
Abstract
The diagnostic criteria for focal cortical dysplasia type I (FCD I) remain to be well and consistently defined. Cortical layer-specific markers (CLM) provide a potential tool for the objective assessment of any dyslamination. We studied expression patterns of recognised CLM using immunohistochemistry for N200, ER81, Otx1, Map1b (subsets of V/VI projection neurones), Pax6, Tbr1, Tbr2 (differentially expressed in cortical neurones from intermediate progenitor cells), Cux 1 (outer cortical layers) and MASH1 (ventricular zone progenitors). Dysplasia subtypes included FCD I and II, dysplasias adjacent to hippocampal sclerosis (HS) or dysembryoplastic neuroepithelial tumours (DNTs); all were compared to neonatal and adult controls. Laminar expression patterns in normal cortex were observed with Tbr1, Map1b, N200 and Otx1. FCDI cases in younger patients were characterised by abnormal expression in layer II for Tbr1 and Otx1. FCDII showed distinct labelling of balloon cells (Pax6, ER81 and Otx1) and dysmorphic neurones (Tbr 1, N200 and Map1b) supporting origins from radial glia and intermediate progenitor cells, respectively. In temporal lobe sclerosis cases with dysplasia adjacent to HS, Tbr1 and Map1b highlighted abnormal orientation of neurones in layer II. Dyslamination was not confirmed in the perilesional cortex of DNT with CLM. Finally, immature cell types (Otx1, Pax6 and Tbr2) were noted in varied pathologies. One possibility is activation of progenitor cell populations which could contribute to the pathophysiology of these lesions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Following the revision to the classification of focal cortical dysplasias (FCD) in epilepsy in 2004 [41], there has been improved consistency in the clinical, radiological and pathological correlative data for the more severe types of dysplasia (FCD II) characterised by abnormal cytomorphology [38]. The criteria for milder dysplasias (FCD I), characterised mainly by abnormalities in cortical lamination, are less well defined [7] and more open to subjective interpretation with poor reproducibility shown between observers [12]. Varied neocortical laminar abnormalities are also reported adjacent to hippocampal sclerosis (HS) [51] and glioneuronal tumours including dysembryoplastic neuroepithelial tumours (DNT) [17, 50]. These dysplasias are also less well defined, including their distinction from isolated FCD.
Cortical layer-specific markers (CLM) are a potential diagnostic tool for the evaluation of mild dysplasias in epilepsy [26]. CLM include transcription factors, cytoskeletal proteins and calcium-binding proteins which show neuronal subtype and/or layer-specific expression patterns reflecting ontogeny and cortical maturity. Largely studied in the rodent cortex [26], there are few studies evaluating these markers in human epilepsy tissues [24, 32]. CLM expression is likely to vary between species, the stage of cortical maturation, between cortical regions and with neuronal activity, including seizures; these factors have to be considered in their evaluation in human pathologies [26].
Our aim was to assess a panel of CLM in the discrimination of commonly encountered dysplasia subtypes in epilepsy surgical tissue. We selected a panel shown by previous studies of cortical development and maturation demonstrated to reflect layer or neuronal lineage specificity [26, 36]. Any abnormalities in their distribution might enable a more accurate classification of these disorders.
Materials and methods
Case selection and histopathological features
The study was approved by the respective ethics committees of the participating centres where informed consent was obtained for the use of brain tissue and for access to medical records for research purposes. All surgical cases underwent therapeutic resection for refractory epilepsy and the pathological tissue was surplus to diagnostic requirement. In all cases we selected larger resection specimens so that adjacent normal cortex was more likely to be included. We aimed to include cases involving the temporal neocortex for all pathology groups where possible. The details of cases, including main histological features, are outlined in Table 1. Within groups similar dysplasia pathology types were studied, including cases from previous published series [28, 51]; 14 cases of FCD IA (henceforth referred to as FCDI), 9 cases of FCD II B (henceforth referred to as FCD II), 8 cases of dysplasia adjacent to DNT, 5 cases of dysplasia associated with HS and temporal lobe sclerosis (TLS) in addition to 14 control cases including epilepsy controls (no dysplasia). We did not include FCD type IB or IIA cases in the present study.
Immunohistochemistry
In all cases a single representative formalin-fixed, paraffin-embedded tissue block from each case was selected and further sections cut at 7 μm thickness. Sections were dewaxed and rehydrated, followed by blockage of endogenous peroxidase activity by 15 min incubation in 0.9% hydrogen peroxide in de-ionised water. A panel of 9 CLM antibodies was selected (as detailed in Table 2) in addition to NeuN (1:100, Chemicon). After washing, the sections were microwaved 12 or 15 min in antigen retrieval solution, then cooled for 20 min. Staining was visualised with diaminobenzidine (DAB) and NovaRed for N200 antibody. Sections where DAB was applied were then enhanced in copper sulphate, and counterstained with haematoxylin. Between each step, sections were washed in phosphate buffer saline with 0.05% Tween 20 (PBS+T). Negative controls were treated identically except that the primary antibody was omitted. For some of the antibodies with developmental regulation, e.g. Pax6, Tbr2, sections of foetal brain including the VZ and SVZ (20–36 gestational weeks) were used as a positive control. The laminar staining pattern with each CLM in all cases was assessed through comparison to the corresponding NeuN-stained section (Fig. 1a).

