
Abstract
Camptocormia is a highly disabling syndrome that occurs in various diseases but is particularly associated with Parkinson’s disease (PD). Although first described nearly 200 years ago, the morphological changes associated with camptocormia are still under debate and the pathophysiology is unknown. We analyzed paraspinal muscle biopsies of 14 PD patients with camptocormia and compared the findings to sex-matched postmortem controls of comparable age to exclude biopsy site-specific changes. Camptocormia in PD showed a consistent lesion pattern composed of myopathic changes with type-1 fiber hypertrophy, loss of type-2 fibers, loss of oxidative enzyme activity, and acid phosphatase reactivity of lesions. Ultrastructurally, myofibrillar disorganization and Z-band streaming up to electron-dense patches/plaques were seen in the lesions. No aberrant protein aggregation, signs of myositis or mitochondriopathy were found, but the mitochondrial content of paraspinal muscles in patients and controls was markedly higher than known from limb biopsies. Additionally, we were able to demonstrate a link between the severity of the clinical syndrome and the degree of the myopathic changes. Because of the consistent lesion pattern, we propose criteria for the diagnosis of camptocormia in PD from muscle biopsies. The morphological changes show obvious parallels to the muscle pathology of experimental tenotomy reported in the 1970s, which depend on an intact innervation and do not occur after interruption of the myotactic reflexes. A dysregulation of the proprioception could be part of the pathogenesis of camptocormia in Parkinson’s disease, particularly in view of the clinical symptoms of rigidity and loss of muscle strength.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Camptocormia (from the Greek “kamptein” = to bend and “kormos” = trunk) is an abnormal flexion of the thoracolumbar spine when standing or walking that disappears in the supine position. The syndrome, also known as “bent spine syndrome”, was first described by Earle in 1815 [13] and in some of the original cases reported by James Parkinson in 1817 [31]. When restricted to the cervical spine, “dropped head syndrome” occurs. A pronounced unilateral involvement of muscles may lead to a laterodeviation called Pisa syndrome. Although muscle changes similar to camptocormia have been described for dropped head syndrome, it is unknown whether these diseases share a common pathomechanism.
Camptocormia can be associated with movement disorders, neuromuscular diseases, spinal or metabolic disorders or found isolated as an idiopathic condition [15, 42]. Among movement disorders it is most frequent in idiopathic Parkinson’s disease [11], where it occurs in roughly 6–7% of patients [41] (ranging from 3 to 17% [1, 3, 26]).
The pathophysiology of camptocormia is as yet unresolved. It is under discussion whether the syndrome may result from a loss of muscle strength, amyotonia of vertebral extensors or preponderance of flexors [8, 19]. MRI shows muscle atrophy and fatty replacement to various degrees [5, 24, 26, 27]. The morphological findings from paraspinal muscle biopsies of camptocormia patients associated with different clinical diseases are heterogenous. Mainly based on single cases or small numbers of patients, signs of myositis, inclusion body myositis or mitochondrial disorders were reported [9, 34, 37, 42]. In some few patients, the camptocormia may be related to a hereditary muscle disease [33]. In the vast majority of cases, non-specific myopathic changes, combined with an increase of connective tissue within muscle fascicles, were described [4, 22, 39, 40].
We investigated paravertebral muscle biopsies from a clinically well-defined group of Parkinson patients suffering from camptocormia. Because muscles are morphologically different depending on their function [32], we compared the paraspinal camptocormia biopsies with paraspinal muscle samples from autopsies. Within the control patient group we compared also the paraspinal sample site with a conventional deltoid sample and different paraspinal sample sites with each other. Autopsy samples are reported as being sufficient for histochemical and immunohistochemical staining as well as electron microscopy within a certain postmortem time range [14] and were widely used, for example, as controls [43] or for generating standard values [20]. In PD camptocormia patients, we found a uniform lesion pattern in the muscle biopsies which implies a common pathophysiological mechanism in camptocormia.
Patients and methods
Characterization of patients
The patients in this study suffered from idiopathic Parkinson’s disease and camptocormia and were prospectively included in this study between 2004 and 2010. The diagnosis of Parkinson’s disease was made according to the British Parkinson’s Disease Society Brain Bank criteria [18]. Camptocormia is defined here as a marked anterior flexion of the thoracolumbar spine of at least 30° (with or without additional laterodeviation), appearing in the standing or walking position and disappearing in the recumbent position, but without any signs of a fixed kyphosis like in osteoporotic kyphosis. Absence of clinically detectable flexor-dystonia is required. All patients were stabilized on optimal anti-Parkinson medication. Informed consent to take part in the study was signed by all patients. The study was approved by the ethical committee of the Medical Faculty of the University Hospital Schleswig-Holstein (D 469/11).
The clinical details of our camptocormia patients were reported by Margraf et al. [27]. Briefly, the Parkinson patients showed mean disease duration of 13.5 years. Mean duration of camptocormia was 25 months (5–72 months). A muscle biopsy was offered to all Parkinson patients who had consulted the UK-SH Department of Neurology because of their camptocormia. The muscle biopsies were performed to determine whether the camptocormia could be related to a treatable cause. The most affected part, according to MR imaging of the paraspinal muscles, was selected for the biopsy. All biopsies that were taken in the patient group defined above were used for the present histopathological study to avoid a possible selection bias. Details of the patients are given in Table 1.
