beta is specific for disorders with extracellular amyloid deposition, one found intraneuronal amyloid beta was also in hippocampal neurons of control cases [20]. Conversely, a mouse model overexpressing APP with the Dutch mutation (E693Q) showed only intraneuronal amyloid accumulation, but no extracellular deposits [102]. Curiously, humans with this mutation suffer mainly from cerebral hemorrhage [112]. In animal models of amyloidosis, intraneuronal accumulation of amyloid beta increases with age [177, 192]. In one animal model, intraneuronal accumulation of APP was correlated with spine loss (Fig. 5) [228]. In another model, intraneuronal accumulation of oligomeric amyloid beta led to altered synapse structure in the hippocampus, while spine densities were not changed [151]. Furthermore, extracellular amyloid was found to be taken up into neurons via receptor for advanced glycosylation end products (RAGE) [193] and hypercholesterinemia accelerated uptake, leading to reduced synaptophysin immunoreactivity, and abnormal tau phosphorylation in the hippocampus [207]. The accumulation of amyloid in neurons also depended on the apolipoprotein E genotype, which is a known genetic risk factor for Alzheimer’s disease. The ε4 (ApoE4) isoform, which confers the highest risk for Alzheimer’s disease, also strongly increased the intraneuronal accumulation of amyloid [226].

The content of intraneuronal APP is negatively correlated with spine density. a APP accumulation in GFP expressing layer V pyramidal neurons from APP23 mice (top). Dendrites that originated from the soma are shown below as high-contrast representations of maximum-intensity projected confocal images. b The dot plot is the corrected intensity of intercellular APP vs. spine density. Straight line is fitted by nonlinear regression. **p < 0.01 (F test). Figure adapted from Ref. [228]
Fibrillar amyloid was observed in dendrites close to plaques [122]. In an animal model with particularly strong pathology, the 5XFAD mouse model, fibrillar amyloid beta was detectable even in cell bodies [139], where it may have acted as seeds for plaques upon cell death [131]. Intracellular amyloid beta in axons, presynapses, as well as in dendritic spines was associated with synaptic pathology [44, 158, 190, 192]. Cytoplasmic amyloid beta was associated with mitochondrial alterations [158], apoptosis markers [61] and with oxidative damage to nucleic acids [138]. However, the latter study claimed that intracellular amyloid may be a compensatory mechanism, as amyloid beta has anti-oxidative properties [138]. Synaptic activity reduced intraneuronal amyloid and protected against amyloid-mediated synaptic alterations, while inhibition of synaptic activity increased intraneuronal amyloid and worsened synaptic damage [194]. It is, however, currently unclear whether intraneuronal amyloid beta contributes to synaptic damage in sporadic Alzheimer’s disease.
Hyperphosphorylated tau protein
The deposition of hyperphosphorylated tau protein in neurons may be secondary to several different types of insults, such as epilepsy [134, 227], chronic traumatic brain injury [71], focal cortical dysplasia [174] or Niemann–Pick disease type C [117, 187]. Tau protein itself seems to be a prerequisite for neuronal damage, as tau knockout mice are immune to neuronal insults mediated by NMDA receptor-dependent excitotoxicity, as well as those caused by amyloid beta [110, 159, 160]. These findings corroborate tau pathology as a secondary effect of amyloid beta in Alzheimer’s disease. On the other hand, the degree of tau pathology is better correlated to cognitive decline than amyloid pathology [3, 13, 72]. Furthermore, a group of neurodegenerative diseases, collectively termed tauopathies, is characterized by mutations in tau protein, which lead to the deposition on hyperphosphorylated tau protein without an associated primary pathology. In experimental animals, the overexpression of wild-type human tau suffices for the formation of neurofibrillary tangles and age-dependent reductions in spine head volumes [57]. Similarly, the introduction of P301S mutant human tau causes inflammation and spine loss [10, 84]. These results suggest that hyperphosphorylated tau on its own may cause synaptic damage. Moreover, a recent study on 5 Alzheimer’s disease patients revealed a reduced spine number in distal parts of the dendritic tree in pyramidal neurons of the parahippocampal cortex and CA1 neurons with intraneuronal neurofibrillary tangles [127]. Another study found that loss of spinophilin-positive puncta in CA1 field and area 9, which are markers of dendritic spines, correlated with cognitive decline and tau pathology [3].
Physiologically, tau protein is primarily located in axons, where it is associated with the cytoskeleton. The physiological phosphorylation pattern of tau determines its subcellular localization: For instance, one specific phosphorylation pattern localizes tau to the nucleus, while another is required during mitosis [137]. LTP induction or pharmacological stimulation of synaptic activity increase translocation of tau to postsynapses [28]. Hyperphosphorylation of tau disrupts this localization pattern [22, 137, 222, 223]. Interestingly, there is also some evidence that phosphorylation of tau may be mediated by APP [137]. The subcellular localization of tau is also influenced by splicing [113] as well as by post-translational enzymatic cleavage [162]. In particular, truncation of tau by specific proteases may increase the toxicity of tau and facilitate hyperphosphorylation [225]. Different patterns of truncation and hyperphosphorylation give rise to different conformations, or “strains”, of tau, which underlie different tauopathies [166]. Furthermore, these tau strains can be propagated from human tissue to susceptible mouse models in a prion-like manner while maintaining their identity [46, 166]. A characteristic form of propagation of pathological tau has also been observed within the brain experimental animals. There, tau pathology spreads synaptically from neuron to neuron [52, 60, 114], which may be the mechanism underlying the specific spreading pattern of tau pathology in Alzheimer’s disease [25].
Tau has been claimed to mediate NMDA receptor-dependent excitotocixity via the Src-family tyrosine kinase Fyn [88] in a phosphorylation-dependent manner [130]. Dendritic tau localizes Fyn to dendrites, where it phosphorylates the GRIN2B subunit of NMDA receptors, thereby enhancing their function [164]. In mice lacking tau, less Fyn is present in dendrites, leading to lower GRIN2B phosphorylation [23]. Fyn, in turn, was found to be activated by oligomeric amyloid beta binding to the cellular prion protein [205]. Amyloid oligomers also disrupt the activity-dependent relocalization of tau to postsynapses [28] in a phosphorylation-dependent manner [128]. These studies provide causal links between the toxicity of amyloid oligomers and the physiological function of tau protein. Furthermore, tau deposits may also activate inflammatory processes, such as increased immunoreactivity for interleukin 1β and cyclooxygenase 2, which in turn activate microglia [10]. Alternatively, dendritic spine loss in Alzheimer’s disease may be simply the consequence of deafferentation and thus a secondary phenomenon that is not at all related to any pathological action of tau at the dendritic spine itself [127]. Hence, dendritic tau hyperphosphorylation and aggregation in sporadic Alzheimer’s disease may be a secondary or even compensatory phenomenon due to slowly progressing deafferentation/disconnection in the aging brain rather than the cause of dendritic spine loss or synaptic failure. The correlation between the detection of hyperphosphorylated tau and human aging is extraordinarily strong—probably stronger than the link to sporadic Alzheimer’s disease.
Similar to amyloid beta, hyperphosphorylated tau may form thioflavin-S binding, fibrillar aggregates which appear microscopically as neurofibrillary tangles, as well as soluble oligomers. In contrast to amyloid beta, however, the verdict seems to be clearer that neurofibrillary tangles themselves are functionally inert [84, 101, 167] and that soluble tau aggregates mediate synaptic damage [148, 219, 223]. Hyperphosphorylated tau was shown to localize to both pre- and postsynapses in multiple studies [81, 86, 88, 148, 188, 189, 191, 219], where it causes synaptic dysfunction by impairing the trafficking or synaptic anchoring [86] as well as the excitability [130]. In THY-Tau22 mice, which express tau with the G272V and P301S mutations, the synaptic enhancement induced by exogenous BDNF was lost due to impaired NMDA receptor function [28]. Electrophysiologically, synaptic dysfunction manifested as a presynaptic deficit in the probability of neurotransmitter release [86, 148] as well as altered excitability of neurons [50, 126, 161]. Our own in vivo imaging studies in P301S mice gave evidence for a postsynaptic accumulation of hyperphosphorylated tau only in spines of CA3 neurons but not within pyramidal neurons of the cerebral cortex [84]. Similar results were found in Alzheimer’s disease patients, where only the thorny excrescences of CA3 neurons, but not spines of cortical neurons, were found to contain hyperphosphorylated tau protein [21, 127]. This may be related to the fact that CA3 thorny excrescences may contain microtubules, whereas dendritic spines of cortical neurons have an actin-based cytoskeleton [42, 181].