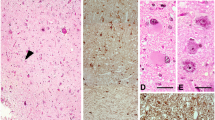
Controls. a Adult control: NeuN labelling in temporal lobe demonstrating distinct cortical laminae. b Adult control: Map1b showing cytoplasmic labelling in layers II, III, V (arrowed) and VI pyramidal cells with an absence of labelling in mid layers. c Adult control: N200 with labelling of isolated pyramidal cells mainly in layer V and VI and to a lesser extent the upper cortical layers (arrows). d Adult control: Tbr1 with nuclear labelling of a proportion of neurones in all cortical layers particularly IV, V and VI. e Paediatric control (day 1): Otx1 with frequent cells visible throughout the cortex but more prominent in layers II and IV. f Tbr2 frequent cytoplasmic labelling of multipolar cells through the white matter were observed in neonatal period. (inset double labelling for Tbr2 and GFAP showing co-localisation in some white matter cells GFAP green, Tbr2 red). g Map1b staining of the same case as in f labelled only occasional cells in cortical layer V at 1 day in contrast to the hippocampus which showed numerous Map1b-positive cells. h Pax6: nuclear labelling of small cells in the periventricular white matter in neonate (inset ER81, nuclear labelling of pyramidal cells and a subset of satellite cells). i Tbr1 staining at 2 years confirming nuclear labelling in proportion of layer V neurones. j Otx1 layer II in adult epilepsy control showing frequent cytoplasmic labelling of small, cells with immature morphology. Bars f, g, i 30 µm; h, j 45 µm, a–e cortical panoramas representing layers I–VI, taken at original magnification (×10 objective) and rescaled to align layers
Immunofluorescence
Cases were selected for double labelling immunofluorescence using combinations of NeuN, GFAP, CD68 CD34, calretinin with CLM, following the initial observations with immunohistochemistry. 7 μm sections were dewaxed and rehydrated and washed in water. Endogenous peroxidase was quenched with 3% hydrogen peroxide and de-ionised water. Sections were microwaved in antigen retrieval buffer. Protein blocking was carried out with normal horse serum followed by incubation of primary antibodies overnight at 4°C. Sections were washed and incubated with secondary anti-rabbit ImmPRESS (Vector Laboratories) for polyclonal antibodies followed by Cy3, or fluorescein tyramide signal amplification (PerkinElmer Life and Analytical Sciences, Boston, MA). Sections then were washed and quenched in 1% hydrogen peroxide in PBS for 20 min in order to prevent any deposited tyramide combining with the second tyramide signal that followed. The sections were incubated with anti-mouse ImmPRESS (Vector Laboratories) for monoclonal antibodies followed by CY3 or fluorescein tyramide with dilutions for monoclonal GFAP (Dako, 1:30), polyclonal GFAP (Dako; 1:300), and polyclonal Calretinin (1:2,000; Swant, Bellinzona, Switzerland). Alexa Fluor chicken anti-rabbit 594, (1:100) and goat anti-mouse 488, (1:70) (Molecular Probe; Eugene, OR, USA) was used. After each step, sections were washed in PBS. Sections were mounted on Vectashield with DAPI (Vector Laboratories) and visualised with a Zeiss LSM 510 Meta or Leica confocal laser microscope.
Results
Controls
CLM expression for control cases is summarized in Table 3 (Fig. 1). The most distinct lamina-specific expression patterns were noted for Map1b (layers V–VI and II), N200 (layers V–VI) and Tbr1 (layers IV–VI) (Fig. 1b–d). For three CLM (N200, Map1b, Tbr1) progressively greater intensity and frequency of cortical neuronal staining was noted in older children and adults compared to infant controls (Fig. 1b, g). In a further three CLM (Otx1, Tbr2, Cux1) the expression reduced with cortical maturation (Fig. 1e, f) and in three (Mash1, ER81, Pax6) no obvious difference was noted between age groups (Fig. 1h and inset). Immature cell types, some with bipolar or multipolar processes were noted with Tbr2, Pax6, Otx1 particularly in paediatric cases (Fig. 1e, f). In epilepsy controls without dysplasia, enhanced labelling with Otx1 and Tbr2 was noted compared to non-epilepsy controls (Table 3; Fig. 1j).
Cortical dysplasias
The patterns of CLM expression specific to each dysplasia type are summarized below (and in Table 4). All markers were studied for each pathology but only detailed where they deviated from control findings.
