
Abstract
Mitochondrial function is maintained by several strictly coordinated mechanisms, collectively termed mitochondrial quality control mechanisms, including fusion and fission, degradation, and biogenesis. As the primary source of energy in cardiomyocytes, mitochondria are the central organelle for maintaining cardiac function. Since adult cardiomyocytes in humans rarely divide, the number of dysfunctional mitochondria cannot easily be diluted through cell division. Thus, efficient degradation of dysfunctional mitochondria is crucial to maintaining cellular function. Mitophagy, a mitochondria specific form of autophagy, is a major mechanism by which damaged or unnecessary mitochondria are targeted and eliminated. Mitophagy is active in cardiomyocytes at baseline and in response to stress, and plays an essential role in maintaining the quality of mitochondria in cardiomyocytes. Mitophagy is mediated through multiple mechanisms in the heart, and each of these mechanisms can partially compensate for the loss of another mechanism. However, insufficient levels of mitophagy eventually lead to mitochondrial dysfunction and the development of heart failure. In this review, we discuss the molecular mechanisms of mitophagy in the heart and the role of mitophagy in cardiac pathophysiology, with the focus on recent findings in the field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mitochondria are not only the powerhouse of the cell but also a hub for signaling activities that determine the cell’s fate and functionality. Thus, the quality and quantity of mitochondria need to be regulated precisely. Various stress conditions that lead to mitochondrial damage result in leakage of mitochondrial proteins and cause cell death [38, 162]. Repairing damaged mitochondria and eliminating dysfunctional mitochondria are critical to maintaining homeostasis and preventing cell death. Mitophagy is a process by which damaged or unnecessary mitochondria are specifically removed through autophagy-mediated lysosomal degradation, and it is the most well-studied type of selective autophagy [6, 135]. Impaired mitophagy and accumulation of damaged mitochondria lead to cell and tissue damage and are associated with a broad spectrum of pathologies, including neurodegeneration, myopathies, metabolic disorders, inflammation, autoimmune disorders, and cancer [135, 205]. Mitophagy is crucial for maintaining cardiovascular homeostasis and protecting the myocardium. Defects in mitophagy are observed in cardiovascular diseases such as myocardial infarction, cardiac hypertrophy, heart failure, ischemia/reperfusion, and diabetic cardiomyopathy (DCM) [10, 112]. In this review, we focus on the molecular mechanisms that orchestrate selective removal of mitochondria and discuss the role of mitochondrial fission and fusion in mitochondrial quality control mechanisms during myocardial ischemia/reperfusion injury, hypertrophy, and diabetic cardiomyopathy.
Molecular mechanism of mitophagy
The general steps mediating the selective degradation of mitochondria through mitophagy include the identification and tagging of damaged mitochondria, compartmentalization of the mitochondria by autophagosomes, fusion of the autophagosomes with lysosomes, and proteolysis of the mitochondrial components in the fused autolysosomes [135].
Mechanism of autophagosome biogenesis
One of the most well-characterized conditions promoting mitophagy is energy stress, where an energy deficiency is sensed and signaled by mammalian target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK) [75]. AMPK and mTORC1 post-translationally modify the unc-51-like kinase 1 (Ulk1) complex (which includes Ulk1, Atg13, Atg101, and FIP200) to initiate formation of the phagophore, a double-membrane structure that sequesters the cargo to be degraded. Class III phosphatidylinositol 3-kinase (PI3KC3) complex I (PI3KC3-C1), consisting of Atg6/Beclin 1, Atg14, PI3KC3/Vps34, and the regulatory subunit Vps15/p150, generates phosphatidyl inositol 3-phosphate (PI3P) to nucleate the phagophore [137]. Autophagosome expansion and nucleation are facilitated by vesicles containing Atg9, the sole multi-membrane-spanning protein in the autophagosome-forming machinery [128]. While autophagosomes are being established, Atg18 and WIPIs (WD-repeat proteins interacting with phosphoinositides), members of the PROPPIN (β-propellors that bind polyphosphoinositides) family of proteins, can directly interact with PI3P via a conserved FRRG motif and facilitate the recruitment of downstream Atg effector proteins Atg12-5-16/Atg12-5-16L1 [128]. Atg8/LC3 is a well-known marker protein for traditional autophagy and a ubiquitin-like (Ubl) protein. Atg4 protease immediately processes newly translated LC3 (pro-LC3) proteins at the C-terminus to form LC3-I [176]. LC3 is then activated by Atg7 (E1 enzyme) and transferred to Atg3 (E2 enzyme), and the Atg12-5-16L1 complex (E3 enzyme) facilitates the transfer of LC3 from Atg3 to phosphatidylethanolamine (PE) (Atg8/LC3 lipidation) [130]. Membrane recruitment of the Atg12-5-16L1 complex and Atg8/LC3 lipidation are controlled by the PI3P-binding protein WIPI2B under starvation conditions [128]. The double-membrane autophagosomes, decorated by Atg8 family proteins LC3/GABARAP (GABA type A receptor-associated protein), form isolation compartments for sequestration of various cargos. The specific endosomal sorting complexes required for transport (ESCRT) proteins are recruited to autophagosomes by Atg8s to keep the membrane impermeable and sealed, allowing the autophagosomes to mature into degradative autophagic compartments [68]. In HEK293 and HeLa cells lacking Atg8s, autophagic organelles are permeable, arrest as amphisomes, and do not progress to functional autolysosomes [68]. The Atg8 family proteins, especially GABARAP, promote Pleckstrin homology domain-containing protein family member 1 (PLEKHM1) recruitment and govern autophagosome–lysosome fusion [103, 124]. Phagophore expansion occurs through the action of Atg9 [102]. Rab7 small GTPase directs the trafficking of autophagosomes along microtubules and facilitates fusion with lysosomes to degrade the cargo materials via acidic hydrolases [63]. The fusion of mature autophagosomes to lysosomes is also facilitated by SNARE proteins [177]. The lysosome-associated membrane proteins (LAMP-1 and LAMP-2) promote acidification of lysosomes by directly interacting with and inhibiting the lysosomal cation channel TMEM175, thereby maintaining the acidic pH required for hydrolase activity [221]. Conventional forms of mitophagy utilize a similar mechanism of autophagosome formation as that used for non-selective autophagy, as described above. However, the mechanisms of phagosome initiation and progression differ in unconventional forms of mitophagy and are discussed in Sect. “Alternative mitophagy and mechanism”.
Mode of selection of mitochondria
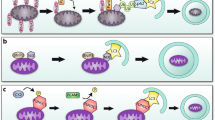
The selection of mitochondria for degradation via mitophagy can be classified as (a) ubiquitin dependent or (b) ubiquitin independent or receptor mediated [73] (Fig. 1).

Mechanisms of mitophagy. A Ubiquitin-dependent mitophagy. In the PINK1–Parkin pathway of mitophagy, PINK1 is stabilized on the OMM following stress, which then stimulates Parkin recruitment. Several components of the outer membrane are ubiquitinated by Parkin. Following this, PINK1 phosphorylates poly-Ub chains, acting as an "eat me" signal for the autophagic machinery. Phosphorylated poly-Ub chains on mitochondrial proteins are recognized by adaptor proteins (p62, OPTN, TAX1BP1, NBR1, NDP52, HDAC6, and ABIN-1 or TNIP1), which then bind to LC3 to start the formation of autophagosomes. TNIP1 has been shown to inhibit TAX1BP1 by competition and downregulate mitophagy. By phosphorylating OPTN, TBK1 increases the protein's ability to bind to Ub chains. A feed-forward mechanism promoting mitochondrial clearance is established by the OPTN-TBK1 complex. Prior to mitophagy, alternative E3 ubiquitin ligases that target OMM proteins include Gp78, SMURF1, MUL1, SIAH1, ARIH1, MARCH5, HUWE1, p62-Keap1-Rbx1, and TRAF2. B Receptor-mediated mitophagy. Mitophagy receptors BNIP3, NIX, FKBP8, BCL2L13, AMBRA1, ATAD3, and FUNDC1 localize to the OMM and interact with LC3 directly to mediate mitochondrial elimination. BCL2L13 has also been shown to activate mitophagy by recruiting the Ulk1/FIP200/Atg13/Atg101 initiation complex and LC3B during starvation stress. Following mitochondrial depolarization, PHB2 and cardiolipin are externalized to the OMM and interact with LC3. Choline dehydrogenase (CHDH) accumulates in the OMM of depolarized mitochondria and interacts with p62 and LC3. FUNDC1 phosphorylation status is influenced by PGAM5 phosphatase, CK2 and Src kinases, all of which control mitochondrial dynamics during hypoxia. The figure was created with Biorender.com
Ubiquitin-dependent mitophagy
-
i.
PINK1–Parkin-mediated mitophagy
One of the most well-studied mechanisms of targeting mitochondria for mitophagy occurs through the mitochondrial membrane kinase PTEN-induced putative kinase-1 (PINK1) and the cytosolic E3 ubiquitin ligase Parkin [139]. In functional mitochondria, PINK1 is transported to the inner mitochondrial membrane (IMM) and cleaved by several proteases [156]. During mitochondrial damage and mitochondrial membrane potential dissipation, the translocation to the IMM does not take place and PINK1 is stabilized on the outer mitochondrial membrane (OMM) [156]. This leads to autophosphorylation and activation of PINK1. PINK1 phosphorylates both polyubiquitin at Ser65 and Parkin at Ser65 in the Ubl domain, which in turn induces mitochondrial recruitment and activation of Parkin E3 ubiquitin ligase activity in a feed-forward loop that polyubiquitinates OMM proteins [55, 133].
Recent studies have shed further light on the molecular mechanism regulating PINK1. Adenine nucleotide translocase 1 and 2 (ANT1 and 2) present on the IMM can induce mitophagy independently of their nucleotide exchange activity by forming a complex with Tim23 and Tim44, leading to stabilization of PINK1 on the OMM [61]. Mitochondrial GSNOR (S-nitrosoglutathione reductase) denitrosylates ANT1 at Cys160, and GSNOR downregulation in cardiomyocytes aggravates mitochondrial dysfunction in the presence of hypertrophic stimuli, accompanied by downregulation of mitophagy [175]. However, how S-nitrosylation of ANT1 at Cys160 negatively regulates mitophagy remains to be clarified. Signaling by RhoA, a small GTPase, promotes stabilization of PINK1 independently of mitochondrial depolarization in cardiomyocytes, thereby promoting mitophagy and protecting the heart against ischemia [163]. In addition, AMPKα2 phosphorylates PINK1 at Ser495 and promotes mitophagy in cardiomyocytes [190].
Parkin ubiquitinates several OMM proteins, including mitofusin1 (MFN1), mitofusin2 (MFN2), MIRO, and voltage-dependent anion channel (VDAC) [45, 46, 141, 174, 192]. Unintentional ubiquitination and targeting for mitophagy is controlled by deubiquitinating enzymes, including USP8, USP15, USP30, and USP35 [24, 35, 47, 193]. However, phosphorylated ubiquitin and polyubiquitin chains are not recognized by deubiquitinases, thus stabilizing the mitophagy signal [55]. It is of great interest to clarify how endogenous deubiquitinating enzymes contribute to the regulation of mitophagy in the heart. PINK1-dependent MFN2 phosphorylation also promotes the recruitment of Parkin to mitochondria and mediates elimination of fetal cardiomyocyte mitochondria through mitophagy during perinatal development. This allows fetal mitochondria to be replaced with adult ones so that metabolic maturation of individual mitochondria is achieved [50].
Taken together, accumulating evidence points to the importance of PINK1 and Parkin in targeting mitochondria for degradation in response to various developmental cues and under stress conditions. However, since some of the studies described above used carbonyl cyanide m-chlorophenyl hydrazone (CCCP), a chemical inducer of mitochondrial depolarization and integrated stress response [77], the degree to which PINK1–Parkin is involved under more physiological stress conditions in various tissues is still largely unknown. It should be noted that Parkin-dependent mechanisms are also utilized in other forms of mitochondrial degradation, including those mediated through Rab5-positive early endosomes [53] and mitochondrial-derived vesicles (MDVs) [104].
Although PINK1 plays a crucial role in Parkin-mediated mitophagy, increasing evidence suggests that Parkin translocation to mitochondria can occur through other molecular mechanisms as well. Heat shock 70 kDa protein 1L (HSPA1L) enhances Parkin recruitment, while BAG Cochaperone 4 (BAG4) inhibits Parkin translocation to mitochondria [57]. Heat shock protein 72 (HSP72) translocates to damaged mitochondria and facilitates Parkin recruitment in skeletal muscles [33]. B cell leukemia/lymphoma-2 (Bcl-2)-interacting death suppressor (BIS), also known as Bcl-2-associated athanogene 3 (BAG3), a co-chaperone of HSP70, co-migrates with Parkin to mitochondria and executes mitophagy in cardiomyocytes [171]. On the other hand, the anti-apoptotic Bcl-2 family proteins, Bcl-xL and Mcl-1 (in HeLa cells), and the tumor suppressor protein, p53 (in liver and heart), prevent Parkin translocation to mitochondria [59, 60, 62, 213]. The significance of these mechanisms relative to the PINK1-dependent mechanisms remains to be clarified.
-
ii.
Parkin-independent ubiquitin-mediated mitophagy
Mice genetically deficient in PINK1 and Parkin exhibit normal mitophagy and cardiac function at young ages, suggesting that PINK1–Parkin-independent forms of mitophagy responsible for the basal turnover of mitochondria may exist in cardiomyocytes [79, 81]. There are several E3 ubiquitin ligases that participate in ubiquitin-dependent mitophagy independently of Parkin. These include glycoprotein 78 (Gp78) [41], SMAD-ubiquitination regulatory factor1 (SMURF1) [27, 134], seven in absentia homolog (SIAH)-1 [169], ARIH1/HHARI [187], MARCH5 [18], HUWE1 [30], p62-keap1-Rbx1 axis [206] and MAPL/MULAN/GIDE/MUL1 [4, 91, 215]. Likewise, TNF-receptor-associated factor 2 (TRAF2) is an E3 ubiquitin ligase that is localized to mitochondria at baseline and during pathological conditions, inducing mitophagy in the heart [98]. TRAF2 is unique in that, besides its involvement in innate immunity through TNF receptor signaling, it activates mitophagy even at baseline and protects the heart against sterile infection by facilitating safe disposal of mitochondrial DNA [98, 150]. These ligases can act independently of Parkin and may be more prominent when the PINK1–Parkin pathway is inhibited [132].
-
iii.
Autophagy adaptors
The ubiquitin chain on OMM proteins triggers the recruitment of a variety of autophagy adaptors, including TAX1 binding protein 1 (TAX1BP1), Nuclear dot protein 52 (NDP52, also known as CALCOCO2), a neighbor of BRCA1 gene 1 (NBR1), Sequestosome-1 (SQSTM1/p62) and optineurin (OPTN) [132]. These adaptor proteins contain a ubiquitin-binding domain, which enables them to recognize and sequester ubiquitinated cargoes, and an LC3-interacting region (LIR) that allows the recruitment of LC3-coated phagophore membrane around the cargo. Histone deacetylase 6 (HDAC6), another ubiquitin-binding protein, is also involved in mitophagy. However, HDAC6 mediates autophagosome–lysosome fusion rather than cargo recruitment [86, 87]. Parkin-induced mitophagy was unaffected by the absence of p62 in transformed mouse embryonic fibroblasts and HeLa cells, suggesting that p62 is not essential, and other adaptors, including NBR1, appear to have redundant roles in the sequestration of damaged mitochondria [123]. In HeLa cells lacking Parkin, PINK1 can promote the recruitment of OPTN, NDP52, and TAXBP1 to induce mitophagy [85]. OPTN is phosphorylated by TANK-binding kinase 1 (TBK1), allowing increased ubiquitin chain binding and mitophagy in HeLa cells treated with CCCP [111]. NDP52 functions as a redox sensor through its redox-sensitive cysteine residues, which promote disulfide bond formation in HeLa cells under H2O2-treated conditions. Oligomerization of NDP52 in damaged mitochondria through these disulfide bonds facilitates the recruitment of the autophagy machinery for mitophagy in response to oxidative stress [72].
Additional LIR-containing proteins, serving as positive or negative regulators of mitophagy, have been reported recently. A20 binding inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1), a polyubiquitin-binding protein, is a positive regulator of mitophagy in HEK293 and HeLa cell lines [107]. In contrast, TNIP3-interacting protein 1 (TNIP1) is an LIR-containing protein that binds to the LC3/GABARAP family of proteins and allosterically to TAX1BP1, a mitophagy receptor, thereby acting as a negative regulator of mitophagy. TNIP1 binding to TAXBP1 prevents the binding of ubiquitinated cargos to TAXBP1. Interestingly, ABIN-1 and TNIP1 are aliases for the same protein; thus, its function appears to be context dependent. It should be noted that TNIP1 can be phosphorylated by TBK1, leading to enhancement of the interaction of TNIP1 with FIP200, a protein in the Ulk1 complex. This interaction then competes with the interaction between FIP200 and TAXBP1, releasing FIP200 from the autophagosome. Thus, phosphorylated TNIP1 can act positively in mitophagy by facilitating the recycling of FIP200 [51]. TNIP1 is one of the few proteins identified as an endogenous negative regulator of mitophagy but its effect appears complex. Further investigation is required to clarify how endogenous TNIP1 function is regulated during stress conditions.
Mitophagy adaptors also have other important functions relevant for mitophagy. For example, p62 has the ability to phase separate ubiquitinated proteins into a larger condensate. How cargo condensation and mitophagy is linked is poorly understood, but it is known that ubiquitinated Nur77 forms condensates that can sequester damaged mitochondria and targets cargo mitochondria for autophagy through interaction with p62 [138]. In addition, p62 serves as a scaffold for the recruitment of the Ulk1 complex, the core autophagy machinery, through interaction with the FIP200 claw domain, thereby promoting autophagosome formation at ubiquitin condensates containing damaged mitochondrial proteins [184].
Ubiquitin-independent or receptor-mediated mitophagy
Mitophagy can also be mediated by mitochondrial integral membrane proteins that have LIRs independently of ubiquitination [132]. Upon mitochondrial depolarization, these receptors accumulate on the OMM and trigger the formation of an isolation membrane around the damaged mitochondria by interacting with Atg8 family proteins (LC3A/B/C, GABARAP, GABARAP-L1/2) on the autophagosome [132].
-
(i) BNIP3 and BNIP3L/NIX
BCL2 interacting protein 3 (BNIP3) is involved in mitochondrial turnover during hypoxia [218]. In response to hypoxia, BNIP3 is upregulated and anchored to the OMM through its C-terminal transmembrane domain. The N-terminal domain carrying the LIR motif is then exposed to the cytosol, allowing it to serve as a mitophagy receptor [54, 80, 146]. Phosphorylation of Ser17 and Ser24 proximal to the LIR region of BNIP3 is critical for LC3 interaction [231]. BNIP3 is upregulated by HIF-1α, thereby mediating mitophagy in response to hypoxia in murine embryonic fibroblast (MEF) cells [217]. BNIP3 is also upregulated by p53, thereby mediating mitochondrial dysfunction and autophagic cell death, the major known function of p53 in the heart [191]. Thus, BNIP3 mediates both adaptive and maladaptive mitophagy in a context-dependent manner.
BNIP3L/NIP3-like protein X (NIX) is 53–56% homologous to BNIP3 and mediates mitochondrial clearance via programmed mitophagy during reticulocyte maturation and erythrocyte differentiation [154]. NIX promotes mitophagy by recruiting GABARAP-L1 in CCCP-treated cells [129]. The NIX-LC3 interaction is stabilized by phosphorylation of NIX at Ser34 and Ser35 near the LIR motif [147]. As with BNIP3, the C-terminal region of NIX is recruited and homodimerizes for efficient mitophagy initiation [101]. Reactive oxygen species (ROS) accumulation caused by oxidative phosphorylation promotes the recruitment of Rheb (a small GTPase of the Ras superfamily) together with NIX and LC3 to promote mitophagosome formation [106]. In cardiac progenitor cells, mitophagy mediated by BNIP3/NIX facilitates their proper differentiation into myocytes through mitochondrial network reorganization and promotes their survival in the infarcted heart [84].
How do BNIP3/NIX mediate mitophagy? Several studies have shown that BNIP3 and NIX can activate PINK1–Parkin-mediated mitophagy. Parkin ubiquitinylates NIX and promotes the recruitment of the selective autophagy adaptor NBR1, which can interconnect ubiquitin and LC3/GABARAP to promote autophagosome formation around mitochondria [43]. Additionally, BNIP3 interacts with PINK1 and promotes its accumulation on the OMM, leading to Parkin recruitment to mitochondria [223]. NIX similarly promotes Parkin recruitment to depolarized mitochondria [31]. However, BNIP3 is also involved in Parkin-independent mitophagy. Myeloid cell leukemia-1 (Mcl-1), an anti-apoptotic Bcl-2 member, interacts with BNIP3 to enhance mitophagy in the heart in the presence of energy stress and mitochondrial damage [113]. Mcl-1 has an LIR motif and acts as an adaptor for BNIP3 to promote mitophagy. Since Mcl-1 inhibits general autophagy through interaction with Beclin 1, it could serve as a mechanism that differentially regulates general autophagy and mitophagy.
BNIP3 may also induce mitophagy through Drp1. As we discuss separately below, Drp1 is a GTPase involved in mitochondrial fission. BNIP3 induces mitochondrial translocation of Drp1, allowing Drp1 to mediate mitochondrial fission and mitophagy in cardiomyocytes [88]. It should be noted that the effect of BNIP3 upon Drp1 is complex and may be detrimental under some conditions through pathological activation of mitochondrial fission and mitophagy. For example, doxorubicin-induced cell death and mitochondrial fission are prevented by downregulation of BNIP3 [29]. A polyphenolic compound, ellagic acid (EA), suppresses mitochondrial injury and necrotic cell death of cardiomyocytes through inhibition of BNIP3 (and subsequent activation of pathological mitophagy), and is thus of therapeutic benefit in lowering oxidative injury and cardiac dysfunction in cancer patients undergoing chemotherapy or under ischemic cardiac stress [29].
Increasing lines of evidence suggest that the level of BNIP3/NIX is regulated by posttranslational mechanisms. For example, SCF-FBXL4 ubiquitin ligase regulates BNIP3 and NIX localization on mitochondria to preserve normal mitochondria levels. The loss of SCF-FBXL4 leads to excessive mitophagy and perinatal lethality [12]. The UBXD8 adaptor regulates mitophagy by aiding in the recruitment of the hexameric AAA-ATPase valosin-containing protein (VCP) to mitochondria and promoting the degradation of BNIP3 through interaction with ubiquitin E3 ligases in mitochondria [226]. In addition, the mitochondrial protein TMEM11 forms a complex with BNIP3 and BNIP3L and is co-enriched at sites of mitophagosome formation. Lack of TMEM11 hyperactivates mitophagy under normoxic and hypoxia-mimetic conditions, suggesting that it provides a spatial restriction effect on mitophagy activation by BNIP3/BNIP3L [206].
-
(ii) FUNDC1
FUN14 domain-containing protein 1 (FUNDC1) is an integral OMM protein with an N-terminal LIR motif that acts as a receptor for hypoxia-induced mitophagy [95]. FUNDC1 is regulated via phosphorylation and dephosphorylation events on Ser13 and Tyr18 near its LIR motif. Normoxic conditions favor Ser13 and Tyr18 phosphorylation of FUNDC1 by casein kinase 2 (CK2) and Src tyrosine kinase, respectively, to negatively regulate FUNDC1 interaction with LC3 [15, 95]. Under hypoxia, Src kinase is inactivated and the FUNDC1–LC3 interaction is stabilized by decreasing Tyr18 phosphorylation. Similarly, removal of Ser13 phosphorylation by PGAM5, a serine/threonine phosphatase, promotes mitophagy [15]. However, the PGAM5–FUNDC1 interaction is prevented by BCL2L1/Bcl-xL (anti-apoptotic BH3 domain-containing molecule) under normoxic conditions [197]. On the other hand, in the presence of hypoxia or mitochondrial depolarization, phosphorylation of Ser17 of FUNDC1 by Ulk1 recruited to fragmented mitochondria promotes mitophagy by increasing interaction with LC3 [198].These studies emphasize the importance of FUNDC1 as a mitophagy receptor under hypoxic stress conditions. FUNDC1-mediated mitophagy also promotes cardiac progenitor cell differentiation and survival in the infarcted heart [84]. Hypoxia acclimation with a low-pressure hypoxic animal chamber alleviated cardiac dysfunction and fibrosis caused by MI injury in mice through activation of FUNDC1-mediated mitophagy [92]. The role of FUNDC1 in ischemia/reperfusion injury in the heart is discussed further later in this review.
-
(iii) BCL2L13
BCL2L13 is a single pass membrane protein anchored on the OMM and contains two LIR motifs. It regulates mitochondrial morphology such that the fragmented state is increased when it is overexpressed and the elongated state is increased when it is knocked down [116]. Involvement of BCL2L13 in inducing mitophagy was first shown in yeast studies, in which BCL2L13 restored mitophagy in yeast cells lacking Atg32, a mitophagy receptor [116]. BCL2L13-dependent mitophagy was shown to be mediated through conventional Atg7 via Atg8 lipidation in Atg32-lacking yeast cells [116]. Specifically, a mutation in one of the LIRs of BCL2L13 inhibited mitophagy in yeast cells lacking Atg32, suggesting the involvement of Atg8 in BCL2L13-mediated mitophagy [116], and phosphorylation at Ser272 near the second LIR motif is important for the BCL2L13–LC3 interaction [116]. In HEK293 and HeLa cells, BCL2L13 recruits the Ulk1 complex (including FIP200, Atg13 and Atg101) along with LC3B to autophagosomes to induce mitophagy under starvation conditions [116].
-
(iv) FKBP8
FK506-binding protein 8 (FKBP8) is an OMM integral protein with an N-terminal LIR motif and a C-terminal transmembrane domain. FKBP8 preferentially binds to LC3A over other Atg8 family proteins in vivo and mediates mitophagy in HeLa cells [9, 94].
-
(v) Prohibitin 2
Prohibitins (PHB) are IMM proteins. PHB2 is exposed to the cytosol in the presence of mitochondrial membrane depolarization and increased proteasomal activity that ruptures the OMM [194]. PHB2 associates with LC3 through its LIR domain and the p62 adaptor to target mitochondria for autophagic degradation [200]. PHB2 also mediates Parkin-induced mitophagy under energetic stress conditions in HeLa cells and MEFs [194].
-
(vi) Cardiolipin
Cardiolipin is a phospholipid in the IMM that is exposed to the cytosol only when the OMM is disrupted by mitochondrial damage and elicits autophagosome formation around damaged mitochondria via its interaction with LC3 [21, 158]. Mesenchymal stem cells (MSCs) derived from high-fat diet (HFD)-induced obese mice (MSC-Ob) have a reduced cardiolipin content and less ability to clear damaged mitochondria compared to MSCs from control mice fed a normal diet [151].
-
(vii) AMBRA1
Autophagy/Beclin 1 regulator 1 (AMBRA1) is an adapter protein in the autophagy signaling network. AMBRA1 is involved in the early steps of autophagosome core complex formation in mTORC1-dependent autophagy [22]. Under basal conditions, AMBRA1 is present in mitochondria and its pro-autophagic activity is inhibited by Bcl-2. However, AMBRA1 directly interacts with LC3 through its LIR motif to recruit mitochondria to autophagosomes upon mitophagy induction. This interaction is crucial for regulating both canonical Parkin-dependent and -independent mitochondrial clearance in fibroblasts of human and PINK1−/− mouse origin, as well as in HEK293 and HeLa cell lines [166].
-
(viii) ATAD3B
ATPase family AAA domain-containing protein 3B (ATAD3B), which is only expressed in primates, is a mitochondrial membrane-bound ATPase that was recently identified as a mitophagy receptor [161]. The ATAD3B protein has an LIR motif that binds to LC3 and stimulates oxidative stress-induced mitophagy without the assistance of PINK1, facilitating the removal of oxidative stress-damaged mtDNA. When ATAD3B and ATAD3A hetero-oligomerize under normal conditions, ATAD3B's C-terminal region is directed toward the mitochondrial intermembrane space, preventing mitophagy. Decreased ATAD3B–ATAD3A hetero-oligomerization as a result of oxidative stress-induced mtDNA damage or mtDNA depletion causes exposure of the ATAD3B C-terminus at the OMM, which then attracts LC3 to stimulate mitophagy [161].
-
(ix) Choline dehydrogenase (CHDH)
CHDH catalyzes the dehydrogenation of choline to betaine aldehyde in mitochondria and participates in glycine, serine, and threonine metabolism. Under normal circumstances, CHDH is found in both the IMM and OMM. When the mitochondrial membrane potential is disrupted, CHDH builds up on the OMM and interacts with p62 via its Phox and Bem1 (PB1) domain, forming a CHDH-p62-LC3 complex and mediating mitophagy [136].
Alternative mitophagy and mechanism
The forms of mitophagy discussed above are all mediated through autophagosomes associated with LC3, requiring the Ubl conjugation system. However, some forms of mitophagy occur independently of the conventional autophagy mechanisms. Using Atg5 knockout cells, Nishida et al. discovered an Atg5- and Atg7-independent mechanism of autophagy, referred to as alternative autophagy [127]. The two Ubl conjugation systems, which involve Atg5, Atg7, and LC3, are not utilized by alternative autophagy. Like conventional autophagy, alternative autophagy utilizes double-membrane autophagosomes to internalize cargo for degradation via fusion with lysosomes. However, autophagosomes in alternative autophagy are produced through a Rab9-dependent, but LC3-independent, mechanism [127].
Although both Ulk1 and Ulk2 help initiate conventional autophagy, Ulk2 function is thought to be redundant, whereas the loss of Ulk1 typically causes disruption of autophagy [56, 216]. Furthermore, Ulk1 is a critical initiator of both the conventional and alternative processes, in contrast to Ulk2, which is only involved in conventional autophagy [83]. Both conventional and alternative autophagy are regulated by phosphorylation of Ulk1 by mTORC1 (at Ser757, inhibition) and AMPK (at Ser317 and Ser777, activation) [75], and Beclin 1 and Vps34 are among the autophagy-related targets that are phosphorylated by Ulk1 [36]. On the other hand, in Atg5-deficient MEF cells treated with etoposide, the necroptosis-regulating kinase RIPK3 interacts with and phosphorylates Ulk1 at Ser746. RIPK3-dependent phosphorylation of Ulk1 at Ser746 only activates alternative autophagy, without activating necroptosis or conventional autophagy, indicating a clear functional distinction between conventional and alternative mechanisms [183, 195].
In alternative autophagy, autophagosomes originate from the trans-Golgi network [127]. At the isolation membrane, WIPI family proteins perform a crucial PIP3 effector function [115]. WIPI3, rather than WIPI1 and WIPI2, is essential for producing alternative isolation membranes [207]. Additionally, a recent study found that Golgi-resident Rab2 participates in the formation of autophagosomes by separating from the Golgi apparatus under autophagy-inducing conditions to interact with Ulk1 [32]. Ulk1-deficient mice are viable, in contrast to mice with knockouts of various important Atg genes, such as Atg5, Atg7, and Atg12, which are fatal to neonates [82]. However, Ulk1-knockout mice exhibit defects in mitochondrial autophagy in primary hepatocytes during erythrocyte development [83, 157].
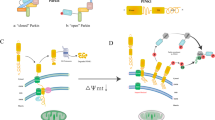
There is a form of mitophagy that uses a molecular mechanism similar to that of alternative autophagy and is, thus, termed alternative mitophagy. Alternative mitophagy can be observed even when conventional autophagy/mitophagy is inhibited, such as in the presence of Atg7 downregulation [120, 152, 180]. In alternative mitophagy, damaged mitochondria are sequestrated in autophagosomes associated with Rab9, but not LC3, through Ulk1-dependent mechanisms. Although depolarized mitochondria can be found in the cargo, how damaged mitochondria are tagged for degradation remains unclear. What is known, however, is that alternative mitophagy is associated with the formation of a large protein complex consisting of Ulk1-Rab9-Rip1-Drp1 near mitochondria-associated endoplasmic reticulum membrane (MAM) [179]. Drp1 is a conserved dynamin GTPase superfamily protein required for mitochondrial fission and various forms of mitophagy, including Parkin-dependent, Parkin-independent, receptor-mediated, and alternative mitophagy [69, 182]. Multiple kinases, such as Cdk1, Erk2, and Rip1, phosphorylate Drp1 at Ser616 and enhance fission [66, 71, 118, 152, 199]. Under stress conditions, Ulk1 interacts with Rab9 and phosphorylates it at Ser179, which is a crucial first step for the formation of the Ulk1-Rab9-Rip1-Drp1 complex [152]. A signaling complex comprising Rip1 and Drp1 is formed in response to Ulk1 activation and phosphorylation of Rab9. Rip1-mediated Drp1 phosphorylation at Ser616 then promotes mitochondrial fission, and Rab9-mediated autophagosomes form around damaged mitochondria [152]. A schematic representation of the molecular mechanisms involved in alternative mitophagy is given in Fig. 2. As mentioned above, the materials for generating alternative autophagosomes are derived from trans-Golgi membranes. Alternative mitophagy is activated after conventional mitophagy is inactivated in diabetic hearts [179, 180]. Thus, inactivation of conventional mitophagy may induce some of the mechanisms that stimulate alternative mitophagy.

Mechanism of alternative mitophagy. Energy stress activates AMPK-mediated phosphorylation of Ulk1 at Ser555. The activated Ulk1 phosphorylates Rab9 at Ser179 and initiates phagophore formation utilizing trans-Golgi-derived membrane. Activated Ulk1-Rab9 recruits Rip1/3 kinase and phosphorylates Drp1 at Ser616, which activates mitochondrial fission to segregate damaged mitochondria and promote alternative mitophagy at the mitochondrial-associated ER membrane. The figure was created with Biorender.com
Other processes also degrade damaged mitochondria, using some of the same molecules as those utilized by conventional mitophagy but via distinct mechanisms. For example, damaged mitochondria with OMM proteins ubiquitinated by Parkin are also sequestrated by Rab5-dependent early endosomes via the ESCRT machinery and degraded in lysosomes [53]. The endosomal-mediated degradation of mitochondria is initiated by the same mechanism as Parkin-mediated mitophagy and requires Beclin 1 [53], suggesting that crosstalk may exist between mitophagy and the endosomal pathway. More investigation is needed to clarify the functional relevance of endosomal-mediated mitochondrial clearance in the heart in vivo.
Micromitophagy
Microautophagy is a mechanism through which cytosolic materials are directly engulfed by lysosomes through membrane invagination. During ischemia–reperfusion (I/R) in the heart, damaged mitochondria can be taken up by lysosomes directly. I/R promotes the association of GAPDH with mitochondria and directs the uptake of damaged mitochondria into multiorganellar lysosomal-like (LL) structures for elimination [209]. Protein kinase Cδ (PKCδ) inhibits this process through phosphorylation of GAPDH at Thr246, thereby leading to accumulation of damaged mitochondria at the edge of the LL structure and causing apoptosis. Inhibition of PKCδ or expression of a phosphorylation resistant GAPDH mutant during I/R rescues mitophagy, promotes clearance of damaged mitochondria, and downregulates apoptosis [209].
MDVs represent another form of microautophagy, in which vesicles enriched in mitochondrial proteins bud off from mitochondria, transit into multivesicular bodies and are engulfed by lysosomes [90, 104, 165]. MDVs are stimulated as an early response to oxidative stress and deliver dysfunctional proteins and lipids from mitochondria as cargo to lysosomes without the involvement of mitochondrial depolarization and fission machinery [104, 165]. Although the process is independent of the macroautophagy machinery, including Atg5 and LC3, it requires PINK1 and Parkin [104, 165].
Taken together, mitochondria can be degraded through multiple mechanisms. How each mechanism of mitochondrial degradation is regulated by stress and whether the activities of the different mechanisms of mitochondrial degradation affect one another remain to be clarified.
Autophagic secretion of mitochondria
Heterophagy is a mechanism whereby damaged mitochondria are ejected from cells and degraded by macrophages [126]. This type of transcellular mitochondrial degradation system exists in the heart, where damaged mitochondria are packed in exophores, large membrane-surrounded vesicles, which are then phagocytized and degraded by resident macrophages after release into the extracellular space [125]. Not only are exophores LC3( +), but the release of exophores is affected by the level of autophagy, suggesting that the exophore-mediated elimination of damaged mitochondria is controlled by mechanisms shared with autophagy. Suppression of exophore phagocytosis or ablation of resident macrophages induces inflammation. Thus, removal of damaged mitochondria through LC3( +) exophores is a mitochondrial quality control mechanism [126]. A recent report showed that damaged mitochondria sequestered into vesicles through PINK1–Parkin-mediated mitophagy are also secreted into the extracellular space, especially when the mAtg8 conjugation system, including Atg3, Atg5, and Atg7, is not functional [172]. Importantly, secreted mitochondria elicit innate immune responses through systemic activation of the cGAS-STING pathway [172]. This highlights the importance of the mAtg8 lipidation system in assuring the completion of mitophagy via lysosomal degradation without activation of inflammatory responses. Thus, it remains to be clarified whether secretion of damaged mitochondria initiated by the PINK1–Parkin-dependent mechanism of mitophagy can play a salutary role in maintaining mitochondrial quality. In addition, cells, including cardiomyocytes, can transfer intact mitochondria from one cell to another, as a cell protective mechanism [39, 64]. The transfer is mediated through several different mechanisms, including tunneling nanotubes and extracellular vesicles [39]. Since extracellular vesicles and MDVs share some properties and the release of extracellular vesicles is affected by lysosomal activity, it is possible that the release of intact mitochondria and eventual improvement of mitochondrial quality control in recipient cells are also controlled by autophagy and mitophagy in host cells. Further investigation is required to clarify this issue.
Mitochondrial fission and fusion
Mitochondria are highly dynamic organelles that constantly undergo fusion and fission to maintain homeostasis in response to changes in the cellular environment. Mitochondrial fusion is coordinated by Mfn1, Mfn2, and optic atrophy 1 (OPA1), whereas fission is regulated by Drp1 [119, 196]. In general, elongated mitochondria are protected from degradation by mitophagy, carry more cristae, and maintain ATP production through increased activity of ATP synthase [49]. Furthermore, mitochondrial fusion rescues a part of mitochondria from reversible damage [185]. On the other hand, isolation of severely damaged mitochondria through fission can protect the healthy portion of mitochondria by preventing the spread of depolarization and ROS [211]. In addition, physiological mitochondrial fragmentation mediated by Drp1 is important to meet the energetic demands during exercise [25]. Thus, understanding the physiological function of fusion and fission is important.
Mfn1 and Mfn2 are specialized proteins localized on the OMM that regulate the fusion of the OMM. They are dynamin-like GTPases that contain N-terminal catalytic GTP-binding domains and C-terminal transmembrane domains [189]. The C-terminal transmembrane domains anchor Mfn1/2 to the OMM, where they interact with adjacent mitochondria via a heptad repeat region [78, 188]. Since individual overexpression of Mfn1 or Mfn2 can rescue the loss of the other protein to promote fusion, Mfn1/2 presumably have redundant functions [16]. However, genetic mutation of Mfn2 was shown to interrupt mitochondrial fusion, leading to a neurodegenerative condition [40]. Following myocardial IR injury, matrix metalloproteinase-2 (MMP-2) activation and cleavage of Mfn2 leads to impaired myocardial contractile function, lowered mitochondrial respiration, and elevated inflammasome response [7]. MORN repeat-containing protein 4 (MORN4) directly binds to Mfn2 and promotes phosphorylation of Mfn2 at Ser442 by Rho-associated protein kinase 2 (ROCK2) to induce mitochondrial dynamics and mitophagy [229]. Downregulation MORN4 during myocardial ischemia accelerated cardiac injury and fibrosis, exacerbating cardiac dysfunction in a murine model [229].
Fusion of the IMM is regulated by OPA1, a dynamin-like GTPase that is localized on the IMM [28] and also regulates crista formation and maintenance. OPA1 is processed to a membrane-bound long isoform (L-OPA1) by mitochondrial processing peptidase (MPP). However, L-OPA1 can also be proteolytically cleaved to a short isoform (S-OPA1) by two IMM peptidases, overlapping proteolytic activity with m-AAA protease 1 (OMA1) and YME1 Like 1 ATPase (YME1L1) [5]. The balance between L-OPA1 and S-OPA1 is regulated by YME1L and OMA1 under basal conditions. When mitochondrial stress occurs, the activity of OMA1 is increased and L-OPA1 is actively cleaved to S-OPA1. The resulting imbalance between L-OPA1 and S-OPA1 promotes mitochondrial fission [99, 188]. Upregulation of OPA1 at appropriate levels protects the heart against ischemia [23, 170, 186]. Furthermore, inhibition of OMA1 protects the heart against heart failure in response to multiple types of cardiac insult [2] by preventing crista remodeling. These findings suggest that OPA1 and inhibition of OMA1 play salutary roles during heart failure, primarily by preventing crista remodeling. However, how degradation of irreversibly damaged mitochondria is affected by these interventions remains unknown.
OMM constriction occurs at mitochondria–ER contact sites where Drp1 is oligomerized. Drp1 is a cytosolic protein with an N-terminal GTPase domain, a middle domain, a variable domain, and a C-terminal GTPase effector domain [142]. Drp1 is recruited to sites of mitochondrial fission in a phosphorylation-dependent manner, through interactions with mitochondrial OMM proteins such as Fis1, MFF, and mitochondrial dynamics proteins of 49 and 51 kDa (MiD49, MiD51) [96, 210]. Fis1 was initially proposed as a Drp1 receptor in yeast, but the role of mammalian Fis1 is less clear [114]. While it may not be required for Drp1 recruitment to the OMM, as shown in Fis1-null MEFs, overexpression of human Fis1 can induce mitochondrial fragmentation in the absence of Drp1 [96, 212]. Fis1 activates mitochondrial fission and suppresses fusion by inhibiting the GTPase activities of fusion proteins Mfn1, Mfn2, and OPA1 [212]. While OMM constriction has been studied extensively, the mechanism of IMM division remains elusive. Recent studies have shown that an intra-mitochondrial influx of Ca2+ activates IMM constriction at mitochondria–ER contact sites [14, 19], but further studies are required to fully understand the fission process of the IMM. A recent study using yeast and HeLa cells showed that mitofissin (Atg44), a mitochondrial intermembrane space protein, mediates fission and contributes to mitophagy [42]. Mitofissin is required for the formation of mitochondrial protrusions that are eventually fragmented and encapsulated by phagosomes for clearance [42].
Mitochondrial fission allows the segregation of unhealthy mitochondria so that they can be eliminated by mitophagy [185]. Although direct involvement of Drp1 in the mitophagy machinery has been shown in yeast [1, 70, 100], the mechanism by which mitochondrial fission is coupled with mitophagy has not been fully elucidated in mammalian cells [65, 69, 164]. Separation of damaged mitochondria during mitophagy takes place even in the absence of Drp1 in yeast and HeLa cells, where the damaged portion of mitochondria can be separated by autophagosomal constriction [208]. However, the loss of Drp1 function does inhibit mitophagy in the heart under some conditions [182]. When Drp1 is phosphorylated at Ser616, it is localized at the MAM, where mitochondrial fission takes place in cardiomyocytes. As noted above, Drp1 associates with a large protein complex at the MAM, thereby mediating alternative mitophagy in cardiomyocytes during ischemia and HFD consumption [179]. It should be noted that Drp1 can regulate mitophagy even without physical contact with the damaged mitochondria. For example, Drp1 positively mediates autophagosome formation by alleviating the inhibition of Beclin 1 by Bcl-2/Bcl-xL [182]. Furthermore, Drp1 also regulates mitochondrial function through fission-independent mechanisms [219]. Thus, it is possible that the effect of loss of Drp1 function upon mitophagy could be secondary to its effect upon global changes in mitochondrial function. Indeed, although Drp1 promotes physiological mitochondrial fragmentation during exercise, mitophagy is inhibited [25]. Although this makes practical sense, in that the mitochondria are not depolarized and it would be undesirable for the content to be decreased when coping with a high energy demand, the mechanism by which Drp1 actively inhibits mitophagy is unknown.
The role of mitophagy during cardiac stress
Here we discuss activation of mitophagy during cardiac stress and its functional significance. We will limit our discussion to three representative stress conditions in which activation of mitophagy plays a critical role in regulating the survival and function of cardiomyocytes.
Mitophagy in ischemia and reperfusion (I/R)
While reperfusion after ischemia is critical for myocardial survival, it dramatically increases oxidative damage through generation of ROS, causing myocardial cell death [20, 58, 117]. Mitochondrial ROS trigger the opening of the mitochondrial permeability transition pore (mPTP) on the IMM [20]. mPTP opening during I/R prevents the recovery of ATP production and causes cardiomyocyte death through necrosis [52].
In general, mitophagy is activated in the heart and plays a protective role during I/R by alleviating the insults from damaged mitochondria and promoting mitochondrial biogenesis for recovery [81]. For example, phosphoglycerate mutase family member 5 (PGAM5) is crucial for mitochondrial homeostasis, promoting mitophagy through stabilization of PINK1; PGAM5 knockout mice showed inhibited PINK1-dependent mitophagy in the heart, resulting in the accumulation of damaged mitochondria, increased oxidative stress, and exacerbated necroptosis during I/R [97]. The mitochondrial Zn2+ transporter (ZIP7 or Slc39a7) is upregulated during the reperfusion phase and negatively regulates mitophagy, contributing to increased reperfusion injury [220]. ZIP7 levels are high in the mitochondria of cardiac tissue from heart failure patients [220]. Cardiac-specific downregulation of ZIP7 decreased ROS production, enhanced mitophagy, and reduced infarct size, conferring protection against reperfusion injury [220]. Transient receptor potential mucolipin 1 (TRPML1) is activated secondary to increased ROS after I/R, thereby inducing lysosomal zinc release into the cytosol and ultimately blocking autophagy in cardiomyocytes, possibly by disrupting fusion between autophagosomes and lysosomes [167, 202]. Pharmacological and genetic inhibition of TRPML1 channels effectively reduced infarct size and rescued heart function in mice subjected to I/R in vivo by restoring impaired myocardial autophagy and reducing accumulation of damaged mitochondria [167, 202]. Downregulation of endogenous Drp1 inhibits mitophagy and increases cardiomyocyte death during I/R [34]. CLOCK, a circadian gene, transcriptionally coordinates genes involved in mitochondrial fission and fusion and mitophagy, thereby promoting cell survival [145]. Thus, interventions that stimulate the direct regulators of mitophagy in cardiomyocytes may alleviate I/R injury by preconditioning the heart and promoting mitochondrial quality control mechanisms. Mitophagy is also activated in platelets during I/R. A synthetic peptide inhibiting the FUNDC1-LC3 interaction inhibits mitophagy in platelets during hypoxia and prevents preconditioning of the heart against reperfusion injury [224]. Thus, interventions that stimulate mitophagy in non-myocyte populations may also be effective in reducing I/R injury. However, caution must be exercised in utilizing these interventions since many molecules have multiple cellular functions and their effects are context dependent. For example, the effect of Mdivi-1, a chemical inhibitor of Drp1, upon I/R injury is dose and context dependent [148, 222]. Myocardial ischemia without reperfusion activates not only conventional mitophagy but also alternative mitophagy in the heart [152]. Although suppression of conventional mitophagy did not increase myocardial damage after 3 h of ischemia, suppression of alternative mitophagy with Rab9(S179A) significantly exacerbated myocardial damage, suggesting that alternative mitophagy may play a more prominent protective role than conventional mitophagy during prolonged ischemia in the heart [152]. The role of alternative mitophagy during reperfusion remains to be elucidated.
Mitophagy during I/R is also negatively regulated by upstream signaling molecules. Casein kinase 2α (CK2α), a constitutive serine/threonine kinase, suppresses FUNDC1 by phosphorylating it at Ser13 [8, 15]. CK2α protein expression is upregulated during acute cardiac I/R injury and inhibits FUNDC1-dependent mitophagy [228]. Cardiac-specific CK2α knockout in mice abrogated cardiac dysfunction after I/R injury [228]. Similarly, genetic deletion of mammalian STE20-like kinase 1 (Mst1), a potent inhibitor of autophagy, activated FUNDC1-induced mitophagy and reduced cardiomyocyte mitochondrial apoptosis, resulting in protected cardiac function [214]. Thus, endogenous Mst1 also inhibits FUNDC1-mediated mitophagy, leading to increased cardiac injury [214]. Myocardial ischemia induces accumulation of CO2 and bicarbonate, the latter of which, in turn, suppresses mitophagy during reperfusion, resulting in exacerbated myocardial injury [143]. Thus, an alternative strategy for protecting the heart against I/R injury could be to inhibit the negative regulators of mitophagy.
It should be noted that some reports showed that mitophagy can be over-activated during I/R and, thus, downregulation of mitophagy may mitigate myocardial injury under some conditions. For example, downregulation of mitophagy during I/R via overexpression of RNA methylation reading protein YTHDF2 relieves myocardial I/R injury by downregulating BNIP3 mRNA expression in hypoxia–reperfusion (H/R)-treated H9c2 cells or myocardial I/R injury (MIRI) rats [11]. Notch1 suppresses the PTEN-PINK1-Parkin signaling and downregulates mitochondrial fusion/fission and mitochondrial autophagy, conferring protection against I/R injury in cardiomyocytes and a rat I/R injury model [203]. In I/R rat models, exercise-induced parasympathetic nerve function increased myocardial M2 acetylcholine receptor (M2AChR) protein expression and effectively reduced mitophagy, endoplasmic reticulum stress (ERS), and apoptosis [17].
We and others have shown that autophagy can become dysregulated during the late stages of I/R injury and that excessive autophagy induces cell death. One mechanism mediating cell death induced by dysregulated autophagy is autosis, a unique form of cell death characterized by specific morphological and biochemical features [121]. Upregulation of Rubicon during the late stage of I/R blocks autophagic flux, thereby inducing excessive accumulation of autophagosomes and dysfunctional intracellular organelles. It is currently unknown whether dysregulation of mitophagy induces autosis. Since, in theory, mitophagy selectively eliminates already damaged mitochondria, it may not participate in autosis. However, further investigation is required to determine whether excessive degradation of mitochondria can take place during I/R through non-selective destruction due to dysregulated autophagy.
An important caveat is that what we currently know about the role of mitophagy during I/R is based upon studies conducted with rodents. Studies with large mammals and humans remain sparse. In a porcine model of I/R, pre-treatment with chloramphenicol succinate (CAPS) (an autophagy activator) greatly reduced infarct size and exerted cardioprotection compared to saline treatment [153]. In patients undergoing coronary artery bypass or valve surgery requiring cardiopulmonary bypass (CPB), an increase in autophagic flux during surgery, indicated by decreases in p62 from right atrial appendage biopsies, was inversely correlated with mortality post-surgery, suggesting that autophagy is cardioprotective in humans [67]. However, another study showed that autophagy, indicated by changes in autophagy markers in left ventricular biopsies, is not activated by early reperfusion or remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. Thus, other mechanisms besides autophagy may predominantly mediate cardioprotection during myocardial ischemia and reperfusion in humans [44]. Further investigation with large mammals and humans is essential. A schematic representation of the effect of various factors discussed here on mitophagy and their influence on the heart during I/R injury is shown in Fig. 3.

Mitophagy in ischemia–reperfusion injury. During myocardial reperfusion following ischemic injury, increased ROS production causes mPTP opening, thereby leading to cardiomyocyte apoptosis and necrosis. In this condition, accumulated CO2 and bicarbonate inhibit mitophagy and promote myocardial injury. ZIP7 and TRPML1 upregulation during I/R inhibits mitophagy thereby contributing to increased reperfusion injury. Upregulated Mst1 and CK2α or downregulated PLK1 inhibit FUNDC1-mediated mitophagy, resulting in exacerbated cardiac injury. Mst1 may inhibit mitophagy through Beclin 1 phosphorylation and inhibition of autophagosome formation. Drp1-mediated and PGAM5-PINK1-mediated mitophagy protects the heart against I/R injury. MORN4 promotes ROCK2-mediated phosphorylation and activation of MFN2 leading to increased mitochondrial dynamics and mitophagy to enhance cardioprotection during I/R injury. Inhibition of overactivation of mitophagy during I/R injury is cardioprotective in certain conditions. M2AChR signaling inhibits excessive mitophagy. Notch1 inhibits PTEN-PINK1-Parkin signaling-mediated mitophagy. YTHDF2 inhibits BNIP3 mRNA expression and downregulates mitophagy. The figure was created with Biorender.com
Mitophagy during cardiac hypertrophy and heart failure
Transverse aortic constriction (TAC) is commonly used to model pressure overload-induced heart failure in mice. Hemodynamic forces induce not only mechanical stress but also neurohormonal and autocrine/paracrine responses, culminating in cellular stress at the level of organelles, including the endoplasmic reticulum and mitochondria. Pressure overload rapidly induces autophagy in cardiomyocytes, which plays both protective and detrimental roles in the heart depending upon the extent of stress and the timepoint [122, 149, 155]. LC3-dependent classical autophagy was shown to be activated acutely post-TAC and rapidly reverted to the baseline level by 24 h [159]. Mitophagy was subsequently activated from Day 3 to Day 7 post-TAC, as demonstrated using electron microscopy and Mito-Keima, a fluorescent indicator of mitophagy [120]. Thus, although mitophagy is activated by pressure overload, its activation is transient, and, importantly, heart failure develops after mitophagy is inactivated. Treatment with TAT-Beclin 1 [160], a peptide that allows mobilization of endogenous Beclin 1 from the intracellular storage site, after mitophagy was inactivated partially rescued mitophagy and improved heart function in the presence of pressure overload. These results suggest that mitophagy plays a protective role in the heart during pressure overload [159]. Consistently, activation of mitophagy is generally protective during other forms of heart failure [60, 168, 190, 230]. It is possible that selective autophagy, including mitophagy and ERphagy [109], may be more consistently protective against heart failure than general autophagy.
General autophagy, evaluated with GFP-LC3, and mitophagy, evaluated with Mito-Keima, appear to take place with distinct time courses. The difference in time course between general autophagy and mitophagy was also observed in the heart in response to other forms of stress, including ischemia and HFD consumption [65, 105, 117, 119, 120, 152, 179,180,181,182], and was also noted in other cell types [3, 48, 108]. One possibility is that selective forms of autophagy, including mitophagy, are mediated through molecular mechanisms distinct from those of conventional autophagy, like alternative mitophagy. Thus, to reactivate mitophagy in failing hearts, it would be essential to clarify possible time-dependent changes in the underlying mechanism of mitophagy and identify molecular interventions, like TAT-Beclin 1, that effectively reactivate mitophagy. For example, Ulk1/Rab9-mediated alternative mitophagy is activated during pressure overload [120]. Cardiac-specific Ulk1-knockout (Ulk1cKO) mice, in which alternative mitophagy is inhibited, developed cardiac dysfunction as early as 3 days post-TAC, as compared to 2 weeks in wild-type mice [120]. However, rescue of LC3-dependent mitophagy using TAT-Beclin 1 in Ulk1cKO mice improved cardiac function after TAC [120]. This suggests that compensation with a TAT-Beclin 1-activatable form of mitophagy is beneficial under pressure overload conditions. Drp1 was found to translocate from the cytosol to mitochondria and activate mitophagy in the heart under pressure overload [110, 159]. Additionally, cardiac-specific heterozygous Drp1 knockout mice showed exacerbated heart failure in response to pressure overload even after activation of autophagy with TAT-Beclin 1, suggesting that Drp1 is required for mitophagy in the heart in response to pressure overload [110, 159]. In addition, Kruppel-like factor 4 (KLF4), a member of the zinc finger family of transcription factors, directly binds to the promoter of Ulk1/2, molecules implicated in mitophagy during pressure overload stress [93].
Volume overload is observed in the chronic phase of myocardial infarction (MI), which also leads to cardiac hypertrophy, chamber dilation, and heart failure. Parkin has been shown to be essential for mitophagy in cardiomyocytes following MI, with Parkin deficiency leading to exacerbation of cardiac remodeling and heart failure after MI. Cardiomyocytes from Parkin knockout mice exhibit accumulation of damaged mitochondria due to reduced mitophagy [81]. However, PINK1 appears not to be essential for mitophagy after MI since Parkin recruitment to damaged mitochondria was unaffected by the loss of PINK1 [79].
When mitophagy takes place, mitochondrial DNA is degraded by DNase II in lysosomes. DNase II hydrolyzes both exogenous DNA and mitochondrial DNA at low pH. Accumulation of mitochondrial DNA due to downregulation of DNase II can accelerate heart failure in the presence of pressure overload by activating TLR9-dependent inflammation [131]. TLR9 receptor-deficient mice have better cardiac function than wild-type mice under pressure overload conditions [131]. Whether interventions that upregulate mitophagy during pressure overload coordinately regulate degradation of mitochondrial DNA by DNase II or whether a mismatch occurs remains to be clarified. The effect of mitophagy during hypertrophy and heart failure is schematically represented in Fig. 4.

Mitophagy in pressure overload-induced hypertrophy. Transient activation of conventional autophagy during pressure overload occurs one day after TAC, whereas activation of mitophagy occurs thereafter, peaking at around 3 and 7 days after TAC. In particular, mitophagy activation after inactivation of conventional autophagy occurs through an Ulk1-Rab9-dependent alternative mitophagy mechanism. Both autophagy and mitophagy contribute to cardioprotection under pressure overload stress. Pressure overload causes mitochondrial dysfunction and inhibits the action of DNase II in the lysosome, leading to non-degradation of mitochondrial DNA. The accumulated mitochondrial DNA provokes inflammation through TLR9-dependent signaling and leads to heart failure. Drp1-mediated mitophagy is critical for the protection of the heart during pressure overload. TAT-Beclin 1 can activate both general and alternative mitophagy. Parkin has also been shown to enhance autophagy during pressure overload. The figure was created with Biorender.com
Mitophagy in diabetic cardiomyopathy
Obesity and diabetes are prevalent metabolic disorders that commonly induce diastolic dysfunction, hypertrophy, and inflammation in the heart, a condition collectively called diabetic cardiomyopathy [173]. Obesity and diabetes also contribute to the pathogenesis of heart failure with preserved ejection fraction (HFpEF), one of the most common forms of heart failure in developed countries [173]. Diabetic cardiomyopathy is associated with a shift in substrate utilization from glucose to fatty acids due to insulin deficiency. Increased utilization of fatty acids for energy production induces more oxidative stress and ultimately causes mitochondrial damage [74]. Mitochondrial dysfunction also causes an imbalance between fatty acid uptake and catabolism, leading to accumulation of lipid droplets and lipotoxicity [26]. Increasing lines of evidence suggest that mitophagy plays a critical role in the maintenance of mitochondrial function in diabetic hearts [227].
In mouse models of type 1 diabetes mellitus (T1DM), including OVE26 mice and streptozotocin (STZ)-treated mice, general autophagy is inhibited in the heart [37, 201, 204]. High glucose levels directly inhibit autophagy in cardiomyocytes [76]. Despite downregulation of general autophagy, however, mitophagy is stimulated in the T1DM mouse heart [204], accompanied by low levels of PINK1 and Parkin and high levels of the small GTPase Rab9, suggesting that alternative mitophagy is activated in these hearts [204]. Activation of mitophagy in the T1DM mouse heart plays an important role in maintaining mitochondrial function. Whether inactivation of general autophagy and activation of mitophagy are coordinately regulated in T1DM hearts, and, if so, how are currently unknown.
In mouse models of type 2 diabetes (T2DM), autophagy is activated in the heart, but its activation is transient and general autophagy is often inhibited during the chronic phase of T2DM [181]. For example, in mice fed a HFD, activation of general autophagy in the heart peaks at 6 weeks and declines thereafter. Interestingly, however, mitophagy remains activated after general autophagy is inactivated [181]. Thus, in both T1DM and T2DM, although general autophagy is inactivated during the chronic phase, mitophagy is activated even after general autophagy is inhibited. How is general autophagy inactivated during the chronic phase of diabetic cardiomyopathy? One mechanism could be activation of a negative regulator of autophagy, such as Mst1. It is also possible that continuous activation of autophagy leads to depletion of the cellular materials required for autophagosome formation. What is the underlying mechanism of mitophagy in T1DM and T2DM hearts that occurs when general autophagy is downregulated? We and others have shown that mitophagy in the T2DM heart is mediated through Parkin-dependent conventional mechanisms during early stages of HFD consumption, when mechanisms commonly used by conventional autophagy are available [144, 181]. However, alternative mitophagy, utilizing Ulk1-Rab9 mechanisms, predominates after conventional autophagy is inactivated [181]. Importantly, loss of mitophagy function consistently exacerbates mitochondrial dysfunction and cardiomyopathy in mice fed a HFD, during both the acute and chronic phases [179,180,181]. Furthermore, we have shown that mitophagy can be stimulated with TAT-Beclin 1 regardless of the timing of HFD consumption and that the enhancement of mitophagy is protective in T2DM hearts [179,180,181]. These results suggest that mitophagy plays an essential role in maintaining mitochondrial function and cardiac function in T2DM hearts. Mitochondrial dysfunction is commonly observed in the T2DM heart despite activation of mitophagy, indicating that the endogenous level of mitophagy is insufficient to prevent the progression of mitochondrial dysfunction and cardiomyopathy in these hearts. By the same token, the fact that stimulation of mitophagy alleviates cardiomyopathy regardless of the timing of HFD consumption indicates that interventions enhancing the level of mitophagy are effective in alleviating the progression of diabetic cardiomyopathy.
Since T2DM is a chronic disease, it is possible that conventional autophagy has already been downregulated by the time diabetic cardiomyopathy develops in these patients. Thus, it is important to understand the molecular mechanism through which alternative mitophagy is activated during the chronic phase of T2DM. We found that transcription factor binding to IGHM enhancer 3 (TFE3), a member of the transcription factor EB (TFEB) family, is upregulated in the heart following 12 weeks of HFD consumption, concurrent with the time of observation of alternative mitophagy [180]. TFE3 is upregulated in the nuclear fraction of cardiomyocytes in response to HFD consumption and associates with the promoter region of the Rab9 gene [180]. Alternative mitophagy is inhibited during the chronic phase of HFD consumption in mice with cardiac-specific knockout of TFE3, indicating that alternative mitophagy requires TFE3 and transcription of Rab9 [180]. Thus, a better understanding of TFE3 may allow development of novel interventions to improve mitochondrial quality control mechanisms during the chronic phase of T2DM. We also found that Drp1 is phosphorylated at Ser616 during the chronic phase of HFD consumption and participates in stimulation of alternative mitophagy at the MAM [179]. Phosphorylation of Drp1 at Ser616 was also observed in human patients with obesity (Body mass index > 30 kg/m2) [179]. Thus, Drp1 could be another promising target to modulate mitophagy during the chronic phase in the T2DM heart. The effects of mitophagy during diabetic cardiomyopathy is schematically represented in Fig. 5.

Mitophagy in diabetic cardiomyopathy. A Accumulated glucose elevates mitochondrial superoxide levels in type 1 diabetes and leads to myocardial cell death. While general autophagy is inhibited by high glucose levels, a compensatory mechanism simultaneously activates alternative autophagy and mitophagy and is cardioprotective. B In a mouse model of type 2 diabetes, mitophagy is stimulated in the heart through multiple mechanisms in a time-dependent manner. During the acute phase of HFD consumption, mitophagy is activated by Atg7- and Parkin-dependent mechanisms. Drp1 is also involved in mitophagy during the acute phase of HFD consumption by inhibiting Bcl-2/Bcl-xL interaction with Beclin 1, which allows activation of Beclin 1. In the chronic phase of HFD consumption, Drp1 is phosphorylated at Ser616 through unknown mechanisms and localized to ER–mitochondria-associated membrane, where it activates Rab9-mediated alternative mitophagy. These mechanisms may be compensatory for the downregulation of conventional mitophagy. Despite activation of conventional mitophagy in the acute phase and alternative mitophagy during the chronic phase, the level of mitophagy appears to be insufficient, and thus mitochondrial dysfunction in the heart develops during the chronic phase of type 2 diabetes. The figure was created with Biorender.com
Perspectives and conclusions
The mechanisms of mitophagy are complex and require tightly regulated molecular signals. The activation mechanisms appear to differ between various cellular stresses and utilize multiple systems to tag and sequester the damaged mitochondria for degradation in the lysosome. Moreover, mitochondrial degradation can also occur through several mechanisms besides the authentic mechanism of mitophagy, including the endosomal–lysosome pathway, mitochondrial-derived vesicles, and autophagic secretion [178]. Therefore, in-depth studies using multiple approaches are required to understand both the mechanisms and the functional consequences of mitophagy in the heart. We believe that it is of high priority to address the following issues:
-
(1)
Are the mechanisms of mitophagy in cardiomyocytes identical to those in other cell types? Owing to the unique characteristics of cardiomyocyte mitochondria, including the presence of interfibrillar mitochondria, which affect mitochondrial dynamics [140], mitophagy in cardiomyocytes may not be identical to that in other cell types.
-
(2)
What is the specific role of ubiquitin-dependent and -independent (receptor-mediated) mitophagy in cardiomyocytes under various physiological and disease/stress conditions? How does the heart coordinate the use of the multiple mechanisms of mitophagy? What is the molecular mechanism mediating non-canonical mechanisms of mitochondrial degradation, including alternative mitophagy, endosomal mechanisms, microautophagy, and autophagic secretion of mitochondria? Although these mechanisms are often compensatory, they can be a major driver of mitochondrial quality control during chronic disease conditions, where conventional mechanisms of mitophagy are no longer available.
-
(3)
Stress-induced activation of the conventional mechanisms of mitophagy, including the PINK1–Parkin mechanism, is often transient. What is the mechanism that induces rapid inactivation of mitophagy in the heart? Investigating the role of signaling molecules negatively affecting autophagy (Mst1, mTOR, deubiquitinases and TNIP), transcriptional regulators of autophagy (CLOCK, KLF4, ATF4 and TFEB), and other mechanisms promoting rapid exhaustion of the autophagy machinery, such as LC3, is of great interest.
-
(4)
What is the consequence of insufficient mitophagy? What are the roles of the mitochondrial DNA sensing mechanism, including ZBAP1 and cGAS [89], and the innate immune responses/inflammation?
-
(5)
Can excessive mitophagy take place? What is the consequence of excessive mitophagy? Unchecked mitochondrial fission and mitophagy exacerbate doxorubicin-induced cardiomyopathy [13]. Whether the detrimental effect of mitophagy takes place under other cardiac stress conditions needs to be clarified.
-
(6)
What is the role of mitophagy in other cell types during heart failure? Suppression of mitophagy in cardiac fibroblasts alleviates cardiac fibrosis [225]. Thus, mitophagy may not be equally salutary in every cell population in the failing heart.
-
(7)
What is the extent and the functional significance of mitophagy in human hearts during cardiac stress?
To address these issues, it is important to establish a more convenient assay system to accurately evaluate the level of mitophagy in the heart and the various cell types therein under stress conditions. Elucidating the underlying signaling mechanism affecting the level of mitophagy should provide important clues to improve mitophagy and mitochondrial quality control mechanisms under given conditions. Eventually, development of small molecule regulators of mitophagy would have a remarkable impact, since insufficient mitophagy appears to be a common trigger of heart failure in almost every cardiovascular condition.
References
Abeliovich H, Zarei M, Rigbolt KT, Youle RJ, Dengjel J (2013) Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat Commun 4:2789. https://doi.org/10.1038/ncomms3789
Acin-Perez R, Lechuga-Vieco AV, Del Mar MM, Nieto-Arellano R, Torroja C, Sánchez-Cabo F, Jiménez C, González-Guerra A, Carrascoso I, Benincá C, Quiros PM, López-Otín C, Castellano JM, Ruíz-Cabello J, Jiménez-Borreguero LJ, Enríquez JA (2018) Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med. 10:eaan4935. https://doi.org/10.1126/scitranslmed.aan4935
Ahn J, Kim J (2013) Nutritional status and cardiac autophagy. Diabetes Metab J 37:30–35. https://doi.org/10.4093/dmj.2013.37.1.30
Ambivero CT, Cilenti L, Main S, Zervos AS (2014) Mulan E3 ubiquitin ligase interacts with multiple E2 conjugating enzymes and participates in mitophagy by recruiting GABARAP. Cell Signal 26:2921–2929. https://doi.org/10.1016/j.cellsig.2014.09.004
Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, Langer T (2014) The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol 204:919–929. https://doi.org/10.1083/jcb.201308006
Ashrafi G, Schwarz TL (2013) The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20:31–42. https://doi.org/10.1038/cdd.2012.81
Bassiouni W, Valencia R, Mahmud Z, Seubert JM, Schulz R (2023) Matrix metalloproteinase-2 proteolyzes mitofusin-2 and impairs mitochondrial function during myocardial ischemia-reperfusion injury. Basic Res Cardiol 118:29. https://doi.org/10.1007/s00395-023-00999-y
Battistutta R, Lolli G (2011) Structural and functional determinants of protein kinase CK2α: facts and open questions. Mol Cell Biochem 356:67–73. https://doi.org/10.1007/s11010-011-0939-6
Bhujabal Z, Birgisdottir ÅB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S, Kirkin V, Lamark T, Johansen T (2017) FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 18:947–961. https://doi.org/10.15252/embr.201643147
Bravo-San Pedro JM, Kroemer G, Galluzzi L (2017) Autophagy and mitophagy in cardiovascular disease. Circ Res 120:1812–1824. https://doi.org/10.1161/CIRCRESAHA.117.311082
Cai X, Zou P, Hong L, Chen Y, Zhan Y, Liu Y, Shao L (2023) RNA methylation reading protein YTHDF2 relieves myocardial ischemia–reperfusion injury by downregulating BNIP3 via m(6)A modification. Hum Cell. https://doi.org/10.1007/s13577-023-00956-w
Cao Y, Zheