
Abstract
Although oligodendroglial neoplasms are traditionally considered purely glial, increasing evidence suggests that they are capable of neuronal or neurocytic differentiation. Nevertheless, ganglioglioma-like foci (GGLF) have not been previously described. Herein, we report seven examples where the primary differential diagnosis was a ganglioglioma with an oligodendroglial component. These five male and two female patients ranged in age from 29 to 63 (median 44) years at initial presentation and neuroimaging features were those of diffuse gliomas in general. At presentation, the glial component was oligodendroglioma in six and oligoastrocytoma in one; one was low-grade and six were anaplastic. A sharp demarcation from adjacent GGLF was common, although some intermingling was always present. The GGLF included enlarged dysmorphic and occasionally binucleate ganglion cells, Nissl substance, expression of neuronal antigens, GFAP-positive astrocytic elements, and low Ki-67 labeling indices. In contrast to classic ganglioglioma, however, cases lacked eosinophilic granular bodies and CD34-positive tumor cells. Scattered bizarre astrocytes were also common and one case had focal neurocytic differentiation. By FISH analysis, five cases showed 1p/19q codeletion. In the four cases with deletions and ample dysmorphic ganglion cells for analysis, the deletions were found in both components. At last follow-up, two patients suffered recurrences, one developed radiation necrosis mimicking recurrence, and one died of disease 7.5 years after initial surgery. We conclude that GGLF represents yet another form of neuronal differentiation in oligodendroglial neoplasms. Recognition of this pattern will prevent a misdiagnosis of ganglioglioma with its potential for under-treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Oligodendroglioma was originally defined by Bailey and Bucy [3] in 1929 as a glioma subtype with cells that morphologically resembled normal oligodendrocytes. To a great extent, that definition still pertains today [30]. Nonetheless, there has been great controversy over the minimal criteria required for this diagnosis. Since the initial discovery that this tumor type is associated with a useful genetic signature predicting favorable prognosis and enhanced therapeutic sensitivity [1, 9, 14, 31, 33], there has been renewed interest in fully characterizing the morphologic and biologic spectra of oligodendroglial neoplasms. One surprising finding from this pursuit has been that despite considering oligodendroglioma a purely glial neoplasm, they can show neuronal features at multiple levels, including immunohistochemically, ultrastructurally, in vitro, and on expression profiling studies [11, 13, 20, 22–25, 27, 28, 36, 38–40]. In most of these cases, the neuronal phenotype is not evident by routine histology alone. Nonetheless, we and others have reported rare examples with overt neurocytic differentiation, associated with slightly smaller darker nuclei, as well as neuropil and rosette formation [20, 22, 28, 36]. When this neurocytic differentiation is extensive, the distinction from extraventricular neurocytoma can be extremely difficult. In such cases, however, the presence of chromosome 1p and 19q codeletion strongly favors the interpretation of oligodendroglioma with neurocytic features. The behavior of these tumors appears to be essentially that of grade matched oligodendrogliomas without neurocytic features based on the limited number of cases reported to date. In the current study, a somewhat analogous situation is described, where the neuronal differentiation, instead, bears striking similarities to ganglioglioma, a significant diagnostic pitfall, since this glioneuronal neoplasm is much more indolent and has completely different therapeutic implications. To our knowledge, this morphologic pattern has not been previously reported.
Materials and methods
The cases reported in this series were encountered by the authors at their home institutions and on the consult service at Washington University, St. Louis, between 2001 and 2010. They were all sent to one of the authors (AP) for second opinion regarding the diagnosis or genetic confirmation of the oligodendroglioma component. Immunohistochemical studies were performed as part of the diagnostic workup using standard antibodies and techniques, either by the referring pathologist or as part of the consult workup.
Dual-color FISH was performed as previously published with slight modification of the interpretation criteria [12]. Deparaffinization of the sections was carried out using two 10-min immersions in Citrisolv, followed by three 3-min immersions in isopropanol. The slides were next rinsed in running water for 5 min, followed by distilled water for 3 min. Target retrieval was achieved by immersing the slides in citrate buffer (pH 6.0) within a plastic Coplin jar which was steamed a total of 20 min, then slowly cooled to room temperature. The slides were then rinsed in running water for 5 min, followed by distilled water for 3 min. Sections were subjected to 0.4% pepsin (P-7012, Sigma-Aldrich, St. Louis, MO) digestion for 15 min (37°C) and then were placed in 2× SSC on a rotator for 5 min. Slides were then air-dried. Paired probes for 1p32/1q42 and 19p13/19q13 were diluted from stock at 1:25 using DenHyb buffer (Insitus Biotechnologies; Albuquerque, NM), applied to each slide and co-denatured with the target DNA at 90°C for 13 min. The slides were incubated overnight at 37°C in a humidified oven and subsequently washed for 5 min each with 50% formamide/1× SSC and two washes in 2× SSC. Depending on the surface area of the section to be covered, 10–20 μL of DAPI in Fluorguard (Insitus) was applied to each slide, which was then coverslipped. Green and red fluorescent signals were enumerated under an Olympus BX60 fluorescence microscope with appropriate filters (Olympus; Melville, NY). For each case, green and red signals were enumerated in 100–200 consecutive non-overlapping nuclei in each of the two basic morphologic patterns: oligodendroglioma and ganglioglioma-like foci (GGLF). In cases where the two phenotypes were intermixed, larger dysmorphic appearing nuclei were counted separately from the smaller oligodendroglioma-like nuclei. In cases where 100 large dysmorphic nuclei were not found or for which there were no adequate signals for reliable interpretation, they were considered non-informative for this portion of the tumor. Similarly, at least 90% of nuclei in any given region had to have interpretable signals for a case to be considered informative. An interpretation of deletion for either 1p or 19q was made when >25% of tumor nuclei harbored twice as many reference probes as test probe signals (e.g., patterns of 2:1, 4:2, 6:3, etc.). This criterion has been used in our clinical FISH lab for several years and is based on similar analyses in non-neoplastic CNS nuclei, using the median percentages for the 2:1 pattern plus three standard deviations. Cutoffs of polysomies (chromosomal gains) were set at 10%. FISH images were captured using a black and white, high-resolution COHU CCD camera, Z-stack motor and CytoVision™ basic workstation (Applied Imaging, Santa Clara, CA), with sequential DAPI (1 level), FITC (5 levels), and rhodamine (5 levels) filter settings. The resulting images were reconstituted with blue, green, and red pseudocolors using CytoVision™ software.
Results
Clinical, radiographic, pathological, and genetic features are summarized in Tables 1, 2 and 3, with more detailed descriptions below.
Case 1
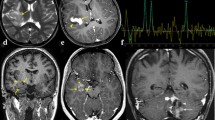
This 44-year-old man presented with a 3-day history of progressive headaches. A head CT revealed a contrast enhancing right frontal mass with a posterior cystic component and associated vasogenic edema. Subsequent MRI similarly revealed a 6 × 3.9 × 5.3 cm heterogeneous, partially enhancing, partially cystic, right frontal mass with moderate cerebral edema, midline shift, and extension into the corpus callosum (Fig. 1a–c). A resection of the enhancing component was performed, followed by adjuvant radiation therapy (6,000 cGy over 30 fractions over 6 weeks). The patient remained stable until 1.3 years post-operatively, when he suffered worsening headaches once again. Neuroimaging revealed a focus of ring enhancement in the right frontal lobe, with several smaller nodular foci of contrast enhancement nearby. Radiologically, recurrent tumor was favored and the patient was therefore treated with temozolomide chemotherapy. Three months later, this focus appeared larger (Fig. 1d), prompting a second resection, which revealed radiation necrosis without any viable tumor. Over the next 6 months, the cavity margins developed further foci of contrast enhancement prompting a third surgery, which again showed only radiation necrosis. The patient was last seen 2.1 years after the initial surgery and is doing well.

Pre-operative MR images from patient 1 showing a partially enhancing ill-defined right frontal lobe mass with extension into the corpus callosum on sagittal post-contrast T1-weighted (a), axial T2-weighted (b), and axial post-contrast T1-weighted (c) images. A rim enhancing mass detected over 1 year post-operatively (d) prompted re-resection for suspected recurrence, but the specimen showed only radiation necrosis on pathological examination
The pathology specimen from the first surgery revealed a neoplasm containing two distinct morphologic appearances with relatively sharp demarcation between them (Fig. 2a), but some intermingling. One portion of the tumor had classic features of anaplastic oligodendroglioma, including an infiltrative growth pattern, uniformly rounded nuclei with delicate chromatin, clear perinuclear haloes, scattered minigemistocytes, nests of slightly larger more epithelioid cells, a rich branching capillary network, increased mitotic activity (up to 10 per 10 high powered fields), microvascular proliferation, and foci of non-palisading tumor necrosis (Fig. 2b, d, f). The second component resembled ganglioglioma and included large dysmorphic cells arranged in nests and sweeping fascicles, subsets of which had discernable Nissl substance and/or showed binucleation (Fig. 2c, e, g). Additionally, there was focal perivascular lymphocytic cuffing and scattered spindled cells with eosinophilic cytoplasm resembling astrocytes. However, no eosinophilic granular bodies (EGBs) were detected. Mitotic figures were hard to find and there was no necrosis or microvascular proliferation. On immunohistochemical studies, GFAP highlighted gliofibrillary oligodendrocytes and minigemistocytes within the oligodendroglioma component and astrocytic cells within the GGLF (Fig. 3a, b). Synaptophysin expression was similarly seen in both components, but yielded a cytoplasmic dot-like pattern in the oligodendroglioma (Fig. 3c) and cytoplasmic and/or membrane staining in the dysmorphic ganglion cells of the GGLF (Fig. 3d). Cytoplasmic neurofilament expression was seen in only a minor subset of oligodendroglioma cells, although entrapped axons were highlighted, consistent with an infiltrative growth pattern (Fig. 3e). Strong neurofilament positivity was seen in ganglion cells and their processes within the GGLF (Fig. 3f). A CD34 stain highlighted endothelial cells, but all tumor cells were immunonegative. The Ki-67 labeling index was markedly elevated in the oligodendroglial portions, reaching up to 44% (Fig. 3g). In contrast, only rare Ki-67 positive nuclei were seen in the GGLF, some of which likely represented perivascular inflammatory cells (Fig. 3h). FISH analysis revealed 1p and 19q codeletion in both components (Fig. 4).