Inflammation
Both amyloid deposition and hyperphosphorylated tau lead to deposition of complement, activation of microglia, invasion of T-cells and release of pro-inflammatory cytokines [6, 10, 67, 116, 119, 198], which in turn may affect dendritic spines by multiple mechanisms. Inflammation also drives tau hyperphosphorylation and aggregation [15, 51], so that a detrimental positive-feedback loop may ensue. Fibrillar amyloid deposits are surrounded by proinflammatory complement complexes [116] and activated microglia which phagocytose protofibrillar amyloid. Increased micgroglial phagocytosis of amyloid attenuates amyloid deposition in animal models [107, 115]. On the other hand, amyloid-independent microglia activation, as it for example occurs in patients with multiple sclerosis or HIV, was found to have no relevant impact on the development of Alzheimer-associated cortical pathology [49, 142]. Furthermore, signs of inflammation have been observed in multiple animal models before plaques [79, 213, 219] or tangles [219] were present, which may suggest that inflammation—if it plays a causative role in sporadic Alzheimer diseases—plays a role early during the development of the disease. We, however, failed to observe significant microglia activation in transgenic amyloid mouse models prior to the occurrence of amyloid plaques [91].
The mechanisms by which inflammation affects dendritic spines include activation of caspases via reactive oxygen species released from inflammatory cells, which causes reductions in dendritic spines [48, 62, 149]. Furthermore, release of interleukin 1β by microglia during the inflammatory process affects dendritic spines by antagonizing the stimulatory effect of BDNF on spine genesis [201]. Microglia themselves also play a role in maintaining dendritic spines in the absence of inflammation by producing BDNF to stimulate spine growth [145] and by pruning of spines during development and plasticity [143, 202]. However, it is unclear whether these mechanisms also play a role in Alzheimer’s disease.
Mechanisms independent of amyloid beta, tau and inflammation
Essentially all animal models of Alzheimer’s disease are mice engineered to express one or several of the proteins which are known to cause familial forms of amyloidosis or tauopathy in humans. However, the expression level of these artificially introduced proteins is manifold higher than naturally occurring levels, so that pathological alterations become apparent within the lifespan of the experimental animals. However, the overexpression of these proteins alone may cause alterations in dendritic spines directly or indirectly, which have to be taken into consideration before translating results from animal models to the human disease. This subsection summarizes the most important mechanisms by which overexpression of Alzheimer’s disease-related genes in mice alters dendritic spines.
In animal models of Down syndrome, which are trisomic for the APP gene locus and thus overexpress APP, spine loss and synaptic damage have been described [9, 208]. These may be the consequence of intraneuronal APP accumulation or the accumulation of BACE1 derived cleavage products rather than due to soluble amyloid species of any type or location [75]. Recently, knock-in animal models expressing physiological quantities of mutant APP have been generated to help differentiate between synaptic and cognitive effects caused by overexpression and those by mutation of APP. Interestingly, only the combination of several mutations caused cognitive deficits [163]. Furthermore, in many models of Alzheimer’s disease, overexpression of mutant human APP is often combined with overexpression of mutant human presenilin 1 (PS1), which speeds up amyloidosis in double transgenic mice, compared to single APP transgenes. In contrast to humans, however, the expression of mutant PS1 alone does not cause an amyloidosis in rodents—a phenomenon that is not well understood, but points to one out of several shortcomings of mouse models in Alzheimer’s disease research. Furthermore, we and others have demonstrated that overexpression of both wild-type and mutated human PS1 actually causes an increase in spine density in young transgenic animals [5, 92, 186]. Consistent with these findings, electrophysiological studies of different mouse lines overexpressing mutant PS1 showed significantly enhanced LTP at hippocampal synapses [5, 7, 55, 144, 173, 211, 221]. Furthermore, a recent study showed that knock-in of L435F mutated PS1, which is a loss-of-function mutation, on a PS2 knockout background led to reduced LTP in comparison to PS2 knockouts with wild type PS1 [215]. These effects are clearly independent of any synaptotoxic effects of amyloid beta, since amyloid beta is not enhanced in these mouse models. Rather, several reports point to an altered calcium homeostasis as underlying mechanism for disturbed dendritic spine plasticity, as presenilin mutations seem to interfere with physiological calcium release from intracellular stores. Different molecular mechanisms have been proposed, ranging from ER leak channel activity of PS1 itself [203, 224] to increased gating probabilities of IP3 receptors [40, 41], elevated expression of ryanodine receptors [37, 180, 185] and most recently to reduced synaptic STIM2 expression and impaired store-operated calcium entry [186]. As an indicator of modified calcium homeostasis, we confirmed upregulated RyR levels in A246E-PS1 overexpressing cortical neurons [92].Given the prominent role of dendritic calcium signalling in dendritic spine plasticity, we, therefore, favour the view that PS1-dependent changes in calcium homeostasis underlie the elevated spine densities in PS1-transgenic mice. It is still a matter of debate how the overexpression of PS1 affects the calcium homeostasis. Based on our cell culture studies and biochemical studies on postmortem brains of patients carrying familial Alzheimer’s disease PS1 mutations, we favour the notion that the disturbed ER calcium homeostasis is mediated by the elevation of PS1 holoprotein levels [85] possibly as a consequence of altered presenilin autocleavage. This hypothesis may have impact on the translation of therapeutic efforts from these familiar forms of Alzheimer’s disease, which are currently used to study the treatment of very early stages of Alzheimer’s disease like the Alzheimer’s Prevention Initiative enrolling members of a Columbian cohort who carry the E280A PS1 mutation, to the treatment of sporadic Alzheimer’s disease.
Presenilins are important constituents of the γ-secretase complex, which is necessary to generate amyloid beta, and hence also amyloid oligomers, from APP. Inhibitors of γ-secretase are, therefore, often used as a research tool to reduce the levels of amyloid beta oligomers in experimental animals and prove putative oligomer-dependent mechanisms. Inhibition of γ-secretase, however, leads to alterations in dendritic spines even in the absence of amyloidosis-related transgenes: By performing chronic in vivo two photon imaging in wild-type mice, we observed reduced spine densities after pharmacological inhibition of γ-secretase for 4 days [18]. This observation is in contradiction to ex vivo studies performed in cell culture or in organotypic cultures by various laboratories including our own where γ-secretase inhibition is used as a tool to inhibit amyloid beta production. There, γ-secretase inhibition had no acute effect on dendrites, spine morphology or excitatory synaptic transmission [152, 153, 221]. However, the duration of γ-secretase inhibition might be critical in order for these detrimental effects on spine plasticity to take effect. These preclinical in vivo findings in rodents might be relevant in the development of Alzheimer’s disease therapies aimed at interfering with the function of the γ-secretase to reduce the production of amyloid beta peptides. Our observation of reducing dendritic spine numbers in vivo following γ-secretase inhibition might offer a potential explanation why Alzheimer’s disease patients treated with a potent γ-secretase inhibitor (semagacestat) showed, among other side effects, a worsening of cognition in the high dose cohort, which caused a phase 3 study to be halted [58].
Because of the complex interplay between the physiological roles of proteins involved in Alzheimer’s disease, it is hard to differentiate disease-specific and hence therapeutically relevant effects from those which are related to the genetic manipulation of experimental models in the first place. This fact may, however, explain why such a large number of treatments which were effective in experimental animals have failed to yield any therapeutic benefit in humans.