FCD I
A striking finding in six cases (between 2 and 6 years of age) was intense nuclear labelling of small cells at the interface of layer I and II with Tbr1 (Fig. 2a). Similar cells also labelled with Otx1 (Fig. 2b). Occasional cells with multipolar processes were noted with Tbr2, scattered in both the upper cortical layers and white matter. These cells were not clearly identified on conventional stained sections and cells of a similar immunophenotype were not seen in older FCDI cases (6–28 years). Enhanced labelling of small cells at the interface of layer I and II was also noted with Pax6 (Fig. 2c); confocal imaging confirmed co-localisation of a proportion with GFAP (Fig. 2c) but not NeuN. In eight cases, labelling of pyramidal neurons for Map1b in layers II–III (Fig. 2d) was greater than in deeper cortical layers, giving the impression of expansion of the superficial cortical layers (Fig. 2e) compared to controls. The Map1b-positive pyramidal neurons remained normally orientated. In regions with enhanced columnar architecture, this organisational pattern was enhanced on Map1b staining with sheaths of perpendicular dendrites separating columns of neurons in the mid-cortical layers.

Focal cortical dysplasia (type Ia). a Tbr1 nuclear labelling of small, immature appearing cells localised to the interface of layer I and II (arrowhead); similar cells are shown at higher magnification from another FCDI case in the inset. b Otx1 labelling of cells at the interface of layer I and II with extension of cytoplasmic processes into layer I (shown at higher magnification in inset). c Double labelling with Pax6 and GFAP in layer I confirmed co-localisation in a proportion of cells. d Map1b the small immature cells in layer II are negative (arrowed) compared to the positive mature pyramidal cells. e Map1b in another case confirming expansion of labelling in upper compared to lower cortical layers. Bars in a, b 30 µm, c 20 µm, d 10 µm, e 500 µm
FCD II
Dysmorphic neurones (DN) and balloon cells (BC) showed distinct CLM expression patterns regardless of laminar location. DN were present through all cortical layers except layer I and showed variable immunopositivity with Tbr1 (Fig. 3a) but more frequently with Map1b and N200 (Fig. 3b). Cytoplasmic, perinuclear or occasional intranuclear labelling patterns for Otx1 in DN was observed (Fig. 3c). Weak to strong cytoplasmic labelling of DN for MASH1 was observed (Fig. 3d), although many were not labelled.

Focal cortical dysplasia type IIB. a Tbr1 moderate labelling of the cytoplasm of dysplastic neurones was observed (arrowhead) compared to negative labelling of other dysplastic cells in proximity (arrow) and strong intensity of interspersed normal neuronal nuclei. b N200 positive labelling of dysmorphic neurones. c Otx1 dysmorphic neurones showing diffuse cytoplasmic positivity and occasional intranuclear inclusions (top-right inset) and perinuclear labelling (bottom-left inset). d Mash1 weak cytoplasmic labelling of dysmorphic neurones is observed in this case. e Pax6 co-localisation with GFAP in balloon cells including a multinucleated balloon cell (arrowed). f Otx1 balloon cell showing strong cytoplasmic positivity and multipolar processes. g Otx1 co-localisation with CD34 in a balloon cell. h ER81 strong nuclear staining of balloon cells is observed. Bars a–d, h 20 µm; e, f 10 µm; g 30 µm
BC, including those located in the white matter and layer I, showed variability of staining with Tbr2 both within and between cases. Strong nuclear labelling of BC was noted for Pax6 in four cases, with co-localisation with GFAP but not NeuN (Fig. 3e). Labelling of the cytoplasm of BC for Otx1 was striking in the majority of cases, highlighting multipolar cellular processes (Fig. 4f). There was co-expression of Otx1 with GFAP and CD34 (Fig. 3g) in a proportion of BC. BC in the white matter or layer I also showed intense nuclear positivity for ER81 (Fig. 3h) in contrast to DN which were more weakly labelled. The majority of BC were Cux1 positive compared to a near absence of labelling of DN with Cux1. In addition, intracortical cytoplasmic labelling of multipolar cells and small immature intracortical cells was noted with Tbr2 and Otx1, as in epilepsy controls.