The histology from case 1 showed relative demarcation of areas resembling oligodendroglioma (a; left) and areas resembling ganglioglioma (a; right). Higher magnification images are shown of the oligodendroglioma (b, d, f) and GGLF (c, e, g). Classical oligodendroglial features included uniform round nuclei, clear haloes, microcalcifications, and chicken-wire like capillaries (b). Anaplastic features included necrosis and microvascular proliferation (d), as well as increased mitotic activity (f, arrow). Ganglioglioma-like features included large dysmorphic appearing cells with abundant eosinophilic to amphophilic cytoplasm, occasional Nissl substance (e, g), scattered perivascular lymphocytes (e), and a paucity of mitotic activity. Occasional binucleate forms were also seen (g, arrow)

Immunohistochemical studies in case 1 with the oligodendroglioma component on the left and the ganglioglioma-like component on the right. GFAP highlighted gliofibrillary oligodendrocytes (a) and astrocytic cells (b). Synaptophysin yielded a cytoplasmic dot-like pattern in the oligodendroglioma (c) and cytoplasmic/membrane pattern staining of dysmorphic ganglion cells (d). Only rare oligodendroglial cells stained with neurofilament protein, although entrapped axons in this component were consistent with an infiltrative growth pattern (e). Many of the dysmorphic ganglion cells and their processes expressed neurofilament (f). The Ki-67 labeling index reached up to 44% in the oligodendroglioma component (g), but was <1% in the ganglioglioma-like foci (h)

FISH studies in the oligodendroglioma component revealed 1p and 19q codeletion with most nuclei being near diploid (not shown). In the ganglioglioma-like foci, there was similarly a codeletion pattern with a mixture of near-diploid (1 test and 2 reference probe signals/cell; arrows) and near-tetraploid (arrowheads) populations, the latter containing up to four red 1q and two green 1p signals (a) and up to four green 19p and two red 19q signals (b) (some cells have fewer signals because they are either out of the plane of focus or truncated during paraffin sectioning)
Case 2
This 58-year-old man presented with progressive dyscalculia and short-term memory loss. MRI revealed a poorly demarcated, non-enhancing left temporo-parietal mass felt radiologically to be consistent with a diffuse glioma. A stereotactic biopsy was performed, but was non-diagnostic. This was followed by a subtotal resection utilizing cavitronic ultrasonic aspiration (CUSA), followed by intraoperative Gliadel® wafer implantation within the surgical cavity. Unfortunately, the resulting specimen was poorly preserved and only a descriptive diagnosis of “diffuse glioma” was possible at that time (slides from outside hospital unavailable for further review). No definite neuronal component was evident by report, although this may have been difficult to discern due to poor specimen quality. The patient was subsequently treated with standard fractionated radiotherapy over 4 months and the tumor remained stable for the next 4.4 years, at which time neuroimaging revealed a new focus of contrast enhancement consistent with recurrence. The enhancing component was resected. The patient was subsequently treated with a temozolomide and marimastat chemotherapy regimen with good response and stable disease for the next 3 years. At this time, temozolomide therapy was reinstituted due to another new focus of contrast enhancement. However, the tumor continued to progress and the patient expired 7.5 years from initial presentation.
The pathology of the recurrent tumor resembled anaplastic oligodendroglioma, WHO grade III in the majority of slides (Fig. 5a). Features included diffuse infiltration, uniform round nuclei with small nucleoli and clear perinuclear haloes, branching “chicken-wire” capillaries, scattered minigemistocytes, increased mitotic activity, and microvascular proliferation. No necrosis was seen. Focally, GGLF were identified and they appeared mostly demarcated from the adjacent oligodendroglioma pattern (Fig. 5b). This portion included a microcystic background and scattered markedly enlarged, mildly dysmorphic ganglion cells with occasional binucleate or multinucleate forms (Fig. 5c). Rare bizarre astrocytes were also noted (Fig. 5d). No EGBs were found. Immunohistochemical studies revealed GFAP positivity in a subset of the oligodendroglioma cells, morphologically consistent with minigemistocytes and gliofibrillary oligodendrocytes. The rare bizarre astrocytes were also positive. Dysmorphic ganglion cells were immunoreactive for synaptophysin and Neu-N, while oligodendroglioma cells were negative. CD34 was negative in tumor cells, but labeled endothelial cells appropriately. The Ki-67 labeling index was estimated at 4% within the oligodendroglioma component and <1% in the GGLF. FISH analysis revealed 1p and 19q codeletion in the oligodendroglioma. There were insufficient numbers of dysmorphic ganglion cells for reliable counts in the GGLF.

The histological features in case 2 were predominantly those of an anaplastic oligodendroglioma, including round regular nuclei, small nucleoli, clear perinuclear haloes, and a rich branching capillary network (a). The oligodendroglioma (b; left) was sharply demarcated from adjacent small GGLF (b; right). The latter contained a neuropil-rich microcystic background, with large mildly dysmorphic ganglion cells, including binucleate forms (c; arrow). Rare, bizarre multinucleate astrocytes were also found (d)
Case 3
This 40-year-old man developed daily headaches and was referred to an ophthalmologist when these were complicated by visual blurring. Fundoscopic exam revealed papilledema, prompting imaging studies. Head CT and MRI scans demonstrated a 5-cm partially calcified, partially enhancing right frontoparietal mass with extension into the splenium of the corpus callosum. There was associated cerebral edema and mass effect with compression of the right lateral ventricle. On the day of the MRI, the patient developed left sided weakness and had a seizure. The patient subsequently underwent a craniotomy with resection of the contrast enhancing component and a portion of the non-enhancing component. Adjuvant therapy included 12 cycles of temozolomide chemotherapy, but this had to be discontinued after the 12th cycle due to severe rash. The last MRI scan performed 4.9 years after initial presentation showed stable disease with residual non-enhancing tumor and otherwise, the patient is doing well.
The pathologic findings in case 3 were nearly identical to those of case 2, except that there was a greater degree of intermingling between the two elements and the zones of infarct-like necrosis within the oligodendroglial component were larger. Synaptophysin staining revealed strong cytoplasmic and Golgi pattern positivity in dysmorphic ganglion cells, while only rare oligodendroglial cells showed cytoplasmic reactivity. There was extensive GFAP positivity with most positive cells showing morphologic features of gliofibrillary oligodendrocytes. However, irregular cells with elongate processes were also seen, suggesting a minor astrocytic component. Both components were Neu-N negative. The CD34 stain highlighted endothelial cells only; tumor cells were negative. The Ki-67 labeling index was low to moderate, focally reaching a maximal staining of 15% in the oligodendroglial component; it was <1% in GGLF. FISH analysis revealed chromosome 19q deletion in both components, while chromosome 1 studies were non-informative due to weak signals. Nevertheless, loss of heterozygosity studies were also performed on this tissue at the Memorial Sloan Kettering Cancer Center and allelic losses of both 1p and 19q were detected. Therefore, this tumor was interpreted as being codeleted.
