
Abstract
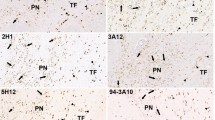
We investigated whether the brainstem is affected by the pathologic process of sporadic Creutzfeldt-Jakob disease (sCJD), with particular attention to brainstem atrophy, neuronal loss, pyramidal tract degeneration, and prion protein (PrP) deposition, in 33 patients with sCJD. Brainstem atrophy, particularly in the pontine base, was relatively prominent in patients with disease of unusually prolonged duration. Neuronal loss and pyramidal tract degeneration were also identified in some but not all patients with prolonged disease. Neuronal loss was relatively prominent in the pontine nucleus and less so in the substantia nigra and inferior olivary nucleus; motor nuclei of the brainstem tegmentum were well preserved. PrP deposition was present in the brainstem in all patients, and was identified predominantly in the substantia nigra, quadrigeminal body, pontine nucleus, and inferior olivary nucleus. PrP deposition was less prominent in the red nucleus and tegmentum of the pons and medulla oblongata. PrP deposition occurred least or not at all in the pyramidal tract. The density of PrP deposition in the sCJD brainstem was not associated with disease duration or neuronal degeneration until the late stage. Our results show that atrophy, neuronal loss, and pyramidal tract degeneration occur in the sCJD brainstem, particularly in patients with an unusually prolonged disease course. These findings are not associated directly with PrP deposition and may reflect end-stage sCJD. No VV1, VV2, or MV2 cases were included in our study; however, we suggest that widespread and relatively stereotypic PrP deposition is a consistent pathologic feature of sCJD, at least in MM1 sCJD patients. Although accumulation of PrP in the brainstem appears to be an early pathologic event in sCJD, and may remain into the late disease stage, the brainstem remains relatively resistant to the pathologic process of sCJD.




Similar content being viewed by others

References
Beck E, Daniel PM (1979) Kuru and Creutzfeldt-Jakob disease: neuropathological lesions and their significance. In: Prusiner SB, Hadlow WJ (eds) Slow transmissible disease of the nervous system, vol 1. Academic Press, New York, pp 253–270
Budka H, Aguzzi A, Brown P, Brucher JM, Bugiani O, Gullotta F, Haltia M, Hauw JJ, Ironside JW, Jellinger K, Kretzschmar HA, Lantos PL, Masullo C, Schlote W, Tateishi J, Waller RO (1995) Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies (prion diseases). Brain Pathol 5:459–466
Castellani R, Parchi P, Stahl J, Capellari S, Cohen M, Gambetti P (1996) Early pathologic and biochemical changes in Creutzfeldt-Jakob disease: study of brain biopsies. Neurology 46:1690–1693
DeArmond SJ, Prusiner SB (1995) Etiology and pathogenesis of prion diseases. Am J Pathol 146:785–811
DeArmond SJ, Dickson DW, DeArmond B (1997) Human prion disease: degenerative disease of the central nervous system. In: Davis RL, Robertson DM (eds) Textbook of neuropathology, 3rd edn. Williams & Wilkins, Baltimore, pp 1111–1125
DeArmond SJ, Kretzschmar HA, Prusiner SB (2002) Prion disease. In: Graham DI, Lantos PL (eds) Greenfield’s neuropathology, vol 2, 7th edn. Arnold, London, pp 273–323
Kascsak RJ, Rubenstein R, Merz PA, Tonna-Demasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H (1987) Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61:3688–3693
Kitamoto T, Tateishi J (1994) Human prion diseases with variant prion protein. Philos Trans R Soc Lond B Biol Sci 343:391–398
Kitamoto T, Shin RW, Doh-ura K, Tomokane N, Miyazono M, Muramoto T, Tateishi J (1992) Abnormal isoform of prion proteins accumulates in the synaptic structures of the central nervous system in patients with Creutzfeldt-Jakob disease. Am J Pathol 140:1285–1294