The severity of the clinical symptoms was scored as described by Margraf et al. [27]. This score encompasses the inclination angle, the circumstances of occurrence, the time of day when the symptoms occur, the degree of back pain and the limitation in daily activities caused by the camptocormia.
Controls
After informed consent from relatives, postmortem muscle specimens were taken from 10 deceased patients, 5 females, 5 males, with an average age of 66.5 years (ranging from 47 to 82 years) who died because of internal diseases (Table 2).
Histological investigations of muscle specimens
Standard muscle evaluation was performed using stainings of cryostat sections. These stainings included hematoxylin and eosin (H&E), elastica van-Giesson, periodic acid Schiff reaction (PAS), oil red O (OrO), modified Gomori trichrome, nicotinamide adenine dinucleotide-tetrazolium reductase (NADH-TR), menadione-linked α-glycerophosphate dehydrogenase (MAG), myoadenylate deaminase (MAD), succinic dehydrogenase (SDH), cytochrome oxidase (COX), combination of COX and SDH, acid phosphatase, non-specific esterase, adenosine triphosphatase (ATPase) at pH values of 4.4, 4.7 and 10.4, which were prepared according to standard procedures [12].
As indicated in the figures, quantification per power field or arbitrary units were used. For counts per field the mean of 5–15 fields was calculated, depending on the size of the biopsy. Nuclear bags and fiber splittings were counted in medium power fields corresponding to a 200-fold magnification. Ragged red fibers, COX-deficient fibers and whorled fibers were counted in low power fields corresponding to a 100-fold magnification. The fiber type distribution was calculated by the relation of type-1 to type-2 fibers. Some changes were calculated in percent of all fibers. These were the number of fibers showing internal nuclei, the number of fibers showing acid phosphatase activity and the number of fibers showing defects in the NADH reaction. If quantification by counting was not applicable, a five-step rating system corresponding to absent, mild, moderate, severe and extensive, was used. The criteria for the semiquantitative rating were—for caliber variation of myofibers: 0 = uniformity, 1 = mild variation, 2 = moderate variation, 3 = variation showing atrophy or hypertrophic fibers, 4 = variation showing atrophy and hypertrophic fibers; for endomysial fibrosis: 0 = absent, 1 = fibrotic sprout into endomysium, 2 = continuous fine endomysial fibrosis, 3 = continuous moderate endomysial fibrosis, 4 = nearly all fibers were trapped by endomysial collagen, for neurogenic atrophy: 0 = no, 1 = single atrophic fibers of both types, 2 = reticular distribution of some atrophic fibers, 3 = several atrophic fibers or small groups, 4 = large groups of atrophic fibers of both types; for fiber type-2 atrophy: 0 = no, 1 = occasional, 2 = scattered but some, 3 = several, 4 = nearly all type-2 fibers.
To compare the myopathic changes with clinical symptoms, we calculated a score that includes the main aspects of the myopathic changes. These are structural defects, acid phosphatase activity in muscle fibers, endomysial fibrosis, caliber variation, whorled fibers, internal nuclei and fiber splitting. The severity of changes for all included parameters was standardized to a scaling system ranging from 0 to 10. To do so, the semiquantitative values were multiplied by 2.5, the percent values divided by 10. All mean values of counts per power fields were within the range of ten and were taken as they were.
Immunohistochemical stainings
All immunohistochemical staining results reported here were obtained from frozen sections that were fixed for 15 min in ice-cold acetone. With the antibody reactions that were performed without epitope retrieval of the slides we examined inflammatory reactions, lysosomal structures, fiber typing, small vessels, mitochondria and protein deposition. The following antibodies were used: CD3 (rat monoclonal, AbD Serotec), CD8 (mouse monoclonal, Dako), CD20 (mouse monoclonal, clone L26, Dako), KiM1P (mouse monoclonal, gift from the Pathology Department of UK-SH), MHC I (mouse monoclonal, clone W6/32, Dako), cathepsin D (rabbit polyclonal, Dianova), LAMP-1 (mouse monoclonal, clone E5, Santa Cruz), SERCA 1 (mouse monoclonal, clone VE121G9, Thermo Scientific), α-5-laminin (mouse monoclonal, clone 4C7, Chemicon), prohibitin (mouse monoclonal, Abcam), sarcomeric α-actinin (mouse monoclonal, clone EA-53, Sigma), α-B-crystallin (rabbit polyclonal, Oncogene), smooth muscle actin (mouse monoclonal, clone 1A4, Beckman Coulter), dystrophin I, rod region, clone Dy4/6D3, dystrophin II, C-terminal, clone Dy8/6C65, dystrophin III, N-terminal, clone Dy10/12B2 (all three were mouse monoclonals from Novocastra), desmin (mouse monoclonal, clone D33, Dako), filamin (mouse monoclonal, clone FLMN 01, Dianova), myotilin (mouse monoclonal, Novocastra), p62 (mouse monoclonal, BD Bioscience), titin (mouse monoclonal, clone T11, Sigma), triadin (mouse monoclonal, clone GE4.90, Acris-Antibodies) and ZASP (rabbit polyclonal, clone LDB-3, Sigma). For the detection of α-synuclein deposits, antibodies LB509 (mouse monoclonal, Signet) and pSer129-α-Syn (rabbit polyclonal, Abcam) were used. As secondary antibodies, the alkaline phosphatase-coupled goat anti-mouse or goat anti-rabbit from Dako were used and vizualized by neufuchsine. Alternatively, the biotinylated sheep anti-mouse or donkey anti-rabbit from GE Healthcare or rabbit anti-rat from Dako were used, detected by horse radish peroxidise coupled extravidin (Sigma) and vizualized by either DAB or AEC as chromogen.