Case 4
This 29-year-old woman was previously healthy until she awoke with a severe left frontal headache associated with nausea and vomiting. A few days later, she had a similar episode, prompting medical attention. A head MRI scan demonstrated a large, partially enhancing, left frontal lesion with some mass effect including mild left-to-right shift and compression of the ipsilateral ventricle. There was some high attenuation on pre-gadolinium images. It also had some hypodense and cystic areas. It did not appear to cross the corpus callosum. A left frontal craniotomy with intraoperative motor and speech mapping was performed and the entire enhancing component was resected. A decision was made not to treat the neoplasm with radiation or chemotherapy, but to follow with frequent MRI scans.
Over the next 2 years, the patient remained asymptomatic and MRI scans were stable. Eventually however, they revealed gradually increasing areas of FLAIR abnormality and a small area of new enhancement situated near the anterior pole of the surgical cavity. The corpus callosum was thickened and the neoplasm had now extended into the right frontal lobe. Therefore, a second resection was performed roughly 2.2 years after her initial presentation. The patient was treated post-operatively with 12 cycles of temozolomide chemotherapy. At her last follow-up visit 3.5 years from the first surgery, she was doing well with stable disease on imaging studies.
The first pathology specimen revealed a neoplasm that was mostly consistent with oligodendroglioma, WHO grade II, but focally showed small foci of enlarged epithelioid cells with increased cytoplasm, nucleolar prominence, and increased mitotic activity of up to 6 mitoses per 10 high powered fields, warranting a designation of focal anaplasia, WHO grade III. Small clusters of minigemistocytes were seen in a few areas. In addition there were multiple small GGLF that were mostly intermingled with the rest of the tumor. These foci included large, dysmorphic, occasionally binucleate ganglion cells embedded within collections of loose to microcystic, delicate fibrillary matrix, consistent with tumor-associated neuropil. A small subset of these dysmorphic cells contained discernable Nissl substance. No EGBs or bizarre astrocytes were seen in either component. The recurrent tumor showed similar features, but had only rare GGLF. Additionally, there was focal microvascular proliferation as well as focal pseudopalisading necrosis, further evidence of its anaplastic nature. Immunohistochemical studies revealed cytoplasmic and Golgi-like synaptophysin positivity in both dysmorphic ganglion cells and a subset of the oligodendroglioma cells. The dysmorphic ganglion cells were also variably immunoreactive for neurofilament protein and Neu-N, while the oligodendroglioma cells were negative. Subsets of tumor cells were strongly GFAP positive, mostly showing morphologic features of minigemistocytes and gliofibrillary oligodendrocytes. Endothelial cells stained appropriately for CD34, although tumor cells were negative. The Ki-67 labeling index was mostly low, but focally reached up to 12% in the oligodendroglial component. Dysmorphic ganglion cells were negative. FISH studies were performed on the recurrent tumor and showed 1p and 19q codeletion. However, there were too few dysmorphic ganglion cells to reliably analyze the GGLF portion.
Case 5
This 55-year-old man was admitted for a several-month history of personality change and more recently had been becoming increasingly drowsy and was noted to be dragging his left foot. A CT scan of the head revealed a large cystic mass in the right frontal lobe with compression of the frontal horn of the right lateral ventricle and right-to-left shift. Decreased signal was seen extending throughout the white matter of the frontal lobe across the genu of the corpus callosum into the left frontal lobe. An MRI of the head confirmed the large complex mass with contrast enhancement of the solid component, as well as a portion of the rim of the cystic component. However, the enhancing component did not extend into the corpus callosum. The differential diagnosis was glioblastoma or possibly a metastatic deposit. While in the hospital, his condition deteriorated and he was given dexamethasone with some improvement. A CT scan of the head revealed a new large intratumoral hemorrhage. An emergent craniotomy was performed with drainage of the cyst and a subtotal resection of the tumor, which appeared to extend into the temporal lobe. The patient received adjuvant external beam radiotherapy with a total dose of 59.4 Gy in 33 fractions. MRI studies performed 6 months post-operatively revealed a new focus of contrast enhancement suspicious for recurrence, which decreased in size on the most recent imaging 2 months later. However, the extent of the surrounding non-enhancing T2/FLAIR signal abnormality had increased. The latest plan was to see him again in 3 months, with the possibility of chemotherapy if imaging revealed tumor progression.
The pathology specimen was very similar in appearance to case 1, except that the oligodendroglial component appeared low-grade (WHO grade II), with low mitotic activity and no evidence of either microvascular proliferation or necrosis (Fig. 6a). Dysmorphic ganglion cells (Fig. 6b) displayed strong cytoplasmic and membrane pattern positivity for synaptophysin (Fig. 6d), but were nearly uniformly negative for Neu-N (Fig. 6f). No EGBs were seen. Weak cytoplasmic positivity for synaptophysin was seen in occasional oligodendroglioma cells as well, but mostly highlighted entrapped neuropil (Fig. 6c); Neu-N stained only entrapped cortical neurons (Fig. 6e). Scattered GFAP-positive cells were seen in both components, although in the GGLF they appeared consistent with tumoral astrocytes (Fig. 6h), while in the oligodendroglioma portion most positive cells looked like reactive astrocytes (Fig. 6g). Occasional gliofibrillary oligodendrocytes were also noted. Tumor cells were CD34 negative, while intratumoral endothelial cells were positive. The Ki-67 stain reached a maximal labeling index of roughly 8% in the oligodendroglioma component, but stained only rare small perivascular cells in the GGLF (similar to Fig. 3h). FISH studies showed chromosome 1p deletion in both components (Fig. 7a, b), while chromosome 19 studies showed a normal disomic (2 copies) dosage in both regions (Fig. 7c, d), i.e., there was no evidence of 19q deletion.

