
Abstract
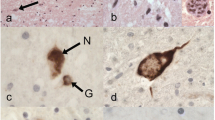
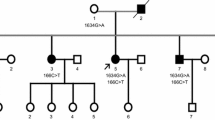
Mutations in the fused in sarcoma gene (FUS) were recently found in patients with familial amyotrophic lateral sclerosis (ALS). The present study aimed to clarify unique features of familial ALS caused by FUS mutation in the Japanese population. We carried out clinical, neuropathological, and genetic studies on a large Japanese pedigree with familial ALS. In six successive generations of this family, 16 individuals of both sexes were affected by progressive muscle atrophy and weakness, indicating an autosomal dominant trait. Neurological examination of six patients revealed an age at onset of 48.2 ± 8.1 years in fourth generation patients, while it was 31 and 20 years in fifth and sixth generation patients, respectively. Motor paralysis progressed rapidly in these patients, culminating in respiratory failure within 1 year. The missense mutation c.1561 C>T (p.R521C) was found in exon 15 of FUS in the four patients examined. Neuropathological study of one autopsied case with the FUS mutation revealed multiple system degeneration in addition to upper and lower motor neuron involvement: the globus pallidus, thalamus, substantia nigra, cerebellum, inferior olivary nucleus, solitary nucleus, intermediolateral horn, Clarke’s column, Onuf’s nucleus, central tegmental tract, medial lemniscus, medial longitudinal fasciculus, superior cerebellar peduncle, posterior column, and spinocerebellar tract were all degenerated. Argyrophilic and basophilic neuronal or glial cytoplasmic inclusions immunoreactive for FUS, GRP78/BiP, p62, and ubiquitin were detected in affected lesions. The FUS R521C mutation in this Japanese family caused familial ALS with pathological features of multiple system degeneration and neuronal basophilic inclusions.




Similar content being viewed by others

References
Baechtold H, Kuroda M, Sok J, Ron D, Lopez BS, Akhmedov AT (1999) Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J Biol Chem 274:34337–34342
Bertrand P, Akhmedov AT, Delacote F, Durrbach A, Lopez BS (1999) Human POMp75 is identified as the pro-oncoprotein TLS/FUS: both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 18:4515–4521
Crozat A, Aman P, Mandahl N, Ron D (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363:640–644
Doi H, Okamura K, Bauer PO et al (2008) RNA-binding protein TLS is a major nuclear aggregate-interacting protein in Huntingtin exon 1 with expanded polyglutamine-expressing cells. J Biol Chem 283:6489–6500
Frank S, Tolnay M (2009) Frontotemporal lobar degeneration: toward the end of conFUSion. Acta Neuropathol 118:629–631
Fujita K, Ito H, Nakano S, Kinoshita Y, Wate R, Kusaka H (2008) Immunohistochemical identification of messenger RNA-related proteins in basophilic inclusions of adult-onset atypical motor neuron disease. Acta Neuropathol 116:439–445
Greenway MJ, Andersen PM, Russ C et al (2006) ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet 38:411–413
Hayashi H, Kato S (1989) Total manifestations of amyotrophic lateral sclerosis. ALS in the totally locked-in state. J Neurol Sci 93:19–35
Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE (1989) Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander’s disease brain. Cell 57:71–78
Kabashi E, Valdmanis PN, Dion P et al (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40:572–574
Kikuchi H, Doh-ura K, Kawashima T, Kira J, Iwaki T (1999) Immunohistochemical analysis of spinal cord lesions in amyotrophic lateral sclerosis using microtubule-associated protein 2 (MAP2) antibodies. Acta Neuropathol 97:13–21
Kusaka H, Matsumoto S, Imai T (1990) An adult-onset case of sporadic motor neuron disease with basophilic inclusions. Acta Neuropathol 80:660–665
Kusaka H, Matsumoto S, Imai T (1993) Adult-onset motor neuron disease with basophilic intraneuronal inclusion bodies. Clin Neuropathol 12:215–218