Temporal lobe sclerosis (TLS) and hippocampal sclerosis with dysplasia and layer marker patterns within dysembryoplastic neuroepithelial tumour (DNT) and the dysplasia in the adjacent cortex. a NeuN TLS case illustrating neuronal loss from layers II and III and an impression of hypercellularity and disorganisation in layer II. b Map1b intense labelling of neurones in upper cortical layers (layer II) is appreciated compared to deeper cortical layers in TLS case. c Tbr1 intense nuclear labelling of remaining, mature appearing neurones in outer layer II was appreciated in some cases; a more horizontal alignment of these neurones was noted in some regions in TLS. d Map1b highlighted the abnormal orientation and clustering or aggregation of neurones and entanglement of processes in layer II in TLS. e Double labelling of Map1b and calretinin in layer II confirmed that the orientation of the calretinin radial processes was normal with only selective misalignment or horizontal orientation of the Map1b positive layer II cells in TLS. f Pax6 frequent nuclear labelling of small immature cells in the layer I/II was observed in TLS. g DNT H&E staining of the perilesional cortex demonstrating hypercellularity in layer I. Inset a cluster of immature cells with vesicular nuclei between more mature neurones in the adjacent cortex which form a small satellite nodule or hamartia (these were CD34 positive). h ER81 hamartia or satellite clusters of oligodendroglial like cells (OLC) were strongly positive in DNT. i Cux1 the layer I hypercellularity adjacent to the DNT was highlighted with Cux1 with the majority of cells showing nuclear positivity. j Map1b DNT tumour nodule in cortex highlighting the pyramidal neurones suspended within the nodule that retain Map1b positivity, normal morphology, orientation and laminar specific distribution. k Map1b higher magnification of case shown in j. Bars a, b 500 µm; b, j 20 µm; d, e 50 µm; f 10 µm; g, h, k 40 µm
Dysplasia adjacent to hippocampal sclerosis [temporal lobe sclerosis (TLS)]
These cases all showed neuronal loss in the supragranular cortex and re-organisational dysplasia of neurons in layer II as previously described [51]. With CLM residual clusters of neurons in layer II in many regions showed intense labelling with Tbr1 (Fig. 4a, c) and Map1b (Fig. 4b, d) highlighting their abnormal, horizontal orientation. Double labelling for calretinin, which labels a subset of fusiform inhibitory interneurones that predominate in upper cortical layers, and Map1b showed that there appeared to be selective mal-alignment of the Map1b-positive cells (Fig. 4e). In addition, labelling of scattered, small immature cells was observed in the upper cortical layers with Tbr2 (layers II, III), Pax6 (layers I, II) (Fig. 4f), Cux1 and ER81 (layer I) and Otx1 (layers I–VI). Co-localisation between Cux1 and GFAP was observed in some cells.
Dysplasia adjacent to DNT
In the perilesional cortex of DNT with apparent dyslamination in conventional stained sections (Table 1), CLM patterns were as in controls, including labelling of white matter neurons. Therefore, abnormal lamination was not confirmed. ER81 and Cux1 highlighted regions with increased cellularity in layer I adjacent to the tumour (Fig. 4g, i), in addition to the small intracortical hamartia-like clusters of immature cells (Fig. 4g, h). No significant expression of Tbr1, Pax6, Map1b or MASH1 by these cells was noted. Small numbers of immature and multipolar cells were noted with Tbr2 (layers I, II), N200 and Otx1. Double labelling with Otx1 and GFAP showed no co-localisation. Within the tumour itself, oligodendrocyte-like cells were ER81, and to lesser extent Cux1, positive but negative for other markers; the astroglia component was negative. The floating pyramidal neurons within the glioneuronal element of the tumour or within cellular nodules showed layer-appropriate expression of Tbr1, Map1b (Fig. 4j, k) or N200 in continuity with the adjacent cortex; this supported their representing entrapped neurons.
Discussion
It is acknowledged that the diagnostic criteria FCD I are less well defined than FCD II [7, 41]. FCD I lacks the distinctive and abnormal cell types that characterise FCD II and usually they are not detected with in vivo MRI. Their histological diagnosis is currently based on standard stains, such as cresyl violet, for assessment of dyslamination and likely includes heterogeneous pathologies. With lack of specific markers there may be subjective interpretive bias which probably explains the variation in diagnosing these lesions between epilepsy centres and pathologists [12, 31, 38]. Consistently applied pathological criteria for FCD I are desirable for reliable clinico-pathological and radiological correlations to emerge, which can guide appropriate surgical management and outcome prediction in these patients.
CLM offer a potential for objective assessment of developmental or acquired dyslamination and heterotopic positioning of neurones. CLM have been more extensively studied in experimental models than in humans. CLM are developmentally, temporally and regionally regulated and specific to different neuronal migratory pathways [26, 32, 36]. CLM expression, including cytoskeletal proteins and transcription factors, are therefore likely to reflect both the intrinsic properties of the cell as well as extrinsic factors specific to locality [25]. Thus, any abnormal cellular expression patterns in dysplasias, as well as diagnostic value, might in theory provide additional information regarding the timing of the insult and cell lineages involved.
In our selected CLM panel, layer specificity was shown for Otx1, N200, Tbr1 and Map1b in keeping with previous data [26, 46]. CLM have been most extensively studied for layer V neurons [26, 35 for review] where diverse neuronal subtypes are identified. ER81 selectively identifies layer V pyramidal neurons in rodent [37, 60] and primate cortex [57] with low levels of expression reported in layers II/III and VI [57, 60]. Although little studied in human brain [26], in our cases ER81 showed no layer specificity, being expressed in a proportion of pyramidal and non-pryamidal neurons (and perineuronal satellite cells) through all layers. Cux1, a putative marker of upper cortical layer neurones in experimental models [39], showed pan-cortical staining, with preferential labelling of layers II and IV and non-pyramidal cell types in keeping with recent data demonstrating expression in subsets of cortical interneurones through all cortical layers [15]. One explanation for these differences may be technical reasons, for example, different methodologies (including antibody source) and different fixation and processing methods between experimental and human tissues. However, it is also possible that CLM expression patterns in experimental models are not always replicated in human tissues.