The oligodendroglioma and GGLF in case 5 are displayed on the left and right sides, respectively. The former appeared low-grade (a), while tight clusters of unmistakably dysmorphic ganglion cells characterized the latter (b). The majority of oligodendroglioma cells were negative for synaptophysin (c) and Neu-N (e), although both these stains highlighted entrapped native elements, consistent with an infiltrative growth pattern. Dysmorphic ganglion cells were strongly synaptophysin positive (d), but mostly Neu-N negative (f). GFAP stains highlighted mostly reactive appearing astrocytes in the oligodendroglioma regions (g) and tumoral astrocytes in the GGLF (h)

FISH images from the oligodendroglioma region and GGLF of case 5 are illustrated on the left and right sides, respectively. Both components showed evidence of chromosome 1p deletion (a, b), but retained chromosome 19 dosages (c, d)
Case 6
This 63-year-old woman presented with a 2-week history of progressive ataxia, falls, confusion, and word finding difficulties. She also had headaches for a few days. After a few falls and increasingly bizarre behavior, her husband brought her to the hospital for medical attention. Head CT and MRI were eventually included in the workup and they showed a large, minimally enhancing left frontal lobe mass with speckled calcifications, an infiltrative pattern, and extension across the corpus callosum to the right frontal lobe (Fig. 8). There was moderate associated edema and considerable mass effect with partial compression of the left lateral ventricle. A large, but subtotal resection was performed. The patient is currently undergoing combined chemoradiotherapy, with temozolomide as the chemotherapeutic agent. No additional follow-up is available given that the patient was only recently diagnosed.

Pre-operative MRI from patient 6 showed a minimally enhancing diffuse left frontal lobe mass with extension across the corpus callosum into the right frontal lobe on axial post-contrast T1-weighted (a) and axial FLAIR (b) images
The pathological findings in this case were striking for at least two distinct morphological appearances, which were sharply demarcated from one another focally (Fig. 9a), but were intimately intermingled in most areas. The predominant cell type was that of an anaplastic oligodendroglioma with classical cytological features (Fig. 9b), up to 6 mitoses per 10 high powered fields, and focal necrosis, but no definite microvascular proliferation. Additionally, scattered enlarged, bizarre and sometimes multinucleated astrocytes were interspersed among the oligodendroglioma cells (Fig. 9b); it was unclear whether these represented reactive, dysplastic, or neoplastic elements. Moreover, there were several small foci of neurocytic differentiation with delicate neuropil formation and neurocytic rosettes (Fig. 9d). The GGLF featured microcystic spaces (Fig. 9c) and large dysmorphic neurons, including binucleate forms (Fig. 9e; arrow). Even in these areas, however, there were scattered smaller oligodendroglioma cells, including minigemistocytes (Fig. 9e; arrowheads). No EGBs were seen. Neuronal markers, such as synaptophysin, chromogranin, neurofilament protein, and Neu-N variably stained the dysmorphic ganglion cells in GGLF, as well as entrapped native elements within the oligodendroglioma regions (Fig. 10). GFAP highlighted scattered astrocytic elements in both foci, including the enlarged bizarre forms (Fig. 10e, f). Intratumoral endothelial cells stained appropriately for CD34, but tumor cells were negative. The Ki-67 labeling index was estimated at roughly 10% in the oligodendroglioma component and <1% in the GGLF. Codeletion of 1p and 19q was seen in both components using FISH (Fig. 11). Most of the dysmorphic ganglion cells appeared near-tetraploid with two test and four reference probe signals, while most of the oligodendroglioma cells appeared near-diploid with one test and two reference probe signals.

The histologic examination of case 6 showed focal demarcation between the oligodendroglioma (a; left) and ganglioglioma-like (a; right) components, but also consisted of intermingled elements. The oligodendroglial cytology had classic features (b, d), but also included scattered large bizarre astrocytes (b) and small foci of neurocytic differentiation with rosette formation (d). The ganglioglioma-like foci had microcystic growth pattern (c), enlarged dysmorphic ganglion cells including occasional binucleate forms (e; arrow), as well as scattered oligodendroglial cells and minigemistocytes (e; arrowheads)

Immunohistochemical results in case 6, with the oligodendroglioma component on the left and the ganglioglioma component on the right. Chromogranin highlighted scattered small entrapped cortical ganglion cells in the oligodendroglial element (a) and the majority of dysmorphic ganglion cells in GGLF (b). Similar results were obtained with the Neu-N stain (c, d), although dysmorphic ganglion cells stained more variably, including occasional negativity in abnormal appearing cells (d; binucleate cell on the right). Most oligodendroglioma cells were negative for GFAP (e), except for patchy staining in minigemistocytes and gliofibrillary oligodendrocytes (not shown). However, the scattered bizarre astrocytes were strongly positive (e). A GFAP-positive astrocytic element was also seen in ganglioglioma-like foci (f)

FISH in case 6 showed a 1p (a) and 19q (b) codeletion pattern similar to that encountered in case #1 (see Fig. 4). In ganglioglioma-like foci, the majority of enlarged dysmorphic ganglion cells showed a tetraploid pattern (2 test and 4 reference probe signals; arrowhead in a and arrows in b), while the adjacent smaller oligodendroglioma cells displayed mostly a diploid pattern (1 test and 2 reference probe signals; arrows in a and arrowhead in b). In these images, the larger nuclei represent the ganglion cells, whereas the smaller nuclei represent oligodendroglioma cells. The delicate fibrillary matrix in between is neuropil
Case 7
This 42-year-old man presented with a 1-month history of progressive headaches, nausea, and vomiting. MRI studies revealed an 8.2 × 6.9 × 5 cm heterogeneously enhancing mass in the right frontal lobe with marked mass effects, including a 16-mm midline shift and compression of both frontal horns of the lateral ventricles. There were several cystic regions and one region with imaging characteristics of intratumoral hemorrhage. The radiographic impression was that of a high-grade diffuse glioma. A subtotal resection was recently performed and there are plans to treat the patient further with combined radiation and temozolomide therapy.