Conclusions
Loss of dendritic spines in Alzheimer’s disease is intimately linked with synaptic dysfunction and loss of memory and cognition—the very functions which define a human being. Understanding the mechanisms of synapse loss may enable us to find an appropriate therapy to halt or even reverse the progress of this debilitating disease. Unfortunately, the scientific findings to date suggest that an extremely complex pathophysiology underlies Alzheimer’s disease with a wide variety of possible mechanisms which may cause synapse loss or dysfunction. At the moment, it is unclear which of the mechanisms covered here (or indeed any of the multitude of mechanisms which we have not covered) is dominantly responsible for synapse dysfunction in human patients. To paraphrase Alzheimer’s own conclusion of his report, “On a peculiar disease of the cerebral cortex” [4]: We are obviously dealing with a peculiar disease process here. These observations should compel us not to content ourselves with forcibly applying the knowledge we have to date to explain insufficiently understood mechanisms. Future study will enable us to gradually untangle specific mechanisms and assess their contribution to the disease.
References
Adalbert R, Nogradi A, Babetto E, Janeckova L, Walker SA, Kerschensteiner M, Misgeld T, Coleman MP (2009) Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain 132:402–416. doi:10.1093/brain/awn312
Ahmad R, Goffin K, Van den Stock J, De Winter FL, Cleeren E, Bormans G, Tournoy J, Persoons P, Van Laere K, Vandenbulcke M (2014) In vivo type 1 cannabinoid receptor availability in Alzheimer’s disease. Eur Neuropsychopharmacol 24:242–250. doi:10.1016/j.euroneuro.2013.10.002
Akram A, Christoffel D, Rocher AB, Bouras C, Kovari E, Perl DP, Morrison JH, Herrmann FR, Haroutunian V, Giannakopoulos P et al (2008) Stereologic estimates of total spinophilin-immunoreactive spine number in area 9 and the CA1 field: relationship with the progression of Alzheimer’s disease. Neurobiol Aging 29:1296–1307. doi:10.1016/j.neurobiolaging.2007.03.007
Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin 64:146–148
Auffret A, Gautheron V, Repici M, Kraftsik R, Mount HT, Mariani J, Rovira C (2009) Age-dependent impairment of spine morphology and synaptic plasticity in hippocampal CA1 neurons of a presenilin 1 transgenic mouse model of Alzheimer’s disease. J Neurosci 29:10144–10152. doi:10.1523/jneurosci.1856-09.2009
Barger SW, Harmon AD (1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388:878–881
Barrow PA, Empson RM, Gladwell SJ, Anderson CM, Killick R, Yu X, Jefferys JG, Duff K (2000) Functional phenotype in transgenic mice expressing mutant human presenilin-1. Neurobiol Dis 7:119–126. doi:10.1006/nbdi.1999.0276
Beaulieu J-M, Gainetdinov RR, Caron MG (2009) Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol 49:327–347. doi:10.1146/annurev.pharmtox.011008.145634
Belichenko PV, Masliah E, Kleschevnikov AM, Villar AJ, Epstein CJ, Salehi A, Mobley WC (2004) Synaptic structural abnormalities in the Ts65Dn mouse model of Down Syndrome. J Comp Neurol 480:281–298. doi:10.1002/cne.20337
Bellucci A, Westwood AJ, Ingram E, Casamenti F, Goedert M, Spillantini MG (2004) Induction of inflammatory mediators and microglial activation in mice transgenic for mutant human P301S tau protein. Am J Pathol 165:1643–1652
Benilova I, De Strooper B (2013) Promiscuous Alzheimer’s amyloid: yet another partner. Science 341:1354–1355. doi:10.1126/science.1244166
Benilova I, Karran E, De Strooper B (2012) The toxic A[beta] oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci 15:349–357
Berg L, McKeel DW Jr, Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM et al (1998) Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55:326–335
Bertram L, Lill CM, Tanzi RE (2010) The genetics of Alzheimer disease: back to the future. Neuron 68:270–281
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT (2010) Regulation of tau pathology by the microglial fractalkine receptor. Neuron 68:19–31
Bitan G, Fradinger EA, Spring SM, Teplow DB (2005) Neurotoxic protein oligomers–what you see is not always what you get. Amyloid 12:88–95. doi:10.1080/13506120500106958
Bittner T, Burgold S, Dorostkar M, Fuhrmann M, Wegenast-Braun B, Schmidt B, Kretzschmar H, Herms J (2012) Amyloid plaque formation precedes dendritic spine loss. Acta Neuropathol 124:797–807. doi:10.1007/s00401-012-1047-8
Bittner T, Fuhrmann M, Burgold S, Jung CK, Volbracht C, Steiner H, Mitteregger G, Kretzschmar HA, Haass C, Herms J (2009) Gamma-secretase inhibition reduces spine density in vivo via an amyloid precursor protein-dependent pathway. J Neurosci 29:10405–10409
Bittner T, Fuhrmann M, Burgold S, Ochs SM, Hoffmann N, Mitteregger G, Kretzschmar H, LaFerla FM, Herms J (2010) Multiple events lead to dendritic spine loss in triple transgenic Alzheimer’s disease mice. PLoS One 5:e15477
Blair JA, Siedlak SL, Wolfram JA, Nunomura A, Castellani RJ, Ferreira ST, Klein WL, Wang Y, Casadesus G, Smith MA et al (2014) Accumulation of intraneuronal amyloid-beta is common in normal brain. Curr Alzheimer Res 11:317–324
Blazquez-Llorca L, Garcia-Marin V, Merino-Serrais P, Avila J, DeFelipe J (2011) Abnormal tau phosphorylation in the thorny excrescences of CA3 hippocampal neurons in patients with Alzheimer’s disease. J Alzheimers Dis 26:683–698. doi:10.3233/jad-2011-110659
Blum D, Herrera F, Francelle L, Mendes T, Basquin M, Obriot H, Demeyer D, Sergeant N, Gerhardt E, Brouillet E et al (2014) Mutant huntingtin alters Tau phosphorylation and subcellular distribution. Hum Mol Genet. doi:10.1093/hmg/ddu421
Boehm J (2013) A ‘danse macabre’: tau and Fyn in STEP with amyloid beta to facilitate induction of synaptic depression and excitotoxicity. Eur J Neurosci 37:1925–1930. doi:10.1111/ejn.12251
Bordji K, Becerril-Ortega J, Nicole O, Buisson A (2010) Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-{beta} production. J Neurosci 30:15927–15942. doi:10.1523/jneurosci.3021-10.2010
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404. doi:10.1007/s00401-006-0127-z
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Thal DR, Ghebremedhin E, Del Tredici K (2011) Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 70:960–969. doi:10.1097/NEN.0b013e318232a379
Burnouf S, Martire A, Derisbourg M, Laurent C, Belarbi K, Leboucher A, Fernandez-Gomez FJ, Troquier L, Eddarkaoui S, Grosjean ME et al (2013) NMDA receptor dysfunction contributes to impaired brain-derived neurotrophic factor-induced facilitation of hippocampal synaptic transmission in a Tau transgenic model. Aging Cell 12:11–23. doi:10.1111/acel.12018
Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, Konnerth A (2012) Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci 109:8740–8745. doi:10.1073/pnas.1206171109
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold K-H, Haass C, Staufenbiel M, Konnerth A, Garaschuk O (2008) Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321:1686–1689. doi:10.1126/science.1162844
Campbell JN, Register D, Churn SB (2011) Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J Neurotrauma 29:201–217. doi:10.1089/neu.2011.1761
Cao L, Schrank BR, Rodriguez S, Benz EG, Moulia TW, Rickenbacher GT, Gomez AC, Levites Y, Edwards SR, Golde TE et al (2012) Aβ alters the connectivity of olfactory neurons in the absence of amyloid plaques in vivo. Nat Commun 3:1009. doi:10.1038/ncomms2013
Castellani RJ, Perry G (2014) The complexities of the pathology–pathogenesis relationship in Alzheimer disease. Biochem Pharmacol 88:671–676. doi:10.1016/j.bcp.2014.01.009
Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C et al (2011) Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3:89ra57. doi:10.1126/scitranslmed.3002156
Castillo-Carranza DL, Guerrero-Munoz MJ, Sengupta U, Hernandez C, Barrett AD, Dineley K, Kayed R (2015) Tau immunotherapy modulates both pathological tau and upstream amyloid pathology in an Alzheimer’s disease mouse model. J Neurosci 35:4857–4868. doi:10.1523/jneurosci.4989-14.2015
Centonze D, Muzio L, Rossi S, Cavasinni F, De Chiara V, Bergami A, Musella A, D’Amelio M, Cavallucci V, Martorana A et al (2009) Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J Neurosci 29:3442–3452. doi:10.1523/jneurosci.5804-08.2009
Chakroborty S, Goussakov I, Miller MB, Stutzmann GE (2009) Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci 29:9458–9470. doi:10.1523/jneurosci.2047-09.2009
Chakroborty S, Stutzmann G (2011) Early calcium dysregulation in Alzheimer’s disease: setting the stage for synaptic dysfunction. Sci China Life Sci 54:752–762. doi:10.1007/s11427-011-4205-7
Cheng IH, Palop JJ, Esposito LA, Bien-Ly N, Yan F, Mucke L (2004) Aggressive amyloidosis in mice expressing human amyloid peptides with the Arctic mutation. Nat Med 10:1190–1192. doi:10.1038/nm1123
Cheung KH, Mei L, Mak DO, Hayashi I, Iwatsubo T, Kang DE, Foskett JK (2010) Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer’s disease-linked presenilin mutants in human cells and mouse neurons. Sci Signal 3:ra22. doi:10.1126/scisignal.2000818
Cheung KH, Shineman D, Muller M, Cardenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK (2008) Mechanism of Ca2+ disruption in Alzheimer’s disease by presenilin regulation of InsP3 receptor channel gating. Neuron 58:871–883. doi:10.1016/j.neuron.2008.04.015
Chicurel ME, Harris KM (1992) Three-dimensional analysis of the structure and composition of CA3 branched dendritic spines and their synaptic relationships with mossy fiber boutons in the rat hippocampus. J Comp Neurol 325:169–182. doi:10.1002/cne.903250204
Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J et al (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570. doi:10.1074/jbc.M100710200
Christensen DZ, Huettenrauch M, Mitkovski M, Pradier L, Wirths O (2014) Axonal degeneration in an Alzheimer mouse model is PS1 gene dose dependent and linked to intraneuronal Abeta accumulation. Front Aging Neurosci 6:139. doi:10.3389/fnagi.2014.00139
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM (2005) Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48:913–922. doi:10.1016/j.neuron.2005.10.028
Clavaguera F, Akatsu H, Fraser G, Crowther RA, Frank S, Hench J, Probst A, Winkler DT, Reichwald J, Staufenbiel M et al (2013) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci 110:9535–9540. doi:10.1073/pnas.1301175110
Coleman PD, Riesen AH (1968) Evironmental effects on cortical dendritic fields. I. Rearing in the dark. J Anat 102:363–374
D’Amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, Ferri A, Diamantini A, De Zio D, Carrara P, Battistini L et al (2011) Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci 14:69–76
Dal Bianco A, Bradl M, Frischer J, Kutzelnigg A, Jellinger K, Lassmann H (2008) Multiple sclerosis and Alzheimer’s disease. Ann Neurol 63:174–183. doi:10.1002/ana.21240
Dalby NO, Volbracht C, Helboe L, Larsen PH, Jensen HS, Egebjerg J, Elvang AB (2014) Altered function of hippocampal CA1 pyramidal neurons in the rTg4510 mouse model of tauopathy. J Alzheimers Dis 40:429–442. doi:10.3233/jad-131358
de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT (2010) Caspase activation precedes and leads to tangles. Nature 464:1201–1204
de Calignon A, Polydoro M, Suárez-Calvet M, William C, Adamowicz David H, Kopeikina Kathy J, Pitstick R, Sahara N, Ashe Karen H, Carlson George A et al (2012) Propagation of Tau pathology in a model of early Alzheimer’s disease. Neuron 73:685–697
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464. doi:10.1002/ana.410270502
DeMattos Ronald B, Lu J, Tang Y, Racke Margaret M, DeLong Cindy A, Tzaferis John A, Hole Justin T, Forster Beth M, McDonnell Peter C, Liu F et al (2012) A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer’s disease mice. Neuron 76:908–920
Dewachter I, Ris L, Croes S, Borghgraef P, Devijver H, Voets T, Nilius B, Godaux E, Van Leuven F (2008) Modulation of synaptic plasticity and Tau phosphorylation by wild-type and mutant presenilin1. Neurobiol Aging 29:639–652. doi:10.1016/j.neurobiolaging.2006.11.019
Dewachter I, Ris L, Jaworski T, Seymour CM, Kremer A, Borghgraef P, De Vijver H, Godaux E, Van Leuven F (2009) GSK3beta, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol Dis 35:193–200. doi:10.1016/j.nbd.2009.04.003
Dickstein DL, Brautigam H, Stockton SD Jr, Schmeidler J, Hof PR (2010) Changes in dendritic complexity and spine morphology in transgenic mice expressing human wild-type tau. Brain Struct Funct 214:161–179. doi:10.1007/s00429-010-0245-1
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG et al (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369:341–350. doi:10.1056/NEJMoa1210951
Dorostkar MM, Burgold S, Filser S, Barghorn S, Schmidt B, Anumala UR, Hillen H, Klein C, Herms J (2014) Immunotherapy alleviates amyloid-associated synaptic pathology in an Alzheimer’s disease mouse model. Brain. doi:10.1093/brain/awu280
Dujardin S, Lecolle K, Caillierez R, Begard S, Zommer N, Lachaud C, Carrier S, Dufour N, Auregan G, Winderickx J et al (2014) Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun 2:14. doi:10.1186/2051-5960-2-14
Eimer W, Vassar R (2013) Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8:2
Ertürk A, Wang Y, Sheng M (2014) Local pruning of dendrites and spines by caspase-3-dependent and proteasome-limited mechanisms. J Neurosci 34:1672–1688. doi:10.1523/jneurosci.3121-13.2014
Esparza TJ, Zhao H, Cirrito JR, Cairns NJ, Bateman RJ, Holtzman DM, Brody DL (2013) Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol 73:104–119. doi:10.1002/ana.23748
Falkenberg T, Mohammed AK, Henriksson B, Persson H, Winblad B, Lindefors N (1992) Increased expression of brain-derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci Lett 138:153–156. doi:10.1016/0304-3940(92)90494-R
Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS (2008) Cell death after spinal cord injury is exacerbated by rapid TNF alpha-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci 28:11391–11400. doi:10.1523/jneurosci.3708-08.2008
Fiala JC, Spacek J, Harris KM (2002) Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Rev 39:29–54. doi:10.1016/s0165-0173(02)00158-3
Fonseca MI, Zhou J, Botto M, Tenner AJ (2004) Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 24:6457–6465. doi:10.1523/jneurosci.0901-04.2004
Fowler SW, Chiang ACA, Savjani RR, Larson ME, Sherman MA, Schuler DR, Cirrito JR, Lesné SE, Jankowsky JL (2014) Genetic modulation of soluble Aβ rescues cognitive and synaptic impairment in a mouse model of Alzheimer’s disease. J Neurosci 34:7871–7885. doi:10.1523/jneurosci.0572-14.2014
Fu M, Zuo Y (2011) Experience-dependent structural plasticity in the cortex. Trends Neurosci 34:177–187
Garcia-Marin V, Blazquez-Llorca L, Rodriguez J-R, Boluda S, Muntane G, Ferrer I, DeFelipe J (2009) Diminished perisomatic GABAergic terminals on cortical neurons adjacent to amyloid plaques. Front Neuroanat. doi:10.3389/neuro.05.028.2009
Geddes JF, Vowles GH, Nicoll JA, Revesz T (1999) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 98:171–178
Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60:1495–1500