Within dysplasia groups, relatively consistent abnormalities were identified with our CLM panel. The FCD I series, with pathology as previously described [28], demonstrated distinct populations of immature cells at the interface of layers I and II with Otx1, Tbr1, and Pax6, primarily in the younger patients. The localization and morphology of these cells was reminiscent of those we recently described in this pathology with doublecortin immunohistochemistry [49]. Similar cells were not identified in controls or other dysplasia types. In FCD I, expansion of the distribution of Map1b-positive mature-appearing neurons in outer cortex was noted, compared to control patterns [14]. The persistent mini-columnar architecture seen on conventional stains was highlighted [28]. These features all suggest a delay in maturation and organisation of upper cortical layers in this FCD I subtype.
FCD II is a well characterised malformation encountered in epilepsy surgical pathology with hallmark features of DN and BC. The CLM patterns we observed were distinct from FCD I supporting the view that this is a different biological entity. Although shared pathways for aberrant neuronal–glial fate specification for the abnormal cell types in FCD II have been suggested [21], distinct immunoprofiles for DN and BC cells were shown with our broader panel of antibodies. This could support origin from different migratory cell subsets during development. DN in all layers, bar layer I, commonly expressed N200, Map1b and Tbr1, albeit Tbr1 expression was weaker compared to normal pyramidal cells. Map1b expression has been previously recognised in dysmorphic neurons in FCD [14]. Otx1 and Mash1 expression were less often observed in dysmorphic neurones, as also noted in a previous study of FCDIIB [32]. No labelling of DN with Tbr2 and Cux1 was seen. The immunophenotype of DN therefore most closely resembles that of mature cortical pyramidal projection neurons [56], derived from intermediate progenitor cells (IPCs), rather than from radial glial cell progenitors as previously suggested [32]. IPCs are present throughout corticogenesis, with developmentally regulated expression of Pax6, Tbr2 and Tbr1 and produce neurons for all cortical layers [30, 43]. It is proposed IPCs play a role in determination of laminar patterns, cortical thickness and regional cyto-architectonics. Local disturbance of IPC populations during the stages of cortical expansion could conceivably be implicated in the causation of FCD IIB.
BC in FCDII showed CLM expression patterns distinct from DN. Cytoplasmic positivity with Otx1 and Tbr2 and nuclear labelling for Pax6, ER81 and Cux1 was shown. The multipolar morphology of these abnormal cell types has previously been demonstrated with immunohistochemistry for GFAP-delta [34] CD34 [22], calbindin [52] and doublecortin [49]. Increased Otx1 mRNA [6] and Pax6 expression [32] but variable Map1b expression [14, 21] has previously been reported in BC, in keeping with our observations. The striking Pax6/GFAP co-expression we observed in BC supports their origin from radial glial cells as previously proposed [10, 32]. A proliferative capacity of BC has also been demonstrated [55], although many appear to be arrested in G1 phase of the cell cycle [54]. Pax6 regulates exit from the cell cycle, controlling cortical cell number [45]. It is conceivable therefore that the observed sustained Pax6 expression in balloon cells also plays a role in determining this cell cycle arrest.
Varied abnormalities in the temporal lobe and pole adjacent to HS in patients with mesial temporal lobe epilepsy have been described, including mild dysplasias (previously called microdysgenesis) to FCD I [23, 53]. It is debated whether these represent independent malformative pathologies or re-organisational or dysmaturational changes linked with temporal lobe damage and HS [7]. One distinct pattern, observed in the context of temporal lobe sclerosis and HS, includes cytoarchitectural changes involving layer II. Neuronal clustering and mal-orientation may be extensive in layer II, although overlooked on conventional stains [51]. We have shown that Map1b and Tbr-1 highlights selective mal-orientation of neurones at the interface of layer I and II. Calretinin-immunopositive interneurones in this lamina, derived from tangential migration [59], remained normally orientated. This specific type of dysplasia, involving the last cortical layer to be formed, may therefore be due to a relatively late event or insult in the process of radial cortical migration, maturation and organisation.
Dysplasias in the vicinity of DNTs are reported as an added component in one-third [29] to 86% of cases [40]. The descriptions are principally cortical dyslamination [16, 18, 19, 29] corresponding to FCD I [41]. FCDII in association with DNT has been less often reported [47, 50]. The perilesional cortex of DNT may be relevant to epileptogenesis [3, 4, 33] and the completeness of surgical resection could potentially influence seizure outcome following surgery [40, 47, 50]. Correct classification of this dysplasia type and discrimination from tumour infiltration zone is therefore desirable for accurate clinico-pathological correlations.