In the majority of slides from the resection specimen, the tumor had features of an anaplastic oligoastrocytoma, WHO grade III, wherein the oligodendroglial (classic and minigemistocytic) and astrocytic (fibrillary and gemistocytic) elements were intermixed. Anaplastic features included up to 9 mitoses per 10 high-powered fields and focal microvascular proliferation. No necrosis was found. GGLF were found in one slide, showing both demarcated and intermixed borders with adjacent glial elements. Multinucleated forms were common and the mitotic index was low in these regions, but no EGBs were seen. Strong GFAP positivity was seen in glial elements from both components. Dysmorphic ganglion cells showed widespread membrane and Golgi-like patterns of immunoreactivity for synaptophysin, while the glial elements were mostly negative. Rare dysmorphic ganglion cells displayed chromogranin positivity, but they were negative for both neurofilament protein and Neu-N. Endothelial cells strongly expressed CD34, but all tumor cells were negative. The Ki-67 labeling index was estimated at 12% in the glioma component, while dysmorphic ganglion cells were essentially negative. FISH studies showed polysomies (gains) of both chromosomes 1 and 19, with no evidence of deletions.
Discussion
The cases presented in this series were novel and created considerable diagnostic difficulties. In most, the diagnosis of ganglioglioma with an oligodendroglial component was originally favored, but the presence of anaplastic features was cause for concern. As such, the possibility of anaplastic ganglioglioma was also considered. On further analysis however, the presence of 1p and 19q codeletion in the majority of our cases, along with the finding of identical genetic alterations in both components suggested the opposite interpretation. In other words, this genetic signature is highly associated with the diagnosis of oligodendroglioma and is fairly specific to oligodendroglial neoplasms in general [1, 18, 29]. In contrast, it has never been reported in gangliogliomas [16, 26, 41]. Therefore, our interpretation was that these were essentially oligodendrogliomas with ganglioglioma-like maturation. We believe that this represents an unusual form of neuronal and to a lesser degree, astrocytic differentiation, somewhat analogous to prior studies showing foci of neurocytic differentiation in oligodendrogliomas [20, 22, 28, 36]. This potential link between these two forms of neuronal maturation was further strengthened by the finding of both neurocytic and gangliocytic maturation in one of our tumors (case 6). Otherwise, however, the potential mimicry between this morphologic pattern and the entity of ganglioglioma was striking. Of note, there were at least a few important differences. Whereas the great majority of gangliogliomas display at least some EGBs, none were found in the GGLF of our tumors. Additionally, roughly 70–80% of gangliogliomas display variable numbers of intratumoral and/or peritumoral CD34-positive cells, often with a characteristic cytology that includes highly ramified cytoplasmic processes [4, 6, 10]. Positivity has even been described in anaplastic gangliogliomas, although it is also known to be lost in some cases during malignant progression [19]. In our cases, this marker was uniformly negative, with intratumoral endothelial cells providing a useful internal control. As such, this may be a particularly useful immunostain in those rare cases where both diagnostic possibilities are being considered. Overall, perivascular lymphocytic cuffing was also less prominent in our cases than in gangliogliomas, although some inflammation was occasionally found. However, similar to gangliogliomas and in contrast to entrapped non-neoplastic cortical neurons, the dysmorphic ganglion cells in our tumors often displayed more variable Neu-N expression or were entirely negative. Therefore, Neu-N may be a useful immunostain for excluding the native ganglion cells that occasionally show mildly dysmorphic features in the midst of an infiltrating glioma. It is possible that MAP2 staining patterns could further aid in this distinction [5, 7, 32, 34], although this marker was not generally available in the primary author’s institution and was also not utilized by any of the referring pathologists. Therefore, future studies are needed to assess its potential utility for this differential diagnosis.
In addition to these histologic and genetic features distinguishing gangliogliomas from oligodendrogliomas with ganglioglioma-like maturation, there were significant clinical and neuroimaging clues in our cases. These patients were generally older than those with ganglioglioma; they lacked the typical long histories of chronic seizure disorder; and neuroimaging studies showed characteristic findings of diffuse gliomas in general, i.e., infiltrative cerebral hemispheric neoplasms with significant mass effects and common involvement of the corpus callosum. In contrast, gangliogliomas are classically demarcated, often cystic, with a mural contrast enhancing nodule, and most commonly involve the mesial temporal lobe [35]. Unlike diffuse gliomas, complete surgical excision is feasible in most cases and is often curative without any need for adjuvant therapy. Although there is insufficient data to determine for certain whether the presence of ganglioglioma-like maturation has any prognostic impact in oligodendrogliomas, our follow-up data to date would suggest that the clinical behavior is analogous to otherwise classical examples without neuronal features and of similar grade and molecular genetic background. This includes a long-term risk of recurrences and eventual death from progressive disease. For instance, the median overall survival times for patients with classical anaplastic oligodendrogliomas at Washington University were recently reported as 11.3 and 5.7 years, corresponding to tumors with and without 1p/19q codeletion, respectively [21]. Progression free survival times are similarly longer in patients with codeleted anaplastic oligodendrogliomas, although they vary considerably from one patient to another [8, 37]. Therefore, although longer follow-up is clearly needed for more definitive comparisons, the presence of one death at 7.5 years and two recurrences at 2–3 years seems par for the course.