PET blot
To avoid any influence of a neurodegenerative disease on changes in muscles of our control group, we used antibodies against β-amyloid (clone 6E10, mouse monoclonal, Chemicon) or phosphorylated tau (AT8, mouse monoclonal, Covance) to exclude individuals who showed intracerebral protein aggregate deposits according to CERAD C or Braak and Braak IV to VI and antibodies LB509 (Signet) and pSer129-α-Syn (Abcam) to exclude individuals in whom α-synuclein aggregates were detectable in the brain tissue. For the detection of these protein aggregates, the most sensitive PET blot method was used according to the protocol by Kramer and Schulz-Schaeffer [23]. For the sensitive detection of α-synuclein aggregates, the autopsy and biopsy muscles were investigated using the regular PET blot protocol [23] and a postfixed frozen tissue blotting protocol as described by Daus et al. [7].
Electron microscopic investigation
For electron microscopy, glutaraldehyde-fixed specimens were postfixed with 1% osmium tetroxide (Serva, Heidelberg, Germany) and embedded in araldite (Serva). Ultrathin sections were contrasted with lead citrate (Serva) and uranyl acetate (Serva). Investigations were performed on a Zeiss EM10 transmission electron microscope (Zeiss, Oberkochen, Germany) equipped with a MegaView III imaging system (Olympus Soft Imaging Systems GmbH, Münster, Germany).
Results
Histologically, paraspinal muscles differ from limb muscles
Because different biopsy sites may vary in their histological picture, we compared the paraspinal muscle specimens of 10 control patients with those of their deltoid muscles. Paraspinal muscles displayed a higher number of ragged red and COX-deficient fibers than deltoid muscles in the same control patients. Ragged red fibers (RRF) were observed in paraspinal muscles as frequently as 0.125–1.6 RRF (mean 0.66) per low power field (LPF), whereas in deltoid muscles, they ranged from 0 to 0.5 RRF (mean 0.2) per LPF. COX-negative fibers varied between 0 and 20 (mean 5.19) per LPF in paraspinal muscles and between 0 and 8.2 deficient fibers (mean 1.58) in deltoid muscles (Fig. 1a). In paraspinal muscles, the fiber type ratio varied among the fascicles more considerably than in the deltoid muscles and more frequently showed a preponderance of type-1 fibers. No systematic differences could be observed when comparing cervical (C7) and lumbar (L2) paraspinal muscle samples (Fig. 1b). Muscle pathology, if present, was always detectable in the paraspinal and deltoid samples (Table 2).

Morphological differences between paraspinal and limb muscles in controls. Paraspinal muscles display a higher number of ragged red and COX-deficient fibers than deltoid muscles of the same control patients (a). No further morphological differences were detectable between paraspinal muscles of cervical or lumbar levels (b)
Camptocormia in Parkinson’s disease presents with uniform muscle pathology
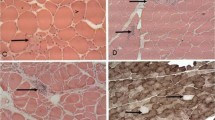
Using light microscopy, all 14 muscle specimens obtained from PD camptocormia patients showed myopathic changes, i.e. increased variability in fiber size (Fig. 2a), hypertrophy of type-1-fibers (defined as significant increase of fiber diameters compared to controls, as demonstrated in Fig. 2g), more than 5% fibers containing internalized nuclei (ranging from 5 to 65%), rounded fiber shape, a distinct number of nuclear clumps and variable, but always detectable, endomysial fibrosis (Fig. 2c) as well as focally detectable replacement by fat cells in severely affected camptocormia muscles. The myopathic changes were predominantly seen in type-1 fibers.

