
Abstract
Rhombencephalosynapsis is an uncommon cerebellar malformation defined by vermian agenesis with fusion of the hemispheres and of the dentate nuclei. Embryologic and genetic mechanisms are still unknown, and to date, no animal models are available. Ultrasound diagnosis is generally suspected after 22 weeks of gestation, and usually the abnormality is suggested by ventriculomegaly. Morphological analysis of 40 fetuses after medical termination of pregnancy allowed us to confirm that rhombencephalosynapsis was always associated with other brain abnormalities or malformations: Purkinje cell heterotopias, fusion of colliculi, forking and/or atresia of the aqueduct and of the third ventricle resulting in a fusion of the thalami, agenesis of the corpus callosum, lobar holoprosencephaly and neural tube defects. Pons and medulla were very infrequently abnormal. Furthermore, complete autopsy made it possible to separate either pure neurologic phenotypes, or associated with extraneural anomalies from syndromic forms: Gomez–Lopez-Hernandez syndrome (1 case) and VACTERL-H syndrome (6 cases). The number of our fetal cases strongly suggests that VACTERL-H association related with rhombencephalosynapsis emerges as a non-random association. Furthermore, recurrence and consanguinity were noted in two different families, which argue for a sporadic or inherited cause. From our results, it could be suggested that rhombencephalosynapsis may be due to defective genes regulating formation of the roof plate and the development of midline cerebellar primordium at the junction of the mesencephalon and of the first rhombomere.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rhombencephalosynapsis (RS) is a rare congenital defect of the cerebellum which was first described by Obersteiner [30] as a sporadic condition about one century ago, from a postmortem examination of a 28-year-old man who committed suicide. The term RS was then introduced by Gross and Hoff [14]. This entity is classically defined as cerebellar hypoplasia, in which cerebellar hemispheres and underlying dentate nuclei are apposed or fused across the midline in the absence of vermis [14, 20]. Its frequency has been estimated at 0.13% by Sener [44] from a series of 3,000 consecutive pediatric brain magnetic resonance imaging (MRI). A number of pediatric and adult cases (less than 80) have been previously reported in the literature and were diagnosed on MRI screening in living patients displaying various degrees of neurological signs. After birth, clinical presentation can vary highly in severity. In post-natal cases as well as in children, typical symptoms consist of swallowing difficulties, delayed motor acquisitions, muscular hypotonia, spastic quadriparesis, cerebellar signs including dysarthria, gait ataxia, abnormal eye movements, nystagmus and head stereotypies [34, 53, 54], seizures and hydrocephalus being inconstant. Ocular abnormalities have recently been extensively described and attributed to a direct consequence of absent cerebellar vermis, an essential component of the neural pathways that controls horizontal vergence [33]. In most cases, cognitive functions are impaired [1, 3, 5, 7, 24, 32, 34, 36, 39–41, 45, 46, 50, 53–56]. Cognitive affective syndrome occurs frequently in children as well as in adults, due to the impairment of limbic cerebellum, i. e., vermis and fastigial nuclei [36]. Most patients die in early life, but survival into adulthood has been reported [2, 20, 29, 30]. In patients alive, clinical prognosis depends on the severity of neurological signs, on mental retardation and long-term cognitive functions which are most often impaired, even though RS is compatible with normal cognitive functions in childhood [34, 53] as well as in adulthood [2]. At the present time, the precise etiological factors responsible for this cerebellar malformation, which is supposed to occur between 28 and 42 post-conceptional days [51, 55] are unknown, though several hypotheses have been proposed including teratogenic [27, 47] or genetic, either chromosomal [24, 54] or genetic ones [58]. Hypothetic genes implicated in early development have also been discussed [28] and up to now, no animal models have been described.
In a few cases only, an extensive neuropathological examination has been described [14, 20, 21, 30, 58]. In adulthood, the main neuropathological features consist of microcephaly, atrophy of optic nerves, agenesis of the corpus callosum and absent septum pellucidum. In case of ventricular dilatation, stenosis of the aqueduct of Sylvius has been observed, and histological examination revealed diffuse subpial gliosis of the tegmentum as well as an immature pattern of the ependymal lining [58]. Although there is an increasing number of prenatal diagnoses, few reports of fetal RS diagnosed on ultrasound and/or MRI screening have been published and, to our knowledge, only eight autopsy cases with neuropathological findings have been reported so far [26, 27, 47–49]. We report herein a series of 40 fetal cases with RS obtained by means of a collaborative study including 5 French centers, which made it possible to analyze the neuropathological abnormalities accurately and to describe the different intra- and extraneural associated anomalies, in an attempt to identify new possible syndromic entities, and to propose from all these data additional etiological hypotheses.
Materials and methods
Fetal population
A total of 40 fetuses (from 39 families) from 14 to 35 weeks of gestation (WG) were selected through a collaborative study involving five French centers between 1992 and 2007. In all cases, pregnancy was terminated for either hydrocephalus or RS discovered by ultrasound (US) or MRI screening, with the informed consent of the parents and in accordance with the French law. Data from familial history, fetal antenatal clinical US and MRI examinations as well as fetal karyotypes were provided in all cases.
Autopsy procedures
All cases underwent a complete autopsy performed by fetopathologists according to standardized protocols, including X-rays, photographs, macroscopical and histological examination of all viscerae. Fetal biometric data were assessed according to the morphometric criteria of Guihard-Costa et al. [16].
Neuropathological examination
The brains were fixed in 10% buffered formalin. Brain growth was evaluated according to the biometric criteria of Guihard-Costa and Larroche [15]. In all cases, multiple tissue samples were embedded in paraffin. Six micrometer sections were stained using hematoxylin–eosin and cresyl violet. All macroscopical and histological brain examinations were reviewed by two neuropathologists. To make the diagnosis of RS, the general definition according to Friede, Utsunomiya and Yachnis [11, 55, 59] was used, RS being characterized by vermian aplasia or hypoplasia with fusion of the cerebellar hemispheres. In case of associated anomalies of the aqueduct of Sylvius, lesions were classified according to Friede [11].