Glabe CG (2008) Structural classification of toxic amyloid oligomers. J Biol Chem 283:29639–29643. doi:10.1074/jbc.R800016200
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H et al (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156:15–20
Gouras GK, Willén K, Faideau M (2014) The inside-out amyloid hypothesis and synapse pathology in Alzheimer’s disease. Neurodegener Dis 13:142–146
Grady CL, Furey ML, Pietrini P, Horwitz B, Rapoport SI (2001) Altered brain functional connectivity and impaired short-term memory in Alzheimer’s disease. Brain 124:739–756
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8:101–112. doi:10.1038/nrm2101
Halpain S, Hipolito A, Saffer L (1998) Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci 18:9835–9844
Hanzel CE, Pichet-Binette A, Pimentel LSB, Iulita MF, Allard S, Ducatenzeiler A, Do Carmo S, Cuello AC (2014) Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol Aging. doi:10.1016/j.neurobiolaging.2014.03.026
Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356. doi:10.1126/science.1072994
Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu G-Q, Masliah E, Mucke L (2012) Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes Tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One 7:e45881. doi:10.1371/journal.pone.0045881
Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP (2001) Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci 21:2393–2403
Hepler RW, Grimm KM, Nahas DD, Breese R, Dodson EC, Acton P, Keller PM, Yeager M, Wang H, Shughrue P et al (2006) Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry 45:15157–15167. doi:10.1021/bi061850f
Hoffmann N, Dorostkar M, Blumenstock S, Goedert M, Herms J (2013) Impaired plasticity of cortical dendritic spines in P301S tau transgenic mice. Acta Neuropathol Commun 1:82
Honarnejad K, Jung CKE, Lammich S, Arzberger T, Kretzschmar H, Herms J (2013) Involvement of presenilin holoprotein upregulation in calcium dyshomeostasis of Alzheimer’s disease. J Cell Mol Med 17:293–302. doi:10.1111/jcmm.12008
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL et al (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081
Irwin SA, Galvez R, Greenough WT (2000) Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb Cortex 10:1038–1044
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA et al (2010) Dendritic function of tau mediates amyloid-[beta] toxicity in Alzheimer’s disease mouse models. Cell 142:387–397. doi:10.1016/j.cell.2010.06.036
Jimenez S, Navarro V, Moyano J, Sanchez-Mico M, Torres M, Davila JC, Vizuete M, Gutierrez A, Vitorica J (2014) Disruption of amyloid plaques integrity affects the soluble oligomers content from Alzheimer disease brains. PLoS One 9:e114041. doi:10.1371/journal.pone.0114041
Jones WH, Thomas DB (1962) Changes in the dendritic organization of neurons in the cerebral cortex following deafferentation. J Anat 96:375–381
Jung CK, Keppler K, Steinbach S, Blazquez-Llorca L, Herms J (2015) Fibrillar amyloid plaque formation precedes microglial activation. PLoS One 10:e0119768. doi:10.1371/journal.pone.0119768
Jung CKE, Fuhrmann M, Honarnejad K, Van Leuven F, Herms J (2011) Role of presenilin1 in structural plasticity of cortical dendritic spines in vivo. J Neurochem 119:1064–1073. doi:10.1111/j.1471-4159.2011.07503.x
Jung CKE, Herms J (2014) Structural dynamics of dendritic spines are influenced by an environmental enrichment: an in vivo imaging study. Cereb Cortex 24:377–384. doi:10.1093/cercor/bhs317
Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37:925–937. doi:10.1016/s0896-6273(03)00124-7
Keck T, Mrsic-Flogel TD, Vaz Afonso M, Eysel UT, Bonhoeffer T, Hubener M (2008) Massive restructuring of neuronal circuits during functional reorganization of adult visual cortex. Nat Neurosci 11:1162–1167. doi:10.1038/nn.2181
Kendziorra K, Wolf H, Meyer PM, Barthel H, Hesse S, Becker GA, Luthardt J, Schildan A, Patt M, Sorger D et al (2011) Decreased cerebral alpha4beta2* nicotinic acetylcholine receptor availability in patients with mild cognitive impairment and Alzheimer’s disease assessed with positron emission tomography. Eur J Nucl Med Mol Imaging 38:515–525. doi:10.1007/s00259-010-1644-5
Kirkwood CM, Ciuchta J, Ikonomovic MD, Fish KN, Abrahamson EE, Murray PS, Klunk WE, Sweet RA (2013) Dendritic spine density, morphology, and fibrillar actin content surrounding amyloid-[beta] plaques in a mouse model of amyloid-[beta] deposition. J Neuropathol Exp Neurol. doi:10.1097/NEN.1090b1013e31829ecc31889 (Publish Ahead of Print)
Koffie RM, Hashimoto T, Tai H-C, Kay KR, Serrano-Pozo A, Joyner D, Hou S, Kopeikina KJ, Frosch MP, Lee VM et al (2012) Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-β. Brain 135:2155–2168. doi:10.1093/brain/aws127
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM et al (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci 106:4012–4017. doi:10.1073/pnas.0811698106
Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu H-Y, Hyman BT, Bacskai BJ (2008) Aβ plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 59:214–225. doi:10.1016/j.neuron.2008.06.008
Kuchibhotla KV, Wegmann S, Kopeikina KJ, Hawkes J, Rudinskiy N, Andermann ML, Spires-Jones TL, Bacskai BJ, Hyman BT (2014) Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc Natl Acad Sci 111:510–514. doi:10.1073/pnas.1318807111
Kumar-Singh S, Dewachter I, Moechars D, Lubke U, De Jonghe C, Ceuterick C, Checler F, Naidu A, Cordell B, Cras P et al (2000) Behavioral disturbances without amyloid deposits in mice overexpressing human amyloid precursor protein with Flemish (A692G) or Dutch (E693Q) mutation. Neurobiol Dis 7:9–22. doi:10.1006/nbdi.1999.0272
Kummer MP, Heneka MT (2014) Truncated and modified amyloid-beta species. Alzheimers Res Ther 6:28. doi:10.1186/alzrt258
Lanz TA, Carter DB, Merchant KM (2003) Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis 13:246–253
Larson ME, Lesné SE (2012) Soluble Aβ oligomer production and toxicity. J Neurochem 120:125–139. doi:10.1111/j.1471-4159.2011.07478.x
Lashuel HA, Lansbury PT Jr (2006) Are amyloid diseases caused by protein aggregates that mimic bacterial pore-forming toxins? Q Rev Biophys 39:167–201. doi:10.1017/s0033583506004422
Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT (2010) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. doi:10.2353/ajpath.2010.100265
Lendvai B, Stern EA, Chen B, Svoboda K (2000) Experience-dependent plasticity of dendritic spines in the developing rat barrel cortex in vivo. Nature 404:876–881
Leonoudakis D, Zhao P, Beattie EC (2008) Rapid tumor necrosis factor alpha-induced exocytosis of glutamate receptor 2-lacking AMPA receptors to extrasynaptic plasma membrane potentiates excitotoxicity. J Neurosci 28:2119–2130. doi:10.1523/jneurosci.5159-07.2008
Leroy K, Ando K, Laporte V, Dedecker R, Suain V, Authelet M, Héraud C, Pierrot N, Yilmaz Z, Octave J-N et al (2012) Lack of Tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am J Pathol 181:1928–1940. doi:10.1016/j.ajpath.2012.08.012
Lesné SE, Sherman MA, Grant M, Kuskowski M, Schneider JA, Bennett DA, Ashe KH (2013) Brain amyloid-beta oligomers in ageing and Alzheimer’s disease. Brain 136:1383–1398. doi:10.1093/brain/awt062
Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B (1990) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248:1124–1126
Liu C, Gotz J (2013) Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS One 8:e84849. doi:10.1371/journal.pone.0084849
Liu L, Drouet V, Wu JW, Witter MP, Small SA, Clelland C, Duff K (2012) Trans-synaptic spread of tau pathology in vivo. PLoS One 7:e31302. doi:10.1371/journal.pone.0031302