Of interest, there have been rare reports of tumors interpreted as “ganglioglioma with an anaplastic oligodendroglial component” or “oligodendroglial ganglioglioma with anaplastic features” [2, 15, 17]. Probably the first was a 45-year-old woman with a heterogeneously enhancing left frontal lobe mass reported in 1990 by Allegranza and colleagues [2]. Unfortunately, this paper was scantly illustrated and documented by today’s standards. Nonetheless, based on the descriptions in the text, it appears very similar to those of the current study. As with our cases, this tumor was subtotally resected due to an ill-defined or infiltrative border. Based on the pathological findings, the interface between the two components was sharp in some areas and intermingled in others. Dysmorphic and binucleate ganglion cells displayed Nissl substance and were neurofilament positive, whereas the oligodendroglial component was Leu-7 positive and GFAP negative. No additional support for the interpretation of ganglioglioma with an anaplastic oligodendroglioma component was given, although it appeared to be partially based on a consultation diagnosis rendered by Dr. Lucien Rubenstein. No long-term clinical follow-up was provided. Another case was reported as part of a larger series of gangliogliomas in adults [15]. Unfortunately, this case was not elucidated in much detail. It represented a 26-year-old man with a frontal lobe mass that was treated with gross total resection, radiation, and chemotherapy. Despite therapy, this patient died 47 months after surgery. A third case was reported in a 17-year-old girl with a massive, partially enhancing, and hemorrhagic thalamic mass that extended into the brainstem [17]. This case also had some overlapping features with ours, although the authors reported finding rare EGBs in their case and the patient was younger than those described in the current series. As such, it is difficult to determine whether this case was the same phenomenon described in our paper or could potentially represent a true ganglioglioma with an anaplastic oligodendroglial component. None of the previously reported cases included genetic studies.
In summary, we report a new form of neuronal and to a lesser extent, astrocytic differentiation in oligodendroglial neoplasms. We refer to this pattern as “ganglioglioma-like maturation” given the combined morphologic and immunohistochemical evidence of ganglion cell differentiation and the loss of proliferative activity in comparison to adjacent oligodendroglial components. However, these GGLF differ from true gangliogliomas in their lack of (1) ganglioglioma-like demographic, clinical, and radiologic features, (2) EGBs, and (3) CD34-positive tumor cells. Additionally, these tumors often harbor the 1p/19q codeletion pattern typical of classical oligodendroglioma, a genetic signature which was found even in dysmorphic ganglion cells. This genetic finding supports a common lineage for the two otherwise strikingly dissimilar histologic patterns. Awareness of this morphologic variant could prevent the misdiagnosis of “ganglioglioma with an oligodendroglioma component”, averting the potential risk of under-treating these patients.
References
Aldape K, Burger PC, Perry A (2007) Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch Pathol Lab Med 131:242–251
Allegranza A, Pileri S, Frank G, Ferracini R (1990) Cerebral ganglioglioma with anaplastic oligodendroglial component. Histopathology 17:439–441
Bailey P, Bucy PC (1929) Oligodendrogliomas of the brain. J Pathol Bacteriol 32:735–751
Blumcke I, Giencke K, Wardelmann E, Beyenburg S, Kral T, Sarioglu N, Pietsch T, Wolf HK, Schramm J, Elger CE, Wiestler OD (1999) The CD34 epitope is expressed in neoplastic and malformative lesions associated with chronic, focal epilepsies. Acta Neuropathol (Berl) 97:481–490
Blumcke I, Becker AJ, Normann S, Hans V, Riederer BM, Krajewski S, Wiestler OD, Reifenberger G (2001) Distinct expression pattern of microtubule-associated protein-2 in human oligodendrogliomas and glial precursor cells. J Neuropathol Exp Neurol 60:984–993
Blumcke I, Wiestler OD (2002) Gangliogliomas: an intriguing tumor entity associated with focal epilepsies. J Neuropathol Exp Neurol 61:575–584
Blumcke I, Muller S, Buslei R, Riederer BM, Wiestler OD (2004) Microtubule-associated protein-2 immunoreactivity: a useful tool in the differential diagnosis of low-grade neuroepithelial tumors. Acta Neuropathol (Berl) 108:89–96
Cairncross G, Berkey B, Shaw E, Jenkins R, Scheithauer B, Brachman D, Buckner J, Fink K, Souhami L, Laperierre N, Mehta M, Curran W (2006) Phase III trial of chemotherapy plus radiotherapy compared with radiotherapy alone for pure and mixed anaplastic oligodendroglioma: Intergroup Radiation Therapy Oncology Group Trial 9402. J Clin Oncol 24:2707–2714
Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, Silver JS, Stark PC, Macdonald DR, Ino Y, Ramsay DA, Louis DN (1998) Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 90:1473–1479
Deb P, Sharma MC, Tripathi M, Sarat Chandra P, Gupta A, Sarkar C (2006) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468
Ducray F, Idbaih A, de Reynies A, Bieche I, Thillet J, Mokhtari K, Lair S, Marie Y, Paris S, Vidaud M, Hoang-Xuan K, Delattre O, Delattre JY, Sanson M (2008) Anaplastic oligodendrogliomas with 1p19q codeletion have a proneural gene expression profile. Mol Cancer 7:41
Fallon KB, Palmer CA, Roth KA, Nabors LB, Wang W, Carpenter M, Banerjee R, Forsyth P, Rich K, Perry A (2004) Prognostic value of 1p, 19q, 9p, 10q, and EGFR-FISH analyses in recurrent oligodendrogliomas. J Neuropathol Exp Neurol 63:314–322
Ferrer-Luna R, Mata M, Nunez L, Calvar J, Dasi F, Arias E, Piquer J, Cerda-Nicolas M, Taratuto AL, Sevlever G, Celda B, Martinetto H (2009) Loss of heterozygosity at 1p–19q induces a global change in oligodendroglial tumor gene expression. J Neurooncol 95:343–354