Cerebellar sections were assessed for immunohistochemical studies using antibodies directed against calbindin 28 kD (diluted 1/100; Tebu Novo-Castra, Le Perray en Yvelines, France) and parvalbumin (1:500; SIGMA, St Louis, USA). Immunohistochemical procedures included a microwave pre-treatment protocol to aid antigen retrieval. Incubations were performed for 1 h at room temperature, using the TECHMATE system (DAKOPATTS, Trappes, France). After incubation, histological slides were processed using the LSAB detection kit (DAKOPATTS, Trappes, France). Peroxidase was visualized by means of 3-3′ diaminobenzidin or amino-ethyl carbazole.
Results
Antenatal data
Prenatal data are summarized in Table 1. Seventeen fetuses were male and 23 female (sex ratio, 0.73). There was only one recurrent case in a sibling (cases 31 and 38), and a notion of parental consanguinity in one other case (case 40). Standard karyotypes were normal in all cases.
Based on US and MRI screening, medical termination of pregnancy was performed for ventriculomegaly in 36 fetuses (90% of cases). In 22 cases (55%), ventriculomegaly was the only ultrasound sign. Ventriculomegaly was associated with a small cerebellum (transverse diameter below the fifth centile) in 13 cases (32.5%), and antenatal MRI made it possible to confirm the diagnosis of RS in these cases. In one case, termination of the pregnancy was performed for associated holoprosencephaly (case 29) or agenesis of the corpus callosum (case 8) and for microcephaly with no ventriculomegaly in one case (case 27). Ventriculomegaly was associated with intra-uterine growth restriction in 4 fetuses (cases 1, 4, 27 and 29), with cardiac malformation in 3 (cases 26, 33 and 34). Medical termination of the pregnancy was achieved at the end of the first trimester in one case, during the second trimester in 31 (77.5% of cases), and during the third trimester in 8 (20%).
General autopsy findings
All but four fetuses were eutrophic, and skeletal measurements as well as the weight and the histological maturation of the different viscerae were concordant with the gestational age. Except for case 27, all fetuses presented with macrocranium with large fontanels, and a characteristic, although not specific, cranio-facial dysmorphism, associating prominent forehead, hypertelorism, small pointed nose with a large nasal bridge and anteverted nostrils, midface hypoplasia, smooth philtrum, small mouth with thin lips, microretrognathism (Fig. 1a, b) as well as posteriorly rotated low set ears with a coarse up-sized lobule (Fig. 1c) and deformation of the external auditory canal (Fig. 1d). In six cases (number 32–37), at least two different skeletal or visceral malformations including vertebral, limb or heart defects and esophageal or anal atresia were found, which led to the diagnosis of VACTERL + Hydrocephalus syndrome (VACTERL-H, MIM276950). Vertebral defects were observed in three cases (cases 38–40) associated with unilateral atresia of the external auditory canal, preauricular tag and microtia in one case (case 40) (Fig. 1e). One fetus (case 25) had bilateral temporal alopecia, strongly suggestive of Gomez–Lopez-Hernandez syndrome (GLH, MIM601853), although craniosynostosis was not observed (Fig. 1f). In one fetus, postmortem examination revealed associated genito-urinary malformations (case 30), and in another, a polymalformative syndrome of unknown etiology associating RS, complete agenesis of the corpus callosum, cardiac septal defect and Meckel diverticula (case 26).

Cranio-facial anomalies in RS. a Cranio-facial dysmorphism with macrocranium, hypertelorism, short nose with anteversed nostrils, small mouth and pointed chin (case 7, 27WG). b Characteristic left side face displaying prominent forehead, midface hypoplasia, smooth philtrum, microretrognathism and posteriorly rotated low set ears (case 16, 25WG). c Posteriorly rotated low set ears with a coarse up-sized lobule (case 29, 30WG). d Flat external ear with abnormal lobule and deformation of the external auditory canal (case 13, 28WG). e Hypoplastic, malformed ear with preauricular tag and auditory external canal atresia (case 40, 26WG). f Gomez–Lopez-Hernandez syndrome (arrowhead indicates the temporal alopecia) (case 25, 29WG)
Neuropathological studies
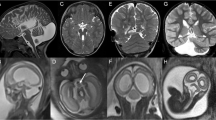
Detailed macroscopic and microscopic findings are summarized in Tables 1 and 2, respectively. On external examination, gyral configuration of the forebrain was normal according to the age in four cases. In one fetus, micropolygyria was noted (case 29). In the other cases, gyral configuration was difficult to assess, due to the severity of hydrocephalus. For the same reason, the ratio infra/supratentorial weight of the brain was not calculable. The weight of the brainstem and cerebellum was available in 33 cases. It was normal in 17 cases (51%). In seven cases, the weight corresponded to a delayed growth of 2 weeks, in six of 4 weeks and in three, of 8 weeks. Transversal diameter of the cerebellum, obtained in 28 cases, was estimated to the 50th centile in 28.5% of cases, to the 25th centile in 14.5% of cases, and below to the 10th centile in 57% of cases. In all cases, the cerebellum appeared to be small and most often pear-shaped (Fig. 2a). In 33 cases (82%), the vermis was absent, with a complete fusion of hypoplastic cerebellar hemispheres, folia running across the midline (Fig. 2b). In seven cases, a partial RS was observed (18%), with a small poorly formed vermis, its rostral part being more severely affected, whereas the caudal vermis was better formed, with a discernible flocculo-nodular lobe (Fig. 2c, d). On cut sections, the fourth ventricle was narrowed, with a characteristic diamond-shaped pattern in some cases and fused or apposed dentate nuclei (Fig. 2e, f). The pons and medulla were macroscopically normal.