Common features observed in conventional stains in our DNT cases in the perilesional region included layer I hypercellularity, impressions of dyslamination and satellite clusters of immature cells (intracortical hamartias). CLM studies, however, showed a normal laminar pattern in the disrupted adjacent cortex. Furthermore, the pyramidal cells within the glioneuronal elements and nodules of the tumour displayed layer-appropriate expression, maturity and orientation with Map1b, N200 and Tbr1 suggesting that they are normal cortical neurones entrapped by the tumour. The layer I hypercellularity and hamartias were highlighted by ER81 and to a lesser extent Cux1, with cell phenotype similar to tumour oligodendrocyte-like cells. These peritumoural changes could represent either tumour extension or precursor lesions from which the tumour arises, as originally proposed [17], for example remnants of the secondary germinal layers or marginal zone [13, 61]. Cortical changes adjacent to DNT are therefore distinct from other FCD types. For perceived dyslamination on H&E, CLM may aid in the distinction of true dysplasia from peritumoural changes which will require verification through larger series. The mature neuronal component within the glio-neuronal element are probably residual entrapped cortical neurones.
In control cases we confirmed variation in expression patterns between neonatal and adult cortex for some CLM, particularly Otx1 which showed strong labelling in cases less than 1 year old. Otx1 does not influence early determination of neuronal cell type and laminar fate but has later roles in maturation and organisation of superficial neurons as well as deeper cortical layers including of GABAergic neurones [1, 42, 44, 48, 58]. We noted increased expression of Otx 1 in small immature cells in epilepsy cases, including those without dysplasia. CLM also highlighted small, immature cell populations, for example with Pax6 and Tbr2 in epilepsy cases, a proportion co-expressing GFAP. These immature cell types may represent activation of residual progenitor cell populations or a delay in normal rates of cell maturation. Recent studies have confirmed a physiological contribution of immature neurones to seizures in FCD lesions [11]. Residual populations of multipotential cells in the maturing cortex may switch to either glio-genic or neuronal pathways (including GABAergic neurones) [25]. The regulation of this process and diversification of cell types may depend on developmental stage, local factors and the maturing environment [9, 25]. This process could conceivably be deregulated by seizures. Therefore, the further delineation of immature cell populations may be of relevance to our understanding of the pathophysiology of dysplasias.
In conclusion, this study highlights the potential application of CLM to the confirmation and categorization of distinct dysplasia types and differentiation from normal cortex. The spectrum of immature cell types highlighted and their contribution to the pathophysiology of these lesions deserves further study.
References
Acampora D, Annino A, Tuorto F et al (2005) Otx genes in the evolution of the vertebrate brain. Brain Res Bull 66:410–420
Arnold SJ, Huang GJ, Cheung AF et al (2008) The T-box transcription factor Eomes/Tbr2 regulates neurogenesis in the cortical subventricular zone. Genes Dev 22:2479–2484
Aronica E, Gorter JA, Jansen GH, Leenstra S, Yankaya B, Troost D (2001) Expression of connexin 43 and connexin 32 gap-junction proteins in epilepsy-associated brain tumors and in the perilesional epileptic cortex. Acta Neuropathol (Berl) 101:449–459
Aronica E, Yankaya B, Jansen GH et al (2001) Ionotropic and metabotropic glutamate receptor protein expression in glioneuronal tumours from patients with intractable epilepsy. Neuropathol Appl Neurobiol 27:223–237
Bayatti N, Moss JA, Sun A et al (2008) A molecular neuroanatomical study of the developing human neocortex from 8 to 17 postconceptional weeks revealing the early differentiation of the subplate and subventricular zone. Cereb Cortex 18:1536–1548
Baybis M, Yu J, Lee A et al (2004) mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 56:478–487
Blumcke I, Vinters HV, Armstrong D, Aronica E, Thom M, Spreafico R (2009) Malformations of cortical development and epilepsies. Epileptic Disord 11:181–193
Britz O, Mattar P, Nguyen L et al (2006) A role for proneural genes in the maturation of cortical progenitor cells. Cereb Cortex 16(Suppl 1):i138–i151
Burns KA, Murphy B, Danzer SC, Kuan CY (2009) Developmental and post-injury cortical gliogenesis: a genetic fate-mapping study with Nestin-CreER mice. Glia 57:1115–1129