Kwiatkowski TJ Jr, Bosco DA, Leclerc AL et al (2009) Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323:1205–1208
Lagier-Tourenne C, Cleveland DW (2009) Rethinking ALS: the FUS about TDP-43. Cell 136:1001–1004
Matsumoto S, Kusaka H, Murakami N, Hashizume Y, Okazaki H, Hirano A (1992) Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol 83:579–583
Mizutani T, Sakamaki S, Tsuchiya N et al (1992) Amyotrophic lateral sclerosis with ophthalmoplegia and multisystem degeneration in patients on long-term use of respirators. Acta Neuropathol 84:372–377
Munoz DG, Neumann M, Kusaka H et al (2009) FUS pathology in basophilic inclusion body disease. Acta Neuropathol 118:617–627
Munoz-Garcia D, Ludwin SK (1984) Classic and generalized variants of Pick’s disease: a clinicopathological, ultrastructural, and immunocytochemical comparative study. Ann Neurol 16:467–480
Nelson JS, Prensky AL (1972) Sporadic juvenile amyotrophic lateral sclerosis. A clinicopathological study of a case with neuronal cytoplasmic inclusions containing RNA. Arch Neurol 27:300–306
Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR (2009) Frontotemporal lobar degeneration with FUS pathology. Brain 132:2922–2931
Neumann M, Roeber S, Kretzschmar HA, Rademakers R, Baker M, Mackenzie IR (2009) Abundant FUS-immunoreactive pathology in neuronal intermediate filament inclusion disease. Acta Neuropathol 118:605–616
Nishihira Y, Tan CF, Hoshi Y et al (2009) Sporadic amyotrophic lateral sclerosis of long duration is associated with relatively mild TDP-43 pathology. Acta Neuropathol 117:45–53
Nishihira Y, Tan CF, Onodera O et al (2008) Sporadic amyotrophic lateral sclerosis: two pathological patterns shown by analysis of distribution of TDP-43-immunoreactive neuronal and glial cytoplasmic inclusions. Acta Neuropathol 116:169–182
Nonaka T, Kametani F, Arai T, Akiyama H, Hasegawa M (2009) Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum Mol Genet 18:3353–3364
Oda M, Akagawa N, Tabuchi Y, Tanabe H (1978) A sporadic juvenile case of the amyotrophic lateral sclerosis with neuronal intracytoplasmic inclusions. Acta Neuropathol 44:211–216
Piao YS, Wakabayashi K, Kakita A et al (2003) Neuropathology with clinical correlations of sporadic amyotrophic lateral sclerosis: 102 autopsy cases examined between 1962 and 2000. Brain Pathol 13:10–22
Rabbitts TH, Forster A, Larson R, Nathan P (1993) Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 4:175–180
Rosen DR, Siddique T, Patterson D et al (1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362:59–62
Shaw CE, Enayat ZE, Powell JF et al (1997) Familial amyotrophic lateral sclerosis. Molecular pathology of a patient with a SOD1 mutation. Neurology 49:1612–1616
Sreedharan J, Blair IP, Tripathi VB et al (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319:1668–1672
Tsuchiya K, Matsunaga T, Aoki M et al (2001) Familial amyotrophic lateral sclerosis with posterior column degeneration and basophilic inclusion bodies: a clinical, genetic and pathological study. Clin Neuropathol 20:53–59
Vance C, Rogelj B, Hortobagyi T et al (2009) Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323:1208–1211
Wang X, Arai S, Song X et al (2008) Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 454:126–130
Yang L, Embree LJ, Tsai S, Hickstein DD (1998) Oncoprotein TLS interacts with serine-arginine proteins involved in RNA splicing. J Biol Chem 273:27761–27764
Yoshida M, Murakami N, Hashizume Y, Itoh E, Takahashi A (1992) A clinicopathological study of two respirator-aided long-survival cases of amyotrophic lateral sclerosis. Rinsho Shinkeigaku 32:259–265
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tateishi, T., Hokonohara, T., Yamasaki, R. et al. Multiple system degeneration with basophilic inclusions in Japanese ALS patients with FUS mutation. Acta Neuropathol 119, 355–364 (2010). https://doi.org/10.1007/s00401-009-0621-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0621-1