Myopathic changes in paraspinal muscles of camptocormia compared to controls: Increased fiber size spectrum (a H&E) and hypertrophy (g), endomysial fibrosis (c elastica van Gieson stain) and lack of type-2 fibers (e SERCA1 immunohistochemistry) in camptocormia compared to controls (b, d, f). Quantification of morphological changes are shown in h. Bar 100 μm
These non-specific changes were accompanied by central disarrangements of the sarcoplasmic reticulum in up to 25% of fibers, leading to a depletion of oxidative enzymes in these fibers (Fig. 3a) which were visible in NADH-TR, SDH, COX and MAG reactions, and in some fibers in ATPases but not in the MAD reactions. Almost exclusively, type-1 fibers were affected, regardless of the amount of type-2 fibers seen in the tissue. The defects were most often centrally located, round to oval in shape and showed a well-defined border. No densifications, fuchsinophilic structures, PAS-positive deposits or vacuoles were visible in the lesions. In all PD camptocormia biopsies, the acid phosphatase reaction showed an intense, finely granular reactivity in the areas of depletion of enzyme reaction (Fig. 3c). The acid phosphatase reaction seemed to have preceded structural defects, as it was seen in more fibers than those showing enzymatic defects.

Camptocormia is characterized by myofibrillar disorganization. Loss of oxidative enzyme reaction (a NADH reaction) accompanied by pathological acid phosphatase reactivity (c) are detectable but not in controls (b, d). Ultrastructurally, myofibrillar disorganization, Z-band streaming and electron-dense patches correspond to the light microscopic lesions (e semithin section, Richardson staining, f electron microscopy). Bars a–d 100 μm, e 25 μm, f 1 μm
By electron microscopy, we were able to observe myofibrillar sarcoplasmic structures being destroyed in those lesions that were identified by light microscopy as having oxidative depletion. Z-band streaming, developing to electron-dense patches/plaques, were seen, but not cytoplasmic bodies (Fig. 3e, f). In some areas, myofibrillar elements were completely disorganized. Triads were displaced and were found among pathological sarcoplasmic structures. Mitochondria were found in reduced numbers within the lesions, but appeared normal in fine structure. The number of lysosomes was variable but not sufficient to explain the acid phosphatase reaction. Inclusions within muscle fiber nuclei were not found.
Additionally, in 9 of 12 evaluable PD camptocormia biopsy specimens, hypertrophic fibers with a whorled, “fingerprint-like” appearance were seen by light microscopy. These fibers were seen at a frequency of one to 40 fibers per 10 LPF (Fig. 2h). Whorled fibers, as well as the fibers with a central loss of enzyme activity, did not stain dark in the non-specific esterase reaction.
Histochemical reactions (MAG) showed type-1 predominance, with more than 90% of type-1 fibers in 6 of 12 biopsy specimens. The type-2 fiber loss could be confirmed using an antibody against SERCA1 (Fig. 2e, f). Small fibers and nuclear clumps were mostly detectable with the SERCA1 antibody.
COX, SDH as well as ATPase reactions in general were reduced in paraspinal muscles of PD camptocormia patients. A marked loss of ATPase activity was observed in 4 of 12 samples. Lipid contents by oil red O staining were reduced compared to controls (Fig. 4e).

Camptocormia shows no evidence for mitochondriopathy or inflammation. Modified Gomori’s trichrome stain sometimes shows a higher content of ragged red and COX-deficient fibers in PD camptocormia biopsy tissues (a, e) than in control limb muscles (see Fig. 1) but the same holds true for the paraspinal control muscles (b, e). No evidence for inflammation was observed (c, d MHC-1 immunohistochemistry). Bars 100 μm
In the control samples, we did not observe a reduction in enzyme activity, myopathic changes such as in PD camptocormia or multiple acid phosphatase-reactive myofibrillar lesions (Figs. 2b, d, 3b, d). Although in 50% of the controls structural alterations in the oxidative enzymes were visible in single fibers and one control patient showed an enhanced acid phosphatase reaction in single fibers, no ultrastructural myofibrillar alterations were seen.
Investigating changes that may reflect the etiology of PD camptocormia
To understand the etiology of the lesions found in all PD camptocormia muscle biopsies, we studied whether the areas of myofibrillar disarrangement accumulate proteins, whether the acid phosphatase-stained structures contained lysosomes and whether we could detect changes in the capillary blood supply.
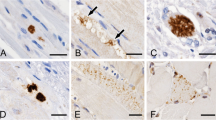
We did not identify aggregation of desmin, dystrophin (C-, N-terminal, rod domain), sarcomeric α-actinin, α-B-crystallin, filamin, myotilin, titin, triadin and ZASP. With antibodies detecting protein aggregates in neurodegenerative diseases, i.e. ubiquitin, α-synuclein, S129-phosphorylated α-synuclein, Aβ, hyperphosphorylated tau and prion protein, no specific staining was seen in muscle fibers. Even with modifications of the PET blot technique for the detection of protein aggregates in muscle tissues [7], no α-synuclein aggregates were detectable in muscle fibers.
The dust-like acid phosphatase-stained structures in abnormal muscle fibers did not react with antibodies against lysosomal markers LAMP1 or cathepsin D and non-specific esterase activity was not enhanced. In single fibers that contained acid phosphatase-reactive myofibrillar lesions, an immunoreaction was observed with the antibody P62 against a peptide of the human ubiquitin interactive motif (UIM).