MacDonald ST, Sutherland K, Ironside JW (1996) Prion protein genotype and pathological phenotype studies in sporadic Creutzfeldt-Jakob disease. Neuropathol Appl Neurobiol 22:285–292
Mahadevan A, Shankar SK, Yasha TC, Santosh V, Sarkar C, Desai AP, Satishchandra P (2002) Brain biopsy in Creutzfeldt-Jakob disease: evolution of pathological changes by prion protein immunohistochemistry. Neuropathol Appl Neurobiol 28:314–324
Malamud N (1979) Creutzfeldt-Jakob’s disease: a clinicopathologic study. In: Prusiner SB, Hadlow WJ (eds) Slow transmissible disease of the nervous system, vol 1. Academic Press, New York, pp 271–285
Masters CL, Richardson EP Jr (1978) Subacute spongiform encephalopathy (Creutzfeldt-Jakob disease). The nature and progression of spongiform change. Brain 101:333–344
Mizutani T (1985) Panencephalopathic type of Creutzfeldt-Jakob disease. In: Mizutani T, Shiraki H (eds) Clinicopathological aspects of Creutzfeldt-Jakob disease. Elsevier, Amsterdam, pp 123–162
Mizutani T, Okumura A, Oda M, Shiraki H (1981) Panencephalopathic type of Creutzfeldt-Jakob disease: primary involvement of the cerebral white matter. J Neurol Neurosurg Psychiatry 44:103–115
Parchi P, Castellani R, Cortelli P, Montagna P, Chen SG, Petersen RB, Manetto V, Vnencak-Jones CL, McLean MJ, Sheller JR, Lugaresi E, Autilio-Gambetti L, Gambetti P (1995) Regional distribution of protease-resistant prion protein in fatal familial insomnia. Ann Neurol 38:21–29
Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG, Farlow M, Dickson DW, Sima AA, Trojanowski JQ, Petersen RB, Gambetti P (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol 39:767-768
Parchi P, Giese A, Capellari S, Brown P, Schulz-Shaeffer W, Windl O, Zerr I, Budka H, Kopp N, Piccardo P, Poser S, Rojiani A, Streichemberger N, Julien J, Vital C, Ghetti B, Gambetti P, Kretzschmar H (1999) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233
Riku S, Okamoto T, Hashizume Y, Koike Y, Sobue I (1983) Panencephalopathic type of Creutzfeldt-Jakob disease and special reference to its pyramidal tract degeneration (in Japanese with English abstract). Rinsho Shinkeigaku 23:147–151
Satoh K, Muramoto T, Tanaka T, Kitamoto N, Ironside JW, Nagashima K, Yamada M, Sato T, Mohri S, Kitamoto T (2003) Association of an 11–12 kDa protease-resistant prion protein fragment with subtypes of dura graft-associated Creutzfeldt-Jakob disease and other prion diseases. J Gen Virol 84:2885–2893
Shimizu S, Hoshi K, Muramoto T, Homma M, Ironside JW, Kuzuhara S, Sato T, Yamamoto T, Kitamoto T (1999) Creutzfeldt-Jakob disease with florid-type plaques after cadaveric dura mater grafting. Arch Neurol 56:357–362
Taguchi Y, Mohri S, Ironside JW, Muramoto T, Kitamoto T (2003) Humanized knock-in mice expressing chimeric prion protein showed varied susceptibility to different human prions. Am J Pathol 163:2585–2593
Tateishi J, Kitamoto T (1993) Developments in diagnosis for prion diseases. Br Med Bull 49:971–979
Watanabe H, Hamada K, Hishikawa N, Iwasaki Y, Ito H, Hirayama M, Sobue G, Ito H, Yoshida M, Hashizume Y (2003) An autopsy case of severe autonomic failure with Parkinson’s disease associated with Creutzfeldt-Jakob disease. Neuropathology 23:A42
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Iwasaki, Y., Hashizume, Y., Yoshida, M. et al. Neuropathologic characteristics of brainstem lesions in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol 109, 557–566 (2005). https://doi.org/10.1007/s00401-005-0981-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-005-0981-0