Cepeda C, Andre VM, Levine MS et al (2006) Epileptogenesis in pediatric cortical dysplasia: the dysmature cerebral developmental hypothesis. Epilepsy Behav 9:219–235
Cepeda C, Andre VM, Wu N et al (2007) Immature neurons and GABA networks may contribute to epileptogenesis in pediatric cortical dysplasia. Epilepsia 48(Suppl 5):79–85
Chamberlain WA, Cohen ML, Gyure KA et al (2009) Interobserver and intraobserver reproducibility in focal cortical dysplasia (malformations of cortical development). Epilepsia 50:2593–2598
Costa MR, Kessaris N, Richardson WD, Gotz M, Hedin-Pereira C (2007) The marginal zone/layer I as a novel niche for neurogenesis and gliogenesis in developing cerebral cortex. J Neurosci 27:11376–11388
Crino PB, Trojanowski JQ, Eberwine J (1997) Internexin, MAP1B, and nestin in cortical dysplasia as markers of developmental maturity. Acta Neuropathol 93:619–627
Cubelos B, Sebastian-Serrano A, Kim S, Redondo JM, Walsh C, Nieto M (2008) Cux-1 and Cux-2 control the development of Reelin expressing cortical interneurons. Dev Neurobiol 68:917–925
Daumas-Duport C (1993) Dysembryoplastic neuroepithelial tumours. Brain Pathol 3:283–295
Daumas-Duport C, Scheithauer BW, Chodkiewicz JP, Laws ER Jr, Vedrenne C (1988) Dysembryoplastic neuroepithelial tumor: a surgically curable tumor of young patients with intractable partial seizures. Report of thirty-nine cases. Neurosurgery 23:545–556
Daumas-Duport C, Varlet P, Bacha S, Beuvon F, Cervera-Pierot P, Chodkiewicz JP (1999) Dysembryoplastic neuroepithelial tumors: nonspecific histological forms—a study of 40 cases. J Neurooncol 41:267–280
Deb P, Sharma MC, Tripathi M, Sarat Chandra P, Gupta A, Sarkar C (2006) Expression of CD34 as a novel marker for glioneuronal lesions associated with chronic intractable epilepsy. Neuropathol Appl Neurobiol 32:461–468
Englund C, Fink A, Lau C et al (2005) Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci 25:247–251
Englund C, Folkerth RD, Born D, Lacy JM, Hevner RF (2005) Aberrant neuronal–glial differentiation in Taylor-type focal cortical dysplasia (type IIA/B). Acta Neuropathol 109:519–533
Fauser S, Becker A, Schulze-Bonhage A et al (2004) CD34-immunoreactive balloon cells in cortical malformations. Acta Neuropathol 108:272–278
Garbelli R, Meroni A, Magnaghi G et al (2006) Architectural (type IA) focal cortical dysplasia and parvalbumin immunostaining in temporal lobe epilepsy. Epilepsia 47:1074–1078
Garbelli R, Rossini L, Moroni RF et al (2009) Layer-specific genes reveal a rudimentary laminar pattern in human nodular heterotopia. Neurology 73:746–753
Guillemot F (2007) Cell fate specification in the mammalian telencephalon. Prog Neurobiol 83:37–52
Hevner RF (2007) Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J Neuropathol Exp Neurol 66:101–109
Hevner RF, Daza RA, Rubenstein JL, Stunnenberg H, Olavarria JF, Englund C (2003) Beyond laminar fate: toward a molecular classification of cortical projection/pyramidal neurons. Dev Neurosci 25:139–151
Hildebrandt M, Pieper T, Winkler P, Kolodziejczyk D, Holthausen H, Blumcke I (2005) Neuropathological spectrum of cortical dysplasia in children with severe focal epilepsies. Acta Neuropathol 110:1–11
Honavar M, Janota I, Polkey CE (1999) Histological heterogeneity of dysembryoplastic neuroepithelial tumour: identification and differential diagnosis in a series of 74 cases. Histopathology 34:342–356
Kowalczyk T, Pontious A, Englund C et al (2009) Intermediate neuronal progenitors (basal progenitors) produce pyramidal-projection neurons for all layers of cerebral cortex. Cereb Cortex 10:2439–2450
Krsek P, Maton B, Korman B et al (2008) Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol 63:758–769
Lamparello P, Baybis M, Pollard J et al (2007) Developmental lineage of cell types in cortical dysplasia with balloon cells. Brain 130:2267–2276
Lee MC, Kang JY, Seol MB et al (2006) Clinical features and epileptogenesis of dysembryoplastic neuroepithelial tumor. Childs Nerv Syst 22:1611–1618
Martinian L, Boer K, Middeldorp J et al (2009) Expression patterns of glial fibrillary acidic protein (GFAP)-delta in epilepsy-associated lesional pathologies. Neuropathol Appl Neurobiol 35:394–405
Molnar Z, Cheung AF (2006) Towards the classification of subpopulations of layer V pyramidal projection neurons. Neurosci Res 55:105–115
Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD (2007) Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci 8:427–437
Moroni RF, Inverardi F, Regondi MC et al (2009) Expression of layer-specific markers in the adult neocortex of BCNU-treated rat, a model of cortical dysplasia. Neuroscience 159:682–691