The small capillaries around muscle fibers were irregular in distribution, as was demonstrated using antibodies against α-5-laminin and MHC I (Fig. 4c). We could observe single fibers or groups of muscle fibers with fewer capillaries around them. However, we were neither able to co-localize a capillary loss to fiber lesions nor able to find a statistical difference in the capillary-to-muscle fiber ratio between PD camptocormia biopsy tissues and controls. It was obvious from our PD camptocormia biopsy specimens that the capillaries around muscle fibers were accompanied by an increased amount of connective tissue compared to those in controls. With these results we cannot rule out that a reduced blood supply to muscle fibers may be responsible for the myopathic changes, but we have no evidence for it.
Paraspinal muscles in camptocormia do not present signs of inflammation or mitochondriopathy
Using antibodies against CD3, CD8 and CD20, no inflammatory infiltrates were identified amongst the camptocormia specimens, whereas in the control group four specimens showed clearly visible lymphocytic infiltrates. Modified Gomori’s trichrome stain illustrating mitochondrial content of muscle fibers showed single ragged red fibers in the PD camptocormia biopsy tissues, but the number was even lower compared to those in controls (mean values 0.66 RRF/LPF vs. 0.3 RRF/LPF, Fig. 4a, b, e). A comparable result was obtained with SDH/COX preparations, which showed more COX-deficient fibers in the control group than in PD camptocormia patients (mean values 5.19 vs. 1.95 COX-deficient fibers per LPF). An increased accumulation of mitochondria in the subsarcolemmal zone was accompanied by a lack of mitochondrial staining in the center of these fibers using trichrome staining and anti-prohibitin immunohistochemistry.
Angular atrophic fibers could not be found in PD camptocormia biopsy muscles (except in one patient), whereas amongst controls, five patients had angular atrophic fibers. Grouping of fiber types as a sign of reinnervation was found in none of the PD camptocormia biopsy muscles.
Correlation of the severity of myopathic changes with clinical parameters of camptocormia
We tested whether the severity of myopathic changes as measured by the amount of acid phosphatase-reactive fibers or the numbers of fibers showing structure defects in the NADH reaction, the fibrosis, the degree of fiber type uniformity measured by the relation of the fiber types or a myopathy score that contains the most relevant parameters of the myopathic changes, correlate with either the angle of flexion, the duration of the camptocormia or the clinical camptocormia score as suggested by Margraf et al. [27]. For most of the parameters no significant correlation could be demonstrated, for example as shown for the influence of the fibrosis on the inclination angle (Fig. 5a). Although our patient numbers are relatively small for statistically analyses, we found a statistical relevant correlation between the degree of fibrosis and the clinical camptocormia score as well as a correlation between our myopathy index and the clinical camptocormia score (Fig. 5b, c; thresholds: r > 0.559; p < 0.05).

Correlating the severity of myopathic changes with clinical parameters of camptocormia, no significant correlation of the endomysial fibrosis could be found with regard to the inclination angle (a). But there was a statistically relevant correlation between the degree of endomysial fibrosis and the clinical camptocormia score, as well as a correlation between our myopathy index which was based on the relevant myopathological changes and the clinical camptocormia score (b, c thresholds: r > 0.559; p < 0.05)
Summarizing our results, we have found evidence that camptocormia in PD is linked to a myopathy characterized by a typical lesion pattern that leads to a muscular insufficiency. Based on our findings, we propose criteria typical of camptocormia in biopsied paraspinal muscles as shown in Table 3.
We do not assert that these myopathological findings can occur exclusively in camptocormia. Together with the clinical symptom of bent spine, we suggest that the diagnosis camptocormia can be made if the main features are fulfilled. The minor features can usually be found to a variable extent but are alone not sufficient for making the diagnosis because of their nonspecificity. The occurrence of ragged red and COX-negative fibers in paraspinal muscles is per se not sufficient for diagnosing a mitochondriopathy in paraspinal muscles because of its frequent occurrence in control muscles. The proposed criteria need to be further evaluated in independent cohorts.
Discussion
In the present study we describe a morphologically well-defined disease pattern in patients suffering from Parkinson’s disease and camptocormia. None of these changes is specific by itself, but the same combination of changes—although to various extents—was seen in all PD camptocormia patients (Table 1).
The muscle changes are sufficient to explain the muscle weakness of camptocormia. We observed in several muscle fibers a loss of the structural integrity of myofibrils. These fibers are characterized by central areas of myofibrillar disorganization which were seen in parallel to a loss of oxidative enzyme reaction. The structural defects showed acid phosphatase reactivity, although these changes were not accompanied by an increased number of lysosomes that were detectable immunohistochemically or by electron microscopy. The reduced number of intact myofibers may lead to an impaired ability to keep the trunk in an upright position.
It is an open question whether the myopathy is the primary cause of camptocormia in PD. We were able to show a correlation between the severity of the endomysial fibrosis alone or a myopathy score containing a rating of different myopathic changes and the severity of the camptocormia measured by the camptocormia score of Margraf. There is no significant correlation between the number of muscle fibers that contain lesions and any clinical parameter. This finding parallels that in which the number of cores in central core disease likewise shows no correlation with the disease [45]—although the cores are linked to the pathogenesis of the central core disease. In conclusion, our findings suggest that the physical impairment by the camptocormia is related to the severity of the myopathic changes and shows for the first time a link between the clinical syndrome and the myopathy.