Macroscopic anomalies in RS. a In situ visualization of complete RS (case 23, 24WG). b External view of complete RS after fixation displaying the characteristic pear-shaped pattern (case 9, 27WG). c Partial RS with fusion of cerebellar hemispheric folia on the midline in the rostral part of the cerebellum (case 27, 35WG). d Postero-inferior view of the same partial RS, with two well separated cerebellar hemispheres (case 27, 35WG). e Macroscopic section passing through the middle cerebellar peduncles, showing a diamond-shaped fourth ventricle and fused dentate nuclei (case 9, 27WG). f Macroscopic section passing through the middle cerebellar peduncles, showing the absence of cerebellar vermis, narrowing of the fourth ventricle and apposed dentate nuclei (case 25, 29WG)
On supratentorial coronal sections, ventricular dilatation affected both lateral and third ventricles in 32 cases (80%), and the lateral ventricles with no patent third ventricle in seven cases (17.5%). There was no dilatation in one single case. Ventricular dilatation was severe in 80% of cases, moderate in 12.5%, and mild in 7.5% of cases. Analysis of the corpus callosum was available in 34 cases. Callosal abnormalities were noted in 35.5% of cases. Complete agenesis of the corpus callosum was found in eight cases (23.5%), with Probst bundles in one fetus (case 20), and partial agenesis in one case (case 9). Corpus callosum was hypoplastic in four (12%), and normal in all other cases. The septum pellucidum was identified and considered as normal in 5 cases, identified but ruptured in 11 cases (Fig. 3a), and not present in the remaining cases. Associated anomalies consisted of lobar holoprosencephaly in two fetuses (cases1 and 29: 5%) (Fig. 3b–d), and of neural tube defects in 3 (cases 11, 24 and 28: 7.5%).

Macroscopic associated supratentorial anomalies. a Severe dilatation of the lateral ventricles with rupture of the septum pellucidum (case 2, 18WG). b Superior view of associated lobar holoprosencephaly, with the presence of interhemispheric scissure (case 29, 30WG). c Inferior view of the brain (same case): absence of cerebral hemispheres cleavage and of olfactory bulbs and tracts. d Superior view of the infratentorial structures (same case). The mesencephalon is replaced by a single nodular mass (arrowhead) corresponding to the fusion of superior and inferior colliculi
Histological examination of sections passing through the brainstem and cerebellum allowed us to confirm the macroscopic diagnosis of complete or partial RS, in which fusion of the folia was observed in the rostral part of the cerebellum. In all cases, the flocculo-nodular lobe was well formed. Dentate nuclei were fused in 54% of cases (Fig. 4a, b), apposed in 38% (Fig. 4c). In some cases, they were fused at the rostral level only, with the classical appearance of inverted U-shaped structure, and well formed at the more caudal levels. Despite the multiplicity of samples, they appeared to be normal in three cases (8%). They were hypoplastic and dysplastic in a single case (case 1) (Fig. 4d). Other cerebellar anomalies consisted of heterotopic Purkinje cell clusters arrested in the white matter, or lying near the roof of the fourth ventricle (7 cases; 18%) (Fig. 4e, f). Brainstem lesions were very infrequently observed, and consisted of inferior olivary nuclei absence in one case (case 22), and of dysmorphic and heterotopic olivary nuclei in another case (case 20). Hypoplastic brainstem nuclei were noted in two cases (cases 1 and 20; 5%), and pyramids, although hypoplastic in two fetuses presenting with lobar holoprosencephaly (cases 1 and 27; 5%), were always present. In the mesencephalon, two major abnormalities were observed: fusion of the colliculi, and lesions of the aqueduct of Sylvius. Fusion of the colliculi (inferior, superior or both), also named mesencephalosynapsis, was noted in 75% of cases (superior: 11 cases, inferior: 20 cases; superior and inferior: 9 cases, 22.5%) (Fig. 5a). Analysis of the aqueduct of Sylvius was available in 36 cases (90%). The aqueduct was histologically normal in two cases only (Fig. 5b). Isolated stenosis (narrowing with no modification of its shape) was observed in five cases (13%) (Fig. 5b). Aqueduct of Sylvius was considered as dysmorphic—multiple indentations of the ependymal lumen without narrowing or atresia—in three cases (12%) (Fig. 5d), with associated stenosis in two cases, or with dilatation in one case. Complex forking—two or more distinct permeable channels, surrounded by several ectopic ependymal tubes, separated from each other by normal brain parenchyma—was observed in 11 cases (31%) (Fig. 5e). Atresia, consisting of absent permeable channel, replaced by several small tubules lined with ependymal cells, was noted in 14 cases (39%) (Fig. 5f). Atresia extended more rostrally to the third ventricle in 5 cases (17.5%) resulting in a fusion of the thalami, corresponding to diencephalo-mesencephalosynapsis (Fig. 5g). Stenosis of the third ventricle was noted in case 22. In the other cases, the third ventricle was either normal (12 cases; 36.5%) or dilated (15 cases; 45.5%), with histological hallmarks of intraventricular raised pressure characterized by abrasion of the ependymal cell lining replaced by micronodular astrocytic foci. In case of third ventricle dilatation, the thalami appeared to be dysmorphic in three cases (cases 1, 20 and 28), or hypoplastic (case 36). Histological examination allowed us to confirm the other lesions observed during macroscopic examination: agenesis of the corpus callosum, lobar holoprosencephaly and encephalocele or meningocele. Internal capsule was fragmented in two cases (22 and 29) (Fig. 5h). Anterior commissure was considered as normal in six cases, and was not identified in the other cases, likely due to severe hydrocephalus. Even though anomalies of cortical lamination consisting of focal festooned cortical ribbon resembling the pattern of polymicrogyria but with no 4-layered cortex were present in two fetuses (cases 24 and 29), no migrational abnormalities were observed at the supratentorial level.