Liu Z, Condello C, Schain A, Harb R, Grutzendler J (2010) CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-{beta} phagocytosis. J Neurosci 30:17091–17101. doi:10.1523/jneurosci.4403-10.2010
Loeffler DA, Camp DM, Bennett DA (2008) Plaque complement activation and cognitive loss in Alzheimer’s disease. J Neuroinflammation 5:9. doi:10.1186/1742-2094-5-9
Love S, Bridges LR, Case CP (1995) Neurofibrillary tangles in Niemann-Pick disease type C. Brain 118(Pt 1):119–129
Madsen K, Neumann WJ, Holst K, Marner L, Haahr MT, Lehel S, Knudsen GM, Hasselbalch SG (2011) Cerebral serotonin 4 receptors and amyloid-beta in early Alzheimer’s disease. J Alzheimers Dis 26:457–466. doi:10.3233/jad-2011-110056
Maezawa I, Zimin P, Wulff H, Jin L-W (2010) A[beta] oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity. J Biol Chem. doi:10.1074/jbc.M110.135244
Magdesian MH, Carvalho MM, Mendes FA, Saraiva LM, Juliano MA, Juliano L, Garcia-Abreu J, Ferreira ST (2008) Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling. J Biol Chem 283:9359–9368. doi:10.1074/jbc.M707108200
Malinow R, Malenka RC (2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25:103–126. doi:10.1146/annurev.neuro.25.112701.142758
Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and alzheimer’s disease. J Neurosci 16:5795–5811
Masliah E, Terry RD, DeTeresa RM, Hansen LA (1989) Immunohistochemical quantification of the synapse-related protein synaptophysin in Alzheimer disease. Neurosci Lett 103:234–239. doi:10.1016/0304-3940(89)90582-X
Matthews MR, Powell TPS (1962) Some observations on transneuronal cell degeneration in the olfactory bulb of the rabbit. J Anat 96(89–102):103
McGeer PL, Akiyama H, Kawamata T, Yamada T, Walker DG, Ishii T (1992) Immunohistochemical localization of beta-amyloid precursor protein sequences in Alzheimer and normal brain tissue by light and electron microscopy. J Neurosci Res 31:428–442. doi:10.1002/jnr.490310305
Menkes-Caspi N, Yamin Hagar G, Kellner V, Spires-Jones Tara L, Cohen D, Stern Edward A (2015) Pathological Tau disrupts ongoing network activity. Neuron. doi:10.1016/j.neuron.2015.01.025
Merino-Serrais P, Benavides-Piccione R, Blazquez-Llorca L, Kastanauskaite A, Rabano A, Avila J, DeFelipe J (2013) The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 136:1913–1928. doi:10.1093/brain/awt088
Miller EC, Teravskis PJ, Dummer BW, Zhao X, Huganir RL, Liao D (2014) Tau phosphorylation and tau mislocalization mediate soluble Abeta oligomer-induced AMPA glutamate receptor signaling deficits. Eur J Neurosci 39:1214–1224. doi:10.1111/ejn.12507
Mizukami K, Ishikawa M, Akatsu H, Abrahamson EE, Ikonomovic MD, Asada T (2011) An immunohistochemical study of the serotonin 1A receptor in the hippocampus of subjects with Alzheimer’s disease. Neuropathology 31:503–509. doi:10.1111/j.1440-1789.2010.01193.x
Mondragón-Rodríguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J (2012) Interaction of endogenous Tau protein with synaptic proteins is regulated by N-Methyl-d-aspartate receptor-dependent Tau phosphorylation. J Biol Chem 287:32040–32053. doi:10.1074/jbc.M112.401240
Moon M, Hong H-S, Nam DW, Baik SH, Song H, Kook S-Y, Kim YS, Lee J, Mook-Jung I (2012) Intracellular amyloid-β accumulation in calcium-binding protein-deficient neurons leads to amyloid-β plaque formation in animal model of Alzheimer’s disease. J Alzheimers Dis 29:615–628. doi:10.3233/JAD-2011-111778
Morawski M, Bruckner G, Jager C, Seeger G, Matthews RT, Arendt T (2012) Involvement of perineuronal and perisynaptic extracellular matrix in Alzheimer’s disease neuropathology. Brain Pathol (Zurich, Switzerland) 22:547–561. doi:10.1111/j.1750-3639.2011.00557.x
Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA (2002) Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9:88–102
Nagaishi M, Arai M, Osawa T, Yokoo H, Hirato J, Yoshimoto Y, Nakazato Y (2011) An immunohistochemical finding in glioneuronal lesions associated with epilepsy: the appearance of nestin-positive, CD34-positive and tau-accumulating cells. Neuropathology 31:468–475. doi:10.1111/j.1440-1789.2010.01188.x
Nhan H, Chiang K, Koo E (2014) The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol. doi:10.1007/s00401-014-1347-2
Nimchinsky EA, Sabatini BL, Svoboda K (2002) Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. doi:10.1146/annurev.physiol.64.081501.160008
Nizzari M, Barbieri F, Gentile MT, Passarella D, Caorsi C, Diaspro A, Taglialatela M, Pagano A, Colucci-D’Amato L, Florio T et al (2012) Amyloid-beta protein precursor regulates phosphorylation and cellular compartmentalization of microtubule associated protein tau. J Alzheimers Dis 29:211–227. doi:10.3233/jad-2011-101590
Nunomura A, Tamaoki T, Tanaka K, Motohashi N, Nakamura M, Hayashi T, Yamaguchi H, Shimohama S, H-g Lee, Zhu X et al (2010) Intraneuronal amyloid β accumulation and oxidative damage to nucleic acids in Alzheimer disease. Neurobiol Dis 37:731–737. doi:10.1016/j.nbd.2009.12.012
Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L et al (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140. doi:10.1523/jneurosci.1202-06.2006
Ochs SM, Dorostkar MM, Aramuni G, Schon C, Filser S, Poschl J, Kremer A, Van Leuven F, Ovsepian SV, Herms J (2014) Loss of neuronal GSK3[beta] reduces dendritic spine stability and attenuates excitatory synaptic transmission via [beta]-catenin. Mol Psychiatry 20:482–489. doi:10.1038/mp.2014.55
Okada H, Ouchi Y, Ogawa M, Futatsubashi M, Saito Y, Yoshikawa E, Terada T, Oboshi Y, Tsukada H, Ueki T et al (2013) Alterations in alpha4beta2 nicotinic receptors in cognitive decline in Alzheimer’s aetiopathology. Brain 136:3004–3017. doi:10.1093/brain/awt195
Ortega M, Ances BM (2014) Role of HIV in amyloid metabolism. J Neuroimmune Pharmacol 9:483–491. doi:10.1007/s11481-014-9546-0
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333:1456–1458. doi:10.1126/science.1202529
Parent A, Linden DJ, Sisodia SS, Borchelt DR (1999) Synaptic transmission and hippocampal long-term potentiation in transgenic mice expressing FAD-linked presenilin 1. Neurobiol Dis 6:56–62. doi:10.1006/nbdi.1998.0207
Parkhurst Christopher N, Yang G, Ninan I, Savas Jeffrey N, Yates Iii John R, Lafaille Juan J, Hempstead Barbara L, Littman Dan R, Gan W-B (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. doi:10.1016/j.cell.2013.11.030
Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, Lo E, Wu D, Saule E, Bouschet T et al (2007) LTP Inhibits LTD in the Hippocampus via Regulation of GSK3[beta]. Neuron 53:703–717. doi:10.1016/j.neuron.2007.01.029
Penzes P, Cahill ME, Jones KA, VanLeeuwen J-E, Woolfrey KM (2011) Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci 14:285–293
Polydoro M, Dzhala VI, Pooler AM, Nicholls SB, McKinney AP, Sanchez L, Pitstick R, Carlson GA, Staley KJ, Spires-Jones TL et al (2014) Soluble pathological tau in the entorhinal cortex leads to presynaptic deficits in an early Alzheimer’s disease model. Acta Neuropathol 127:257–270. doi:10.1007/s00401-013-1215-5
Pozueta J, Lefort R, Ribe EM, Troy CM, Arancio O, Shelanski M (2013) Caspase-2 is required for dendritic spine and behavioural alterations in J20 APP transgenic mice. Nat Commun 4:1939. doi:10.1038/ncomms2927
Pozueta J, Lefort R, Shelanski ML (2013) Synaptic changes in Alzheimer’s disease and its models. Neuroscience 251:51–65. doi:10.1016/j.neuroscience.2012.05.050
Price KA, Varghese M, Sowa A, Yuk F, Brautigam H, Ehrlich ME, Dickstein DL (2014) Altered synaptic structure in the hippocampus in a mouse model of Alzheimer’s disease with soluble amyloid-beta oligomers and no plaque pathology. Mol Neurodegener 9:41. doi:10.1186/1750-1326-9-41