Type-1 predominance, fiber defects lacking oxidative enzyme activities and the myofibrillar disorganization may lead to the differential diagnoses of central core disease, myofibrillar myopathy or nemaline myopathy. Using a battery of antibodies, we could not find protein aggregation like in myofibrillary myopathies within the lesions. The myopathological changes in camptocormia differ from those of central core disease by lacking the type-1 fiber hypotrophy—in contrast, we observe a type-1 fiber hypertrophy—and the acid phosphatase reactivity of the lesions that cannot be seen in central core disease. In nemaline myopathy, an acid phosphatase reactivity within lesioned fibers has been described although in a completely different distribution than in camptocormia [29], even when a nemaline myopathy may be the cause of camptocormia in PD, as described in a single case [30]. With electron microscopy, numerous small autophagic vacuoles have been detected in the cores [29], which cannot be seen in camptocormia. Nevertheless, a degradation of proteins may take place in the muscle fiber lesions seen in camptocormia. In single fibers a reactivity with an antibody against p62, also known as sequestome 1, was observed. This is a polyubiquitin chain binding protein that shuttles substrates for proteasomal degradation [36].
As we could find a consistent lesion pattern in PD camptocormia, we checked whether these changes have been seen by others. In most reports of camptocormia the muscle biopsy results were not presented in detail. When histochemical investigations were reported, they frequently highlighted single aspects of the lesion pattern we report here. Lava and Factor [25] reported moth-eaten fibers and disruption of the intermyofibrillar network; Schabitz et al. [34] found myopathic changes in all and selective type II fiber loss, disturbance of mitochondrial distribution and sarcomere destruction in a subset of patients; Spuler et al. [39] described the same changes, including loss of NADH enzyme activity and myofibrillar disarray in half of their biopsy specimens. In our biopsy tissues there was no evidence that the defects of muscle structure reflected a protein surplus myopathy as suggested by Spuler et al. [39].
Why is the interpretation of paraspinal muscle biopsies so heterogenous?
It may contribute to the confusion in the field that only a few results have been published on the “normal appearance” of paraspinal muscles in the elderly. Only Wharton et al. [43] investigated cervical paraspinal muscles in 23 consecutive necropsies and found ragged red fibers in 60–70%, and also core-targetoid fibers in some patients over 60 years of age as well as mild focal endomysial chronic inflammation in 13%. Although it is known that the number of ragged red fibers increases with age [28], we and others [4] found a higher number of ragged red fibers in paraspinal muscles than in the limb muscles usually biopsied. We speculate that this physiologically high number of ragged red fibers in paraspinal muscles is the reason why the diagnosis of a mitochondriopathy was made in several camptocormia patients [17, 34, 46]. The relative frequency of incidental focal chronic endomysial inflammation reported for paraspinal necropsy muscles [43] shows that inflammatory infiltrates should not be overestimated. This is of importance since the justification for biopsies in camptocormia patients, in most cases, is to confirm or rule out a treatable myositis that causes the clinical symptoms. We conclude that ragged red fibers, rimmed vacuoles, COX-deficient fibers and sparse infiltrates of lymphocytes may be found randomly in paraspinal muscles and are not contradictory to the diagnosis of camptocormia myopathy.
Is camptocormia due to an overuse myopathy?
Because the camptocormia-associated paraspinal muscles show an increase in muscle tone, exercise-induced muscle damage may be the cause of camptocormia. Exercise-induced muscle damage was studied in the 1980s and Armstrong et al. [2] observed myofibrillar destruction, fiber necroses, macrophage reaction or inflammation as related pathological changes, whereas Z-disk streaming and A-band disorganization was shown by Friden et al. [16]. These changes differ from those in our PD camptocormia patients, as the hallmark of the myopathy is not fiber necrosis, inflammation or macrophage reaction.
Is camptocormia due to proprioceptive dysregulation?
The function of paraspinal muscles is to support our upright body position. This posture is largely regulated by involuntary reflex mechanisms involving mono- and polysynaptic reflexes. Additionally, central mechanisms can influence the muscle tone via the polysynaptic reflexes [35]. It has been suggested that movement disturbances in PD may be influenced by an impaired load-receptor mechanism. It is known that PD patients underestimate the amplitude of joint motion as a consequence of abnormal proprioception. They demonstrated poorer accuracy and a higher detection threshold for passive movement of a joint as compared to controls (reviewed in [6]). It is further suggested that an impaired supraspinal control of spinal interneuronal circuits influences muscular load-receptors [10]. Consistently, an impaired proprioception of the axial musculature was recently found in PD patients that correlates with the severity of the UPDRS motor score [44]. This led us to the hypothesis that camptocormia may be due to a reduced load sensitivity or proprioceptive dysregulation of paraspinal muscles.