Giannini C, Burger PC, Berkey BA, Cairncross JG, Jenkins RB, Mehta M, Curran WJ, Aldape K (2008) Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol 18:360–369
Hakim R, Loeffler JS, Anthony DC, Black PM (1997) Gangliogliomas in adults. Cancer 79:127–131
Hoischen A, Ehrler M, Fassunke J, Simon M, Baudis M, Landwehr C, Radlwimmer B, Lichter P, Schramm J, Becker AJ, Weber RG (2008) Comprehensive characterization of genomic aberrations in gangliogliomas by CGH, array-based CGH and interphase FISH. Brain Pathol 18:326–337
Johnson MD, Jennings MT, Toms ST (2001) Oligodendroglial ganglioglioma with anaplastic features arising from the thalamus. Pediatr Neurosurg 34:301–305
Kouwenhoven MC, Gorlia T, Kros JM, Ibdaih A, Brandes AA, Bromberg JE, Mokhtari K, van Duinen SG, Teepen JL, Wesseling P, Vandenbos F, Grisold W, Sipos L, Mirimanoff R, Vecht CJ, Allgeier A, Lacombe D, van den Bent MJ (2009) Molecular analysis of anaplastic oligodendroglial tumors in a prospective randomized study: a report from EORTC study 26951. Neuro Oncol 11:737–746
Majores M, von Lehe M, Fassunke J, Schramm J, Becker AJ, Simon M (2008) Tumor recurrence and malignant progression of gangliogliomas. Cancer 113:3355–3363
Makuria AT, Henderson FC, Rushing EJ, Hartmann DP, Azumi N, Ozdemirli M (2007) Oligodendroglioma with neurocytic differentiation versus atypical extraventricular neurocytoma: a case report of unusual pathologic findings of a spinal cord tumor. J Neurooncol 82:199–205
Miller CR, Dunham CP, Scheithauer BW, Perry A (2006) Significance of necrosis in grading of oligodendroglial neoplasms: a clinicopathologic and genetic study of newly diagnosed high-grade gliomas. J Clin Oncol 24:5419–5426
Mrak RE, Yasargil MG, Mohapatra G, Earel J Jr, Louis DN (2004) Atypical extraventricular neurocytoma with oligodendroglioma-like spread and an unusual pattern of chromosome 1p and 19q loss. Hum Pathol 35:1156–1159
Mukasa A, Ueki K, Ge X, Ishikawa S, Ide T, Fujimaki T, Nishikawa R, Asai A, Kirino T, Aburatani H (2004) Selective expression of a subset of neuronal genes in oligodendroglioma with chromosome 1p loss. Brain Pathol 14:34–42
Mut M, Guler-Tezel G, Lopes MB, Bilginer B, Ziyal I, Ozcan OE (2005) Challenging diagnosis: oligodendroglioma versus extraventricular neurocytoma. Clin Neuropathol 24:225–229
Ng HK, Ko H, Tse C (1994) Immunohistochemical and ultrastructural studies of oligodendrogliomas revealed features of neuronal differentiation. Int J Surg Pathol 2:47–56
Pandita A, Balasubramaniam A, Perrin R, Shannon P, Guha A (2007) Malignant and benign ganglioglioma: a pathological and molecular study. Neuro Oncol 9:124–134
Patt S, Labrakakis C, Bernstein M, Weydt P, Cervos-Navarro J, Nisch G, Kettenmann H (1996) Neuron-like physiological properties of cells from human oligodendroglial tumors. Neuroscience 71:601–611
Perry A, Scheithauer BW, Macaulay RJ, Raffel C, Roth KA, Kros JM (2002) Oligodendrogliomas with neurocytic differentiation. A report of 4 cases with diagnostic and histogenetic implications. J Neuropathol Exp Neurol 61:947–955
Perry A, Fuller CE, Banerjee R, Brat DJ, Scheithauer BW (2003) Ancillary FISH analysis for 1p and 19q status: preliminary observations in 287 gliomas and oligodendroglioma mimics. Front Biosci 8:a1–a9
Reifenberger G, Kros JM, Louis DN, Collins VP (2007) Oligodendroglioma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumours of the central nervous system. IARC, Lyon, pp 54–59
Reifenberger J, Reifenberger G, Liu L et al (1994) Molecular genetic analysis of oligodendroglial tumors shows preferential allelic deletions on 19q and 1p. Am J Pathol 145:1175–1190
Shafit-Zagardo B, Davies P, Rockwood J, Kress Y, Lee SC (2000) Novel microtubule-associated protein-2 isoform is expressed early in human oligodendrocyte maturation. Glia 29:233–245
Smith JS, Perry A, Borell TJ, Lee HK, O’Fallon J, Hosek SM, Kimmel D, Yates A, Burger PC, Scheithauer BW, Jenkins RB (2000) Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 18:636–645
Suzuki SO, Kitai R, Llena J, Lee SC, Goldman JE, Shafit-Zagardo B (2002) MAP-2e, a novel MAP-2 isoform, is expressed in gliomas and delineates tumor architecture and patterns of infiltration. J Neuropathol Exp Neurol 61:403–412
Urbach H (2008) MRI of long-term epilepsy-associated tumors. Semin Ultrasound CT MR 29:40–46
Vajtai I, Arnold M, Vassella E (2005) Oligodendroglioma with neurocytic differentiation and characteristic loss of heterozygosity on chromosomes 1p and 19q. Acta Neuropathol (Berl) 110:520–522
van den Bent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, Sipos L, Haaxma-Reiche H, Kros JM, van Kouwenhoven MC, Vecht CJ, Allgeier A, Lacombe D, Gorlia T (2006) Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 24:2715–2722
Vyberg M, Ulhoi BP, Teglbjaerg PS (2007) Neuronal features of oligodendrogliomas—an ultrastructural and immunohistochemical study. Histopathology 50:887–896
Wharton SB, Chan KK, Hamilton FA, Anderson JR (1998) Expression of neuronal markers in oligodendrogliomas: an immunohistochemical study. Neuropathol Appl Neurobiol 24:302–308
Wolf HK, Buslei R, Blümcke I et al (1997) Neural antigens in oligodendrogliomas and dysembryoplastic neuroepithelial tumors. Acta Neuropathol (Berl) 94:436–443
Yin XL, Hui AB, Pang JC, Poon WS, Ng HK (2002) Genome-wide survey for chromosomal imbalances in ganglioglioma using comparative genomic hybridization. Cancer Genet Cytogenet 134:71–76
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Perry, A., Burton, S.S., Fuller, G.N. et al. Oligodendroglial neoplasms with ganglioglioma-like maturation: a diagnostic pitfall. Acta Neuropathol 120, 237–252 (2010). https://doi.org/10.1007/s00401-010-0695-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0695-9