Microscopic patterns of RS and infratentorial associated lesions. a Whole mounted section of the brainstem and cerebellum showing complete RS with fused dentate nuclei (case 26, 22WG). b Fused but normally gyrated dentate nuclei (case 28, 26WG). c Apposed dentate nuclei (case 18, 26WG). d Hypoplastic and non-gyrated apposed dentate nuclei (case 1, 23WG). e Purkinje cell clusters in the cerebellar white matter (HE stain) (case 25, 29WG). f Immunoreactivity of Purkinje cell clusters for calbindin antisera (same case)

Histological associated lesions in the mesencephalon and supratentorial structures. a Characteristic dome-shaped pattern of fused inferior colliculi, with severe stenosis of the aqueduct of Sylvius (case 2, 18WG). b Normal aqueduct of Sylvius (case 9, 27WG). c Stenosis of the aqueduct of Sylvius (case 26, 22WG). d Dysmorphism of the aqueduct of Sylvius (case 27, 35WG). e Dysplasia of the aqueduct of Sylvius (case 32, 24WG). f Atresia-forking of the aqueduct of Sylvius (case 2, 18WG). g Atresia-forking of the third ventricle with fusion of the thalami (case 7, 23WG). h Fragmentation of the internal capsule (case 29, 30WG)
Discussion
Rhombencephalosynapsis is still considered as a rare congenital defect of the cerebellum, generally associated with a poor outcome, either when diagnosed after birth due to neurologic and cognitive impairment [1, 3, 5, 7, 24, 32, 34, 36, 39–41, 45, 46, 50, 53–56], or when suspected during pregnancy, due to the severity of hydrocephalus [26].
Embryopathology
The vermian maldevelopment in complete RS is characterized by an absence of anterior and posterior vermis, whereas the flocculo-nodular lobe is present. In case of partial RS, the rostral part is mostly affected. The cerebellum develops from the cerebellar primordium, an unpaired structure located at the junction of the metencephalon (first rhombomere) and the mesencephalon. The cerebellar primordium becomes divided into a median part, the vermis, and a pair of hemispheres which grow rapidly from the rhombic lips. In previous studies, it has been postulated that the failure of vermian differentiation in the anterior part of the midline cerebellar primordium occurred between the fourth and seventh week, and is considered by most authors as resulting from a fusion of cerebellar hemispheres over a maldeveloped vermis [51]. Other histogenetic hypotheses have also been proposed; RS resulting from an absence of division of the cerebellar hemispheres, since the vermis develops 9 weeks onwards when developing hemispheres are still fused, implying that absence of vermian development results from a nonseparation of cerebellar hemispheres [55]. Otherwise, RS has never been described with dysplastic cerebellar cortex, implying that the lateral parts of the rhombic lips are not involved in the pathogenetic mechanism of RS. On the contrary, the existence of migrational anomalies of Purkinje cells, which were observed in 20% of our cases, argues for a defective patterning of the roof of the fourth ventricle. Although dysplastic or absent olivary nuclei could be interpreted as a secondary phenomenon, they could be more likely related with a migration defect of olivary neurons from the fourth ventricle floor plate. Most of the time RS is associated with other infratentorial anomalies (fused dentate nuclei, fusion of the colliculi, aqueductal abnormalities), which may also be related to an early defect of dorsal midline patterning of the first rhombomere and mesencephalon. Associated supratentorial lesions also consist in midline defects (CCA, holoprosencephaly), which suggests that embryopathological mechanisms responsible for RS could also alter the downstream signalling cascade from the mesencephalo–metencephalic junction at more rostral levels.
Clinical and MRI diagnosis
The diagnosis of RS is increasingly made by MRI, which appears superior to US examination for the delineation of posterior anatomy in fetuses as well as in children [26, 29, 31, 51, 55], which could explain the very low number of previously reported cases in the literature. This cerebellar malformation had probably been missed for years on radiological and/or pathological exams, and in our series, pathological criteria of RS have sometimes been identified several years after the first examination of fetuses displaying hydrocephalus and vermian anomalies. Regarding the diagnosis of partial or total vermian agenesis or hypoplasia, several pathological conditions are now easily distinguished from RS by both brain MRI and morphology, in particular Dandy–Walker continuum, Joubert syndrome, tectocerebellar dysraphy, cerebellar dysplasia of lissencephaly type II or pontocerebellar hypoplasias, and histological analysis makes it possible to distinguish clearly RS from cerebellar atrophy. To date about 70 cases have been extensively described by imaging, two-thirds of them by MRI, the majority of cases having non-syndromic RS [34]. The major antenatal MRI signs consist, on axial planes, of fused cerebellar hemispheres, absent or hypoplastic vermis, narrow diamond-shaped fourth ventricle and fused dentate nuclei. Sagittal planes display a lack of primary vermian fissure and of normal fastigial point [26]. In a minority of cases, partial RS has been identified by MRI [9, 32]. Global similar findings have been reported in children and adults, and as in fetuses, associated anomalies such as rupture or absence of septum pellucidum, dysgenesis or agenesis of the corpus callosum, fused fornices and thalami have been observed [5, 8, 21, 53–55]. In children, atrophy of the white matter with gyral abnormalities, and fusion of the inferior and superior colliculi have also been reported [8, 26, 32, 33, 44]. Other features have also been described, such as malrotated hippocampi, hypoplastic chiasm, fused cerebral peduncles and thalami [2, 9, 32, 34, 54], parietal and temporo-parietal cortical dysplasia [32, 33], and schizencephalic cleft [33]. Using MRI, associated Dandy–Walker malformation has been observed in four fetuses [36, 45] and Chiari I or II malformations have also been reported in two children [46, 54], but these malformations were not identified using prenatal US or MRI in our cases.