Priller C, Bauer T, Mitteregger G, Krebs B, Kretzschmar HA, Herms J (2006) Synapse formation and function is modulated by the amyloid precursor protein. J Neurosci 26:7212–7221. doi:10.1523/jneurosci.1450-06.2006
Priller C, Dewachter I, Vassallo N, Paluch S, Pace C, Kretzschmar HA, Van Leuven F, Herms J (2007) Mutant presenilin 1 alters synaptic transmission in cultured hippocampal neurons. J Biol Chem 282:1119–1127. doi:10.1074/jbc.M605066200
Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S et al (2006) A[beta]42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7:940–946
Renner M, Lacor PN, Velasco PT, Xu J, Contractor A, Klein WL, Triller A (2010) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66:739–754. doi:10.1016/j.neuron.2010.04.029
Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Müller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU et al (2011) The cellular prion protein mediates neurotoxic signalling of β‐sheet‐rich conformers independent of prion replication. EMBO J 30:2057–2070. doi:10.1038/emboj.2011.86
Richardson JC, Kendal CE, Anderson R, Priest F, Gower E, Soden P, Gray R, Topps S, Howlett DR, Lavender D et al (2003) Ultrastructural and behavioural changes precede amyloid deposition in a transgenic model of Alzheimer’s disease. Neuroscience 122:213–228. doi:10.1016/S0306-4522(03)00389-0
Rijal Upadhaya A, Scheibe F, Kosterin I, Abramowski D, Gerth J, Kumar S, Liebau S, Yamaguchi H, Walter J, Staufenbiel M et al (2013) The type of Abeta-related neuronal degeneration differs between amyloid precursor protein (APP23) and amyloid beta-peptide (APP48) transgenic mice. Acta Neuropathol Commun 1:77. doi:10.1186/2051-5960-1-77
Roberson ED, Halabisky B, Yoo JW, Yao J, Chin J, Yan F, Wu T, Hamto P, Devidze N, Yu G-Q et al (2011) Amyloid-{beta}/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31:700–711. doi:10.1523/jneurosci.4152-10.2011
Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu G-Q, Mucke L (2007) Reducing endogenous Tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science 316:750–754. doi:10.1126/science.1141736
Rocher AB, Crimins JL, Amatrudo JM, Kinson MS, Todd-Brown MA, Lewis J, Luebke JI (2010) Structural and functional changes in tau mutant mice neurons are not linked to the presence of NFTs. Exp Neurol 223:385–393. doi:10.1016/j.expneurol.2009.07.029
Rosenmann H (2014) Asparagine endopeptidase cleaves tau and promotes neurodegeneration. Nat Med 20:1236–1238. doi:10.1038/nm.3749
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC (2014) Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17:661–663. doi:10.1038/nn.3697
Salter MW, Kalia LV (2004) Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 5:317–328. doi:10.1038/nrn1368
Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu G-Q, Palop JJ et al (2012) Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci 109:E2895–E2903. doi:10.1073/pnas.1121081109
Sanders DW, Kaufman SK, DeVos SL, Sharma AM, Mirbaha H, Li A, Barker SJ, Foley AC, Thorpe JR, Serpell LC et al (2014) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82:1271–1288. doi:10.1016/j.neuron.2014.04.047
SantaCruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E et al (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309:476–481. doi:10.1126/science.1113694
Scheff SW, Price DA, Ansari MA, Roberts KN, Schmitt FA, Ikonomovic MD, Mufson EJ (2015) Synaptic change in the posterior cingulate gyrus in the progression of Alzheimer’s disease. J Alzheimers Dis 43:1073–1090. doi:10.3233/jad-141518
Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ (2007) Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68:1501–1508. doi:10.1212/01.wnl.0000260698.46517.8f
Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384. doi:10.1016/j.neurobiolaging.2005.09.012
Scheff SW, Price DA, Schmitt FA, Scheff MA, Mufson EJ (2011) Synaptic loss in the inferior temporal gyrus in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 24:547–557. doi:10.3233/jad-2011-101782
Schilling S, Lauber T, Schaupp M, Manhart S, Scheel E, Böhm G, Demuth H-U (2006) On the seeding and oligomerization of pGlu-amyloid peptides (in vitro). Biochemistry 45:12393–12399. doi:10.1021/bi0612667
Schneider I, Reverse D, Dewachter I, Ris L, Caluwaerts N, Kuiperi C, Gilis M, Geerts H, Kretzschmar H, Godaux E et al (2001) Mutant presenilins disturb neuronal calcium homeostasis in the brain of transgenic mice, decreasing the threshold for excitotoxicity and facilitating long-term potentiation. J Biol Chem 276:11539–11544. doi:10.1074/jbc.M010977200
Sen A, Thom M, Martinian L, Harding B, Cross JH, Nikolic M, Sisodiya SM (2007) Pathological Tau tangles localize to focal cortical dysplasia in older patients. Epilepsia 48:1447–1454. doi:10.1111/j.1528-1167.2007.01107.x
Shastry BS (1998) Molecular genetics of familial Alzheimer disease. Am J Med Sci 315:266–272
Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298:776–780. doi:10.1126/science.1075333
Shie F-S, LeBoeur RC, Jin L-W (2003) Early intraneuronal Aβ deposition in the hippocampus of APP transgenic mice. NeuroReport 14:123–129
Shirao T, González-Billault C (2013) Actin filaments and microtubules in dendritic spines. J Neurochem 126:155–164. doi:10.1111/jnc.12313
Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H et al (2014) Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84:10. doi:10.1016/j.neuron.2014.10.024
Smith IF, Hitt B, Green KN, Oddo S, LaFerla FM (2005) Enhanced caffeine-induced Ca2+ release in the 3xTg-AD mouse model of Alzheimer’s disease. J Neurochem 94:1711–1718. doi:10.1111/j.1471-4159.2005.03332.x
Sorra KE, Harris KM (2000) Overview on the structure, composition, function, development, and plasticity of hippocampal dendritic spines. Hippocampus 10:501–511. doi:10.1002/1098-1063(2000)10:5<501:aid-hipo1>3.0.co;2-t
Sperling R, Dickerson B, Pihlajamaki M, Vannini P, LaViolette P, Vitolo O, Hedden T, Becker JA, Rentz D, Selkoe D et al (2010) Functional alterations in memory networks in early Alzheimer’s disease. NeuroMol Med 12:27–43. doi:10.1007/s12017-009-8109-7
Sperling RA, LaViolette PS, O’Keefe K, O’Brien J, Rentz DM, Pihlajamaki M, Marshall G, Hyman BT, Selkoe DJ, Hedden T et al (2009) Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63:178–188. doi:10.1016/j.neuron.2009.07.003
Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold K-H, Mistl C, Rothacher S, Ledermann B, Bürki K, Frey P, Paganetti PA et al (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci 94:13287–13292
Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I (2006) Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer’s disease mice. J Neurosci 26:5180–5189. doi:10.1523/jneurosci.0739-06.2006
Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL 3rd, Bezprozvanny I (2014) Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin mice. Neuron 82:79–93. doi:10.1016/j.neuron.2014.02.019
Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D’Agostino AN, Carstea ED (1995) Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol 89:227–238
Tai H-C, Serrano-Pozo A, Hashimoto T, Frosch MP, Spires-Jones TL, Hyman BT (2012) The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol 181:1426–1435. doi:10.1016/j.ajpath.2012.06.033
Tai H-C, Wang B, Serrano-Pozo A, Frosch M, Spires-Jones T, Hyman B (2014) Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol Commun 2:146
Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK (2013) Accumulation of intraneuronal β-amyloid 42 peptides is associated with early changes in microtubule-associated protein 2 in neurites and synapses. PLoS One 8:e51965. doi:10.1371/journal.pone.0051965
Takahashi RH, Capetillo-Zarate E, Lin MT, Milner TA, Gouras GK (2010) Co-occurrence of Alzheimer’s disease [beta]-amyloid and tau pathologies at synapses. Neurobiol Aging 31:1145–1152
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK (2002) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161:1869–1879
Takuma K, Fang F, Zhang W, Yan S, Fukuzaki E, Du H, Sosunov A, McKhann G, Funatsu Y, Nakamichi N et al (2009) RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc Natl Acad Sci 106:20021–20026. doi:10.1073/pnas.0905686106
Tampellini D, Rahman N, Gallo EF, Huang Z, Dumont M, Capetillo-Zarate E, Ma T, Zheng R, Lu B, Nanus DM et al (2009) Synaptic activity reduces intraneuronal Aβ, promotes APP transport to synapses, and protects against aβ-related synaptic alterations. J Neurosci 29:9704–9713. doi:10.1523/jneurosci.2292-09.2009
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580. doi:10.1002/ana.410300410
Thal DR, Rub U, Orantes M, Braak H (2002) Phases of A{beta}-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Thomas MG, Pascual ML, Maschi D, Luchelli L, Boccaccio GL (2014) Synaptic control of local translation: the plot thickens with new characters. Cell Mol Life Sci 71:2219–2239. doi:10.1007/s00018-013-1506-y
Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, Oda T, Tsuchiya K, Kosaka K (2002) Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol 124:83–92
Tolino M, Köhrmann M, Kiebler MA (2012) RNA-binding proteins involved in RNA localization and their implications in neuronal diseases. Eur J Neurosci 35:1818–1836. doi:10.1111/j.1460-9568.2012.08160.x
Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T et al (2010) A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci 30:4845–4856. doi:10.1523/jneurosci.5825-09.2010
Tong L, Prieto GA, Kramár EA, Smith ED, Cribbs DH, Lynch G, Cotman CW (2012) Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J Neurosci 32:17714–17724. doi:10.1523/jneurosci.1253-12.2012
Tremblay M-È, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8:e1000527
Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I (2006) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126:981–993. doi:10.1016/j.cell.2006.06.059
Um JW, Kaufman AC, Kostylev M, Heiss JK, Stagi M, Takahashi H, Kerrisk ME, Vortmeyer A, Wisniewski T, Koleske AJ et al (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79:887–902. doi:10.1016/j.neuron.2013.06.036
Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012) Alzheimer amyloid-[beta] oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15:1227–1235. doi:10.1038/nn.3178
Umeda T, Maekawa S, Kimura T, Takashima A, Tomiyama T, Mori H (2014) Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice. Acta Neuropathol 127:685–698. doi:10.1007/s00401-014-1259-1
Umeda T, Tomiyama T, Kitajima E, Idomoto T, Nomura S, Lambert MP, Klein WL, Mori H (2012) Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci 91:1169–1176. doi:10.1016/j.lfs.2011.12.022
Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ (2005) Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(171)65Dn and mouse chromosome 12. Mamm Genome 16:79–90
Walsh DM, Hartley DM, Condron MM, Selkoe DJ, Teplow DB (2001) In vitro studies of amyloid beta-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala692→Gly) Alzheimer’s disease. Biochem J 355:869–877
Wang D, Govindaiah G, Liu R, De Arcangelis V, Cox CL, Xiang YK (2010) Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J 24:3511–3521. doi:10.1096/fj.10-156661
Wang Y, Greig NH, Yu QS, Mattson MP (2009) Presenilin-1 mutation impairs cholinergic modulation of synaptic plasticity and suppresses NMDA currents in hippocampus slices. Neurobiol Aging 30:1061–1068. doi:10.1016/j.neurobiolaging.2007.10.009
Wilcock DM, Griffin WS (2013) Down’s syndrome, neuroinflammation, and Alzheimer neuropathogenesis. J Neuroinflammation 10:84. doi:10.1186/1742-2094-10-84
Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP, Clark IA, Abdipranoto A, Vissel B (2013) Neuroinflammation and neuronal loss precede aβ plaque deposition in the hAPP-J20 mouse model of Alzheimer’s disease. PLoS One 8:e59586. doi:10.1371/journal.pone.0059586
Wu H-Y, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL et al (2010) Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci 30:2636–2649. doi:10.1523/jneurosci.4456-09.2010
Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, Kelleher RJ 3rd (2015) Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85:967–981. doi:10.1016/j.neuron.2015.02.010
Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ (2013) Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J Neurosci 33:17042–17051. doi:10.1523/jneurosci.1836-13.2013
Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y (2009) Rapid formation and selective stabilization of synapses for enduring motor memories. Nature 462:915–919. doi:10.1038/nature08389
Yang G, Pan F, Gan W-B (2009) Stably maintained dendritic spines are associated with lifelong memories. Nature 462:920–924. doi:10.1038/nature08577
Yoshiyama Y, Higuchi M, Zhang B, Huang S-M, Iwata N, Saido Takaomi C, Maeda J, Suhara T, Trojanowski JQ, Lee VMY (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337–351
Yuste R (2013) Electrical compartmentalization in dendritic spines. Annu Rev Neurosci 36:429–449. doi:10.1146/annurev-neuro-062111-150455
Zaman SH, Parent A, Laskey A, Lee MK, Borchelt DR, Sisodia SS, Malinow R (2000) Enhanced synaptic potentiation in transgenic mice expressing presenilin 1 familial Alzheimer’s disease mutation is normalized with a benzodiazepine. Neurobiol Dis 7:54–63. doi:10.1006/nbdi.1999.0271
Zempel H, Luedtke J, Kumar Y, Biernat J, Dawson H, Mandelkow E, Mandelkow E-M (2013) Amyloid-[beta] oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. EMBO J. doi:10.1038/emboj.2013.207 (advance online publication)
Zempel H, Mandelkow EM (2012) Linking amyloid-β and Tau: amyloid-β induced synaptic dysfunction via local wreckage of the neuronal cytoskeleton. Neurodegener Dis 10:64–72
Zhang H, Sun S, Herreman A, De Strooper B, Bezprozvanny I (2010) Role of presenilins in neuronal calcium homeostasis. J Neurosci 30:8566–8580. doi:10.1523/jneurosci.1554-10.2010
Zhang Z, Song M, Liu X, Kang SS, Kwon I-S, Duong DM, Seyfried NT, Hu WT, Liu Z, Wang J-Z et al (2014) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20:1254–1262. doi:10.1038/nm.3700
Zhao W, Dumanis SB, Tamboli IY, Rodriguez GA, Jo LaDu M, Moussa CEH, William Rebeck G (2014) Human APOE genotype affects intraneuronal Aβ1–42 accumulation in a lentiviral gene transfer model. Hum Mol Genet 23:1365–1375. doi:10.1093/hmg/ddt525
Zheng P, Shultz SR, Hovens CM, Velakoulis D, Jones NC, O’Brien TJ (2014) Hyperphosphorylated tau is implicated in acquired epilepsy and neuropsychiatric comorbidities. Mol Neurobiol 49:1532–1539. doi:10.1007/s12035-013-8601-9
Zou C, Montagna E, Shi Y, Peters F, Blazquez-Llorca L, Shi S, Filser S, Dorostkar MM, Herms J (2015) Intraneuronal APP and extracellular Abeta independently cause dendritic spine pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. doi:10.1007/s00401-015-1421-4
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dorostkar, M.M., Zou, C., Blazquez-Llorca, L. et al. Analyzing dendritic spine pathology in Alzheimer’s disease: problems and opportunities. Acta Neuropathol 130, 1–19 (2015). https://doi.org/10.1007/s00401-015-1449-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-015-1449-5