Surprisingly, a lesion pattern in muscles, similar to what we have found in our PD camptocormia patients, has been observed after tenotomy. Experimental tenotomy leads to core-like lesions in the center or periphery of almost exclusively type-1 fibers, which show an increase in acid phosphatase activity but no autophagic vacuoles [21]. Fibers of the tenotomised muscle lack ATPase and SDH activity [38]. Ultrastructurally, the core-like lesions have no normal register of sarcomeres and often thick filaments appear more disintegrated than thin ones. Z-band streaming and electron-dense patches/plaques can be observed. Mitochondria are reduced in the lesions [38]. These changes cannot be observed in denervated tenotomised muscles or in tenotomised muscles after chordotomy [21]. Obviously, the experimental lesions depend on disturbances of muscle tension reflex mechanisms, but the reflexes need to be functionally intact. The lesions described in the tenotomy experiments are strikingly similar to the lesions that we describe here for camptocormia. They show not only the lack of oxidative enzymes, but also the acid phosphatase reactivity and ultrastructurally the myofibrillar disorganization. These similarities point to the likelihood that camptocormia may also be related to a proprioceptive dysregulation. A proprioceptive dysregulation may explain the clinical symptoms of rigidity and loss of muscle strength in camptocormia.
We examined whether we could find changes in muscle spindles, as they are involved in the reflex mechanisms. Unfortunately, it was not possible to analyze them in sufficient numbers in the current biopsies. We intend to do this in further studies on autopsy specimens.
Conclusion
Our findings suggest that camptocormia in PD is most frequently related to a myopathy. The myopathy can be defined by pathological core features that were present in all our cases. The myopathological changes may explain the loss of muscle strength as they show a disorganization of myofibers. Additionally, the myopathological changes seem to correlate with the severity of camptocormia symptoms. The cause of this myopathy, however, still needs to be discovered.
References
Abe K, Uchida Y, Notani M (2010) Camptocormia in Parkinson’s disease. Parkinsons Dis. doi:10.4061/2010/267640
Armstrong RB, Ogilvie RW, Schwane JA (1983) Eccentric exercise-induced injury to rat skeletal muscle. J Appl Physiol 54:80–93
Ashour R, Jankovic J (2006) Joint and skeletal deformities in Parkinson’s disease, multiple system atrophy, and progressive supranuclear palsy. Mov Disord 21:1856–1863
Askmark H, Eeg-Olofsson K, Johansson A, Nilsson P, Olsson Y, Aquilonius S (2001) Parkinsonism and neck extensor myopathy: a new syndrome or coincidental findings? Arch Neurol 58:232–237
Bonneville F, Bloch F, Kurys E et al (2008) Camptocormia and Parkinson’s disease: MR imaging. Eur Radiol 18:1710–1719
Carpenter MG, Bloem BR (2011) Postural control in Parkinson patients: a proprioceptive problem? Exp Neurol 227:26–30
Daus ML, Breyer J, Wagenfuehr K et al (2011) Presence and seeding activity of pathological prion protein (PrP(TSE)) in skeletal muscles of white-tailed deer infected with chronic wasting disease. PLoS One 6:e18345
Deuschl G, Margraf N, Spuler S, Kupsch A, Schulz-Schaeffer WJ (2010) Camptocormia and myopathy. Mov Disord 25:2689–2690
Diederich NJ, Goebel HH, Dooms G et al (2006) Camptocormia associated with focal myositis in multiple-system atrophy. Mov Disord 21:390–394
Dietz V (2003) Spinal cord pattern generators for locomotion. Clin Neurophysiol 114:1379–1389
Djaldetti R, Mosberg-Galili R, Sroka H, Merims D, Melamed E (1999) Camptocormia (bent spine) in patients with Parkinson’s disease—characterization and possible pathogenesis of an unusual phenomenon. Mov Disord 14:443–447
Dubowitz V, Sewry CA (2007) Histological and histochemical stains and reactions. Muscle biopsy. A practical approach. Saunders Elsevier, Philadelphia, pp 21–39
Earle H (1815) Reply to the review of Mr. Bayrton’s essay on the cure of crooked spine. Edinburgh Med Surg J 11:35–51
Eriksson O, Eriksson A, Ringqvist M, Thornell LE (1980) The reliability of histochemical fibre typing of human necropsy muscles. Histochemistry 65:193–205
Finsterer J, Strobl W (2010) Presentation, etiology, diagnosis, and management of camptocormia. Eur Neurol 64:1–8
Friden J, Sjostrom M, Ekblom B (1983) Myofibrillar damage following intense eccentric exercise in man. Int J Sports Med 4:170–176
Gdynia HJ, Sperfeld AD, Unrath A et al (2009) Histopathological analysis of skeletal muscle in patients with Parkinson’s disease and ‘dropped head’/’bent spine’ syndrome. Parkinsonism Relat Disord 15:633–639