Neuropathological descriptions
Neuropathological descriptions concern mainly pediatric, 11 cases, from birth to the age of 16 [14, 20, 21, 23, 42, 48] and four adult cases [20, 21, 30, 58]. Fetal neuropathology has seldom been reported, and to our knowledge, only eight autopsy cases with neuropathological analysis have been described [26, 27, 47–49]. In all reports, neuropathological descriptions of RS are similar and distinguish complete and partial RS, the latter suggesting that RS should be considered as a malformation with a variable degree of severity. It is worth noting that a partial RS was found in less than 20% of our cases. In the whole literature, the cerebellum is described as more or less hypoplastic, as confirmed by our study where biometric data were normal in 50% of cases, and in the most severe forms fastigium and flocculi are not visualized, which we did not find in our series. In some cases, the dentate nuclei are unfolded [47, 48], as in one of our cases. Previously described cerebellar histological anomalies consist of dysplastic deep cerebellar roof nuclei and lack of cerebellar folia [47], and in one case, of dysplastic large ectopic neurons [48] which could correspond to the nonmigrated calbindin-positive Purkinje cell clusters we observed in the cerebellar white matter, and which are likely to be a frequent associated anomaly, since observed in about 20% of our cases. In our cases as in those described in the literature, brainstem anomalies are very infrequent. In post-natal cases, they include hypoplasia of the pons [47] and absence of dorsal accessory olivary nuclei [21]. In fetal cases, no brainstem anomalies have been observed until now, and are as infrequent as in post-natal cases, since noted in two of our cases only, and consisting in complete absence or heterotopia of inferior olivary nuclei, brainstem nuclei hypoplasia, and hypoplastic cortico-spinal tracts. Coexistence of RS and Dandy–Walker malformation has been documented in a single case [47], and was observed in one of our cases. In the fetus, hydrocephalus is the most frequently supratentorial associated anomaly and has been attributed to the narrowing of the fourth ventricle, or to lesions of the Aqueduct of Sylvius, usually described as stenoses. In two of our cases only, the aqueduct was normal, and hydrocephalus was moderate. In some previous reports, atresia-forking of the aqueduct and/or of the third ventricle has been described [23, 47, 48]. This anomaly appears to be relatively frequent, since it was present in 39% of our cases. Nevertheless, atresia-forking is the most severe lesion of the aqueduct and probably present mostly in fetal cases in association with massive hydrocephalus. Lesions of the aqueduct of Sylvius, a mesencephalic abnormality which is present in nearly all cases, are generally associated with fusion of the inferior colliculi [21]. Fusion of the colliculi was identified in 75% of our cases, affecting superior, inferior colliculi or both. It is worth noting that sporadic or recurrent cases of isolated atresia-forking or stenosis of the aqueduct of Sylvius have been described (MIM236635) could belong to the RS spectrum and represent the less severe form.
Before birth, supratentorial midline abnormalities are identified with difficulty owing to the severity of hydrocephalus, in particular true agenesis of the septum pellucidum which is most often impossible to confirm. Otherwise, as in our observations, thin or dysmorphic corpus callosum as well as complete or partial agenesis have been also documented [26, 48]. Cortical malformations have been well described by MRI, and confirmed in rare autopsy cases. They encompass a variety of gyral anomalies, including diffuse polymicrogyria [48], or restricted to the occipital lobes [26]. Other lesions have been reported, such as hypoplasia of the temporal lobes [26, 48], and gray matter heterotopias with calcifications of the frontal lobes [26]. Absence of olfactory bulbs and tracts has been reported by Shachenmayr and Friede [42], but were never observed in our series. Eye or visual pathways anomalies have also been reported, consisting of microphthalmia [26], optic nerve atrophy or hypoplasia [21, 58], septo-optic dysplasia [27] and absence of optic chiasm [47], which were not identified in any of our cases. Microcephaly, which is exceptional in our series (one single case), has been reported by others in children and adults [26, 47, 58]. In our series, we found two cases where RS was associated with lobar holoprosencephaly. Interestingly, RS was reported for the first time 20 years ago in a fetus presenting with extreme form of holoprosencephaly, called aventriculi [47], then by Shaw [48], Siebert [49] and recently by McAuliffe [26]. In all these cases including ours, several associated intra- and extraneural abnormalities were noted. At last, we identified associated neural tube defects in three cases. Neural tube defects have never been previously reported in fetuses, and only in one adult and two pediatric cases, consisting of RS and cervicothoracic or lumbosacral myelomeningocele [40, 46], but not of encephalocele as in our three fetuses.
Associated anomalies
Extraneural associated anomalies have been rarely reported and concerned mainly pediatric and adult cases. Regarding the existence of cranio-facial dysmorphism, low set ears and hypertelorism have been reported [54], midface hypoplasia being observed by McAuliffe et al. [26]. Based on the largest series ever reported of fetal cases, we were able to describe a common dysmorphic pattern associating macrocranium and large fontanels with characteristic cranio-facial anomalies, consisting of prominent forehead, hypertelorism, small pointed nose with a large nasal bridge and anteverted nostrils, midface hypoplasia, smooth philtrum, small mouth with thin lips, microretrognathism as well as posteriorly rotated low set ears with a coarse up-sized lobule and deformation of the external auditory canal. Without being specific, this pattern, in particular midface and ear anomalies should alert pathologists to search for RS. The vast majority of extraneural anomalies cited in the literature concern skeletal abnormalities, and consist of segmentations and fusions of the spine, as well as of radial ray defects and anomalies of upper extremities. The latter include polydactyly, syndactyly, phalangeal hypoplasia, and duplication of the thumbs [1, 32]. As in six of our cases, associated, perhaps syndromic but with no definite diagnosis, visceral malformations have also been reported [1, 9, 21, 26, 32]. Gomez–Lopez-Hernandez syndrome, the best characterized syndrome whose key feature is RS which was reported in fewer than 20 isolated cases [3, 25, 35, 43, 52, 57], was suspected in one fetus. Although craniosynostosis was absent and trigeminal nerve involvement