Najm IM, Tilelli CQ, Oghlakian R (2007) Pathophysiological mechanisms of focal cortical dysplasia: a critical review of human tissue studies and animal models. Epilepsia 48(Suppl 2):21–32
Nieto M, Monuki ES, Tang H et al (2004) Expression of Cux-1 and Cux-2 in the subventricular zone and upper layers II–IV of the cerebral cortex. J Comp Neurol 479:168–180
Nolan MA, Sakuta R, Chuang N et al (2004) Dysembryoplastic neuroepithelial tumors in childhood: long-term outcome and prognostic features. Neurology 62:2270–2276
Palmini A, Najm I, Avanzini G et al (2004) Terminology and classification of the cortical dysplasias. Neurology 62:S2–S8
Panto MR, Zappala A, Tuorto F, Cicirata F (2004) Role of the Otx1 gene in cell differentiation of mammalian cortex. Eur J Neurosci 19:2893–2902
Pontious A, Kowalczyk T, Englund C, Hevner RF (2008) Role of intermediate progenitor cells in cerebral cortex development. Dev Neurosci 30:24–32
Puelles E, Acampora D, Gogoi R et al (2006) Otx2 controls identity and fate of glutamatergic progenitors of the thalamus by repressing GABAergic differentiation. J Neurosci 26:5955–5964
Quinn JC, Molinek M, Martynoga BS et al (2007) Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol 302:50–65
Riederer BM (2007) Microtubule-associated protein 1B, a growth-associated and phosphorylated scaffold protein. Brain Res Bull 71:541–558
Sakuta R, Otsubo H, Nolan MA et al (2005) Recurrent intractable seizures in children with cortical dysplasia adjacent to dysembryoplastic neuroepithelial tumor. J Child Neurol 20:377–384
Simeone A, Puelles E, Acampora D (2002) The Otx family. Curr Opin Genet Dev 12:409–415
Srikandarajah N, Martinian L, Sisodiya SM et al (2009) Doublecortin expression in focal cortical dysplasia in epilepsy. Epilepsia 50:2619–2628
Takahashi A, Hong SC, Seo DW, Hong SB, Lee M, Suh YL (2005) Frequent association of cortical dysplasia in dysembryoplastic neuroepithelial tumor treated by epilepsy surgery. Surg Neurol 64:419–427
Thom M, Eriksson S, Martinian L et al (2009) Temporal lobe sclerosis associated with hippocampal sclerosis in temporal lobe epilepsy: neuropathological features. J Neuropathol Exp Neurol 68:928–938
Thom M, Harding BN, Lin WR et al (2003) Cajal–Retzius cells, inhibitory interneuronal populations and neuropeptide Y expression in focal cortical dysplasia and microdysgenesis. Acta Neuropathol 105:561–569
Thom M, Holton JL, D’Arrigo C et al (2000) Microdysgenesis with abnormal cortical myelinated fibres in temporal lobe epilepsy: a histopathological study with calbindin D-28-K immunohistochemistry. Neuropathol Appl Neurobiol 26:251–257
Thom M, Martinian L, Sen A et al (2007) An investigation of the expression of G1-phase cell cycle proteins in focal cortical dysplasia type IIB. J Neuropathol Exp Neurol 66:1045–1055
Thom M, Martinian L, Sisodiya SM et al (2005) Mcm2 labelling of balloon cells in focal cortical dysplasia. Neuropathol Appl Neurobiol 31:580–588
Voelker CC, Garin N, Taylor JS, Gahwiler BH, Hornung JP, Molnar Z (2004) Selective neurofilament (SMI-32, FNP-7 and N200) expression in subpopulations of layer V pyramidal neurons in vivo and in vitro. Cereb Cortex 14:1276–1286
Watakabe A, Ichinohe N, Ohsawa S et al (2007) Comparative analysis of layer-specific genes in mammalian neocortex. Cereb Cortex 17:1918–1933
Weimann JM, Zhang YA, Levin ME, Devine WP, Brulet P, McConnell SK (1999) Cortical neurons require Otx1 for the refinement of exuberant axonal projections to subcortical targets. Neuron 24:819–831
Xu Q, Cobos I, De La Cruz E, Rubenstein JL, Anderson SA (2004) Origins of cortical interneuron subtypes. J Neurosci 24:2612–2622
Yoneshima H, Yamasaki S, Voelker CC et al (2006) Er81 is expressed in a subpopulation of layer 5 neurons in rodent and primate neocortices. Neuroscience 137:401–412
Zecevic N, Rakic P (2001) Development of layer I neurons in the primate cerebral cortex. J Neurosci 21:5607–5619
Acknowledgments
We are grateful for the Tbr1 and Tbr2 antibodies from Dr Robert Hevner and we acknowledge Hans Holthausen for his help with the cortical dysplasia cases from Erlangen. The authors declare that they have no conflict of interest. This work has been supported by MRC grant number G79059. This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hadjivassiliou, G., Martinian, L., Squier, W. et al. The application of cortical layer markers in the evaluation of cortical dysplasias in epilepsy. Acta Neuropathol 120, 517–528 (2010). https://doi.org/10.1007/s00401-010-0686-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0686-x