Hughes AJ, Daniel SE, Kilford L, Lees AJ (1992) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55:181–184
Jankovic J (2010) Camptocormia, head drop and other bent spine syndromes: heterogeneous etiology and pathogenesis of Parkinsonian deformities. Mov Disord 25:527–528
Johnson MA, Polgar J, Weightman D, Appleton D (1973) Data on the distribution of fibre types in thirty-six human muscles. An autopsy study. J Neurol Sci 18:111–129
Karpati G, Carpenter S, Eisen AA (1972) Experimental core-like lesions and nemaline rods. A correlative morphological and physiological study. Arch Neurol 27:237–251
Katz JS, Wolfe GI, Burns DK, Bryan WW, Fleckenstein JL, Barohn RJ (1996) Isolated neck extensor myopathy: a common cause of dropped head syndrome. Neurology 46:917–921
Kramer ML, Schulz-Schaeffer WJ (2007) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J Neurosci 27:1405–1410
Laroche M, Delisle MB, Aziza R, Lagarrigue J, Mazieres B (1995) Is camptocormia a primary muscular disease? Spine (Phila Pa 1976) 20:1011–1016
Lava NS, Factor SA (2001) Focal myopathy as a cause of anterocollis in Parkinsonism. Mov Disord 16:754–756
Lepoutre AC, Devos D, Blanchard-Dauphin A et al (2006) A specific clinical pattern of camptocormia in Parkinson’s disease. J Neurol Neurosurg Psychiatry 77:1229–1234
Margraf NG, Wrede A, Rohr A et al (2010) Camptocormia in idiopathic Parkinson’s disease: a focal myopathy of the paravertebral muscles. Mov Disord 25:542–551
Muller-Hocker J (1990) Cytochrome c oxidase deficient fibres in the limb muscle and diaphragm of man without muscular disease: an age-related alteration. J Neurol Sci 100:14–21
Nonaka I, Ishiura S, Arahata K, Ishibashi-Ueda H, Maruyama T, Ii K (1989) Progression in nemaline myopathy. Acta Neuropathol 78:484–491
Ozer F, Ozturk O, Meral H, Serdaroglu P, Yayla V (2007) Camptocormia in a patient with Parkinson disease and a myopathy with nemaline rods. Am J Phys Med Rehabil 86:3–6
Parkinson J (2002) An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci 14:223–236
Polgar J, Johnson MA, Weightman D, Appleton D (1973) Data on fibre size in thirty-six human muscles. An autopsy study. J Neurol Sci 19:307–318
Sakiyama Y, Okamoto Y, Higuchi I et al (2011) A new phenotype of mitochondrial disease characterized by familial late-onset predominant axial myopathy and encephalopathy. Acta Neuropathol 121:775–783
Schabitz WR, Glatz K, Schuhan C et al (2003) Severe forward flexion of the trunk in Parkinson’s disease: focal myopathy of the paraspinal muscles mimicking camptocormia. Mov Disord 18:408–414
Schomburg ED (1990) Spinal sensorimotor systems and their supraspinal control. Neurosci Res 7:265–340
Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol 24:8055–8068
Serratrice G, Pouget J, Pellissier JF (1996) Bent spine syndrome. J Neurol Neurosurg Psychiatry 60:51–54
Shafiq SA, Gorycki MA, Asiedu SA, Milhorat AT (1969) Tenotomy. Effect on the fine structure of the soleus of the rat. Arch Neurol 20:625–633
Spuler S, Krug H, Klein C et al (2010) Myopathy causing camptocormia in idiopathic Parkinson’s disease: a multidisciplinary approach. Mov Disord 25:552–559
Suarez GA, Kelly JJ Jr (1992) The dropped head syndrome. Neurology 42:1625–1627
Tiple D, Fabbrini G, Colosimo C et al (2009) Camptocormia in Parkinson disease: an epidemiological and clinical study. J Neurol Neurosurg Psychiatry 80:145–148
Umapathi T, Chaudhry V, Cornblath D, Drachman D, Griffin J, Kuncl R (2002) Head drop and camptocormia. J Neurol Neurosurg Psychiatry 73:1–7
Wharton SB, Chan KK, Pickard JD, Anderson JR (1996) Paravertebral muscles in disease of the cervical spine. J Neurol Neurosurg Psychiatry 61:461–465
Wright WG, Gurfinkel VS, King LA, Nutt JG, Cordo PJ, Horak FB (2010) Axial kinesthesia is impaired in Parkinson’s disease: effects of levodopa. Exp Neurol 225:202–209
Wu S, Ibarra MC, Malicdan MC et al (2006) Central core disease is due to RYR1 mutations in more than 90% of patients. Brain 129:1470–1480
Yoshiyama Y, Takama J, Hattori T (1999) The dropped head sign in Parkinsonism. J Neurol Sci 167:22–25
Acknowledgments
We thank Tatjana Pfander, Margit Kollhoff and Kerstin Brekerbohm for their skilful technical assistance. This work was supported by the VolkswagenStiftung (grants ZN 1294 and ZN 2168 to W.J.S.S).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Wrede, A., Margraf, N.G., Goebel, H.H. et al. Myofibrillar disorganization characterizes myopathy of camptocormia in Parkinson’s disease. Acta Neuropathol 123, 419–432 (2012). https://doi.org/10.1007/s00401-011-0927-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0927-7