could not be diagnosed, scalp biopsies revealed the presence of underdeveloped pilosebaceous structures, which does not correspond to a form of aplasia cutis congenita [3]. This syndrome, which could result from dominant neomutations or de novo chromosomal rearrangements [35], was delineated in the late 1970s [13, 25], associating mental retardation, partial parietal and occipital scalp alopecia, craniosynostosis, gait ataxia, facial anesthesia, dysmorphic features and RS on MRI. In fact, this association was initially described in 1959 in a 2-year-old boy showing similar dysmorphic features, bitemporal alopecia and RS on neuropathological examination [14]. In our series, six cases fulfilled at least two specific criteria necessary to conclude to the diagnosis of VACTERL association [38]. VACTERL is a sporadic non-random co-occurrence of the following defects: vertebral anomalies (V), anal atresia (A), cardiovascular defects (C), esophageal atresia and/or tracheo-esophageal fistula (TE), renal (R) and limb/radial (L) anomalies. Since 1984, hydrocephalus has been recognized in a number of individuals with VACTERL [4]. This combination has emerged as a distinct entity named VACTERL-H association. Its genetic heterogeneity is suggested by both recessive autosomal and X-linked mode of inheritance and related to Fanconi anemia genes [6, 19]. However, no association between Fanconi anemia and RS has been reported so far. One major result of our study was to identify the cause of hydrocephalus in VACTERL-H association observed in 15% of our fetal population, which had never been previously reported. The number of our fetal cases strongly suggests that VACTERL-H association due to RS emerges as a non-random association. Furthermore, Michaud et al. [27], who described a case of RS with septo-optic dysplasia, described associated anomalies strongly suggestive of VACTERL-H, as well as in the cases reported by Briard [4], where “stenosis” of the aqueduct of Sylvius without further neuropathological description was noted. In term of genetic counselling, this VACTERL-H/RS association has to be recognized owing to the theoretical risk of recurrence [18, 21, 23, 48]. Oculo-Auriculo-Vertebral (OAV) spectrum (MIM164210) was suspected in two cases because of vertebral defects and unilateral atresia of the external auditory canal but no other specific criteria were found [22]. These two cases could preferably be included in the VACTERL-H-RS association spectrum, as far as VACTERL-H association has been reported with bilateral atresia of the external auditory canal, and OAV spectrum has been statistically related to VACTERL-H association, indicating similarities in pathogenesis or etiology [12, 22]. At last, one patient with GLH syndrome and esophageal atresia has been reported [35], which confirms the existence of a wide phenotypical spectrum, ranging from isolated RS to GLH and VACTERL-H association with several overlaps between all these syndromes.
Etiological factors
Rhombencephalosynapsis is still considered by most authors as a sporadic condition, and etiological factors remain poorly understood. Environmental teratogenic effects have been suggested, in particular insulin-dependent diabetes mellitus [47] and phenylcyclidine intake at the beginning of the pregnancy [27]. Cytogenetic abnormalities appear to be exceptional since only two different chromosomal anomalies involving interstitial deletion of chromosome (2q) [54] and an unbalanced subtelomeric translocation t(2p;10q) [24] have been reported. Rare genetic causes have been identified in syndromic forms. A mutation in the PTEN gene was found in one child [37] as well as a mutation in FAC and FANC B genes in two cases of VACTERL + H [6, 19]. In two fetuses, RS was associated with lobar holoprosencephaly (HPE), which has already been described in a single fetal case with absent left radius and thumb, as well as tetralogy of Fallot, representing a unique association of HPE, RS and VACTERL with unknown genetic background [49]. HPE is now known as a multihit process including mutations or quantitative anomalies throughout mainly SHH, ZIC2, SIX3 and TGIF genes [10]. Our two cases were screened for these genes and were found to be negative. In one fetus, GLH was suspected. Interestingly, microarray-based comparative genomic hybridization (array-CGH) had previously been carried out in one patient showing typical GLH syndrome, but DNA variations were not considered as relevant genomic aberrations [43].
In our study, recurrence was noted in one family and consanguinity in another, which has been already reported, suggesting a possible genetic inherited cause in some cases [5, 39, 40, 53]. Molecular mechanisms are still hardly known even though several genes involved in midbrain–hindbrain patterning have been discovered. All studies using animal models invalidated for several genes contributing to the isthmic organizer region signalling pathway, failed to show their implication in RS: Lmx1a, Lmx1b, Gbx2, Otx2, Fgf8, Wnt1, 2 and 3, and Pax2 [17, 28].
Even though a chromosomal abnormality was excluded in all our cases using conventional cytogenetic methods, a cryptic chromosomal rearrangement could be suspected (microdeletions or microduplications). In addition, since it was impossible to perform linkage analyses due to the too small sampling in familial cases, we carried out a preliminary study on 15 fetal cases using a pangenomic approach by means of array-CGH, which enabled us to identify 2 different de novo microrearrangements: one microduplication on chromosome 1p and one microdeletion on chromosome 7q. Further DNA tests are being carried out in our whole fetal population in order to confirm the pathogenic effect of these genetic alterations and to search abnormal chromosomal recurrent loci.
In conclusion, RS occurs most of the time as a complex condition associated with a variety of intra- and/or extraneural malformations, as proved in our study including 40 fetal cases. Isolated RS without mesencephalosynapsis or aqueductal anomalies were never observed. RS was always associated with intraneural lesions restricted to the infratentorial structures in 33, 3% of cases. In the other cases, various supratentorial abnormalities were observed, the most frequent being agenesis of the corpus callosum and atresia of the third ventricle. Moreover, syndromic forms were observed in this group and representing one-third of these cases. With the systematic use of MRI which has considerably improved its antenatal diagnosis, the frequency of RS appears now to be much higher than previously thought. From our results, it could be suggested that RS is possibly due to a defective gene regulating formation of the roof plate and the development of midline cerebellar primordium at the junction of the mesencephalon and of the first rhombomere.
References
Aydingoz U, Cila A, Aktan G (1997) Rhombencephalosynapsis associated with hand anomalies. Br J Radiol 70:764–766
Bell BD, Stanko HA, Levine RL (2005) Normal IQ in a 55-year-old with newly diagnosed rhombencephalosynapsis. Arch Clin Neuropsychol 20:613–621
Bowdin S, Phelan E, Watson R, McCreery KM, Reardon W (2007) Rhombencephalosynapsis presenting antenatally with ventriculomegaly/hydrocephalus in a likely case of Gomez–Lopez-Hernandez syndrome. Clin Dysmorphol 16:21–25
Briard ML, le Merrer M, Plauchu H, Dodinval P, Lambotte C, Moraine C, Serville F (1984) Association of VACTERL and hydrocephalus: a new familial entity. Ann Genet 27:220–223
Chemli J, Abroug M, Tlili K, Harbi A (2007) Rhombencephalosynapsis diagnosed in childhood: clinical and MRI findings. Eur J Paediatr Neurol 11:35–38
Cox PM, Gibson RA, Morgan N, Brueton LA (1997) VACTERL with hydrocephalus in twins due to Fanconi anemia (FA): mutation in the FAC gene. Am J Med Genet 68:86–90
Danon O, Elmaleh M, Boukobza B, Fohlen M, Hadjnacer K, Hassan M (2000) Rhombencephalosynapsis diagnosed in childhood: clinical and MRI findings. Magn Reson Imaging 18:99–101
Demaerel P, Kendall BE, Wilms G, Halpin SF, Casaer P, Baert AL (1995) Uncommon posterior cranial fossa anomalies: MRI with clinical correlation. Neuroradiology 37:72–76
Demaerel P, Morel C, Lagae L, Wilms G (2004) Partial rhombencephalosynapsis. AJNR Am J Neuroradiol 25:29–31
Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V (2007) Holoprosencephaly. Orphanet J Rare Dis 2:8
Friede RL (1989) Hydrocephalus. Developmental neuropathology, 2nd revised and expanded edn. Springer Verlag. Heidelberg, Berlin, pp 231–246
Froster UG, Wallner SJ, Reusche E, Schwinger E, Rehder H (1996) VACTERL with hydrocephalus and branchial arch defects: prenatal, clinical, and autopsy findings in two brothers. Am J Med Genet 62:169–172
Gomez MR (1979) Cerebellotrigeminal and focal dermal dysplasia: a newly recognized neurocutaneous syndrome. Brain Dev 1:253–256
Gross H (1959) Die Rhombencephalosynapsis, eine systemisierte Kleinhirnfehlbildung. Arch Psychiatr Z 199:537–552
Guihard-Costa AM, Larroche JC (1990) Differential growth between the fetal brain and its infratentorial part. Early Hum Dev 23:27–40
Guihard-Costa AM, Menez F, Delezoide AL (2002) Organ weights in human fetuses after formalin fixation: standards by gestational age and body weight. Pediatr Dev Pathol 5:559–578
Guo C, Qiu HY, Huang Y, Chen H, Yang RQ, Chen SD, Johnson RL, Chen ZF, Ding YQ (2007) Lmx1b is essential for Fgf8 and Wnt1 expression in the isthmic organizer during tectum and cerebellum development in mice. Development 134:317–325
Herman TE, Siegel MJ (2002) VACTERL-H syndrome. J Perinatol 22:496–498
Holden ST, Cox JJ, Kesterton I, Thomas NS, Carr C, Woods CG (2006) Fanconi anaemia complementation group B presenting as X linked VACTERL with hydrocephalus syndrome. J Med Genet 43:750–754
Isaac M, Best P (1987) Two cases of agenesis of the vermis of cerebellum, with fusion of the dentate nuclei and cerebellar hemispheres. Acta Neuropathol 74:278–280
Jellinger KA (2002) Rhombencephalosynapsis. Acta Neuropathol 103:305–306
Kallen K, Robert E, Castilla EE, Mastroiacovo P, Kallen B (2004) Relation between oculo-auriculo-vertebral (OAV) dysplasia and three other non-random associations of malformations (VATER, CHARGE, and OEIS). Am J Med Genet A 127A:26–34
Kepes JJ, Clough C, Villanueva A (1969) Congenital fusion of the thalami (atresia of the third ventricle) and associated anomalies in a 6-months-old infant. Acta Neuropathol 13:97–104
Lespinasse J, Testard H, Nugues F, Till M, Cordier MP, Althuser M, Amblard F, Fert-Ferrer S, Durand C, Dalmon F, Pourcel C, Jouk PS (2004) A submicroscopic unbalanced subtelomeric translocation t (2p;10q) identified by fluorescence in situ hybridization: fetus with increased nuchal translucency and normal standard karyotype with later growth and developmental delay, rhombencephalosynapsis (RES). Ann Genet 47:405–417
Lopez-Hernandez A (1982) Craniosynostosis, ataxia, trigeminal anaesthesia and parietal alopecia with pons-vermis fusion anomaly (atresia of the fourth ventricle). Report of two cases. Neuropediatrics 13:99–102
McAuliffe F, Chitayat D, Halliday W, Keating S, Shah V, Fink M, Nevo O, Ryan G, Shannon P, Blaser S (2008) Rhombencephalosynapsis: prenatal imaging and autopsy findings. Ultrasound Obstet Gynecol 31:542–548
Michaud J, Mizrahi EM, Urich H (1982) Agenesis of the vermis with fusion of the cerebellar hemispheres, septo-optic dysplasia and associated anomalies. Report of a case. Acta Neuropathol 56:161–166
Millonig JH, Millen KJ, Hatten ME (2000) The mouse Dreher gene Lmx1a controls formation of the roof plate in the vertebrate CNS. Nature 403:764–769
Montull C, Mercader JM, Peri J, Martinez Ferri M, Bonaventura I (2000) Neuroradiological and clinical findings in rhombencephalosynapsis. Neuroradiology 42:272–274
Obersteiner H (1914) Ein Kleinhirn ohne Wurm. Arb Neurol Inst (Wien) 21:124–136
Patel S, Barkovich AJ (2002) Analysis and classification of cerebellar malformations. AJNR Am J Neuroradiol 23:1074–1087
Pavone P, Incorpora G, Ruggieri M (2005) A complex brain malformation syndrome with rhombencephalosynapsis, preaxial hexadactyly plus facial and skull anomalies. Neuropediatrics 36:279–283
Phillips PH, Glasier CM, Brodsky MC (2008) Neuro-ophthalmologic findings in patients with rhombencephalosynapsis. J AAPOS 12:97–99
Poretti A, Alber FD, Burki S, Toelle SP, Boltshauser E (2008) Cognitive outcome in children with rhombencephalosynapsis. Eur J Paediatr Neurol
Poretti A, Bartholdi D, Gobara S, Alber FD, Boltshauser E (2008) Gomez–Lopez-Hernandez syndrome: an easily missed diagnosis. Eur J Med Genet 51:197–208
Poretti A, Boltshauser E (2008) “Rhombencephalosynapsis Associated with Dandy–Walker Malformation” is a molar tooth malformation. J Neuroimaging
Reardon W, Zhou XP, Eng C (2001) A novel germline mutation of the PTEN gene in a patient with macrocephaly, ventricular dilatation, and features of VATER association. J Med Genet 38:820–823
Rittler M, Paz JE, Castilla EE (1996) VACTERL association, epidemiologic definition and delineation. Am J Med Genet 63:529–536
Romanengo M, Tortori-Donati P, Di Rocco M (1997) Rhombencephalosynapsis with facial anomalies and probable autosomal recessive inheritance: a case report. Clin Genet 52:184–186
Sandalcioglu IE, Gasser T, van de Nes JA, Menken U, Stolke D, Wiedemayer H (2006) Fusion of the cerebellar hemispheres ventral to the brainstem: a rare hindbrain-related malformation. Childs Nerv Syst 22:73–77
Savolaine ER, Fadell RJ, Patel YP (1991) Isolated rhombencephalosynapsis diagnosed by magnetic resonance imaging. Clin Imaging 15:125–129
Schachenmayr W, Friede RL (1982) Rhombencephalosynapsis: a Viennese malformation? Dev Med Child Neurol 24:178–182
Schell-Apacik CC, Cohen M, Vojta S, Ertl-Wagner B, Klopocki E, Heinrich U, von Voss H (2008) Gomez–Lopez-Hernandez syndrome (cerebello-trigeminal-dermal dysplasia): description of an additional case and review of the literature. Eur J Pediatr 167:123–126
Sener RN (2000) Unusual MRI findings in rhombencephalosynapsis. Comput Med Imaging Graph 24:277–282
Sener RN (2007) Rhombencephalosynapsis associated with Dandy–Walker malformation. J Neuroimaging 17:355–357
Sener RN, Dzelzite S (2003) Rhombencephalosynapsis and a Chiari II malformation. J Comput Assist Tomogr 27:257–259
Sergi C, Hentze S, Sohn C, Voigtlander T, Jung C, Schmitt HP (1997) Telencephalosynapsis (synencephaly) and rhombencephalosynapsis with posterior fossa ventriculocele (‘Dandy–Walker cyst’): an unusual aberrant syngenetic complex. Brain Dev 19:426–432
Shaw CM, Alvord AC (1996) Hydrocephalus. In: Duckett S (ed) Pediatric neuropathology. Williams and Wilkin, Philadelphia, pp 149–216
Siebert JR, Schoenecker KA, Resta RG, Kapur RP (2005) Holoprosencephaly and limb reduction defects: a consideration of Steinfeld syndrome and related conditions. Am J Med Genet A 134:381–392
Silit E, Mutlu H, Ozturk T (2002) A rare cerebellar malformation: rhombencephalosynapsis. J Neuroradiol 29:208–210
Takanashi J, Sugita K, Barkovich AJ, Takano H, Kohno Y (1999) Partial midline fusion of the cerebellar hemispheres with vertical folia: a new cerebellar malformation? AJNR Am J Neuroradiol 20:1151–1153
Tan TY, McGillivray G, Goergen SK, White SM (2005) Prenatal magnetic resonance imaging in Gomez–Lopez-Hernandez syndrome and review of the literature. Am J Med Genet A 138:369–373
Toelle SP, Yalcinkaya C, Kocer N, Deonna T, Overweg-Plandsoen WC, Bast T, Kalmanchey R, Barsi P, Schneider JF, Capone Mori A, Boltshauser E (2002) Rhombencephalosynapsis: clinical findings and neuroimaging in 9 children. Neuropediatrics 33:209–214
Truwit CL, Barkovich AJ, Shanahan R, Maroldo TV (1991) MR imaging of rhombencephalosynapsis: report of three cases and review of the literature. AJNR Am J Neuroradiol 12:957–965
Utsunomiya H, Takano K, Ogasawara T, Hashimoto T, Fukushima T, Okazaki M (1998) Rhombencephalosynapsis: cerebellar embryogenesis. AJNR Am J Neuroradiol 19:547–549
Wan SM, Khong PL, Ip P, Ooi GC (2005) Partial rhombencephalosynapsis and Chiari II malformation. Hong Kong Med J 11:299–302
Whetsell W, Saigal G, Godinho S (2006) Gomez–Lopez-Hernandez syndrome. Pediatr Radiol 36:552–554
Yachnis AT (2002) Rhombencephalosynapsis with massive hydrocephalus: case report and pathogenetic considerations. Acta Neuropathol 103:301–304
Yachnis AT (2004) Cerebellar heterotopia and dysplasia. In: GJaH BN (ed) Pathology and genetics, developmental neuropathology. Neuropath Press, Basel, pp 100–104
Acknowledgments
The authors would like to thank the families and geneticists who participated in this study and Dr. Catherine Fallet-Bianco for her contribution in neuropathological examinations, Robert Faure for his help in editing the manuscript, and Siegfried Le Roy for the iconography. This work is supported by grants from the French Hospital Clinical Research Program 2008 (PHRC inter-régional ARM 08/242).
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pasquier, L., Marcorelles, P., Loget, P. et al. Rhombencephalosynapsis and related anomalies: a neuropathological study of 40 fetal cases. Acta Neuropathol 117, 185–200 (2009). https://doi.org/10.1007/s00401-008-0469-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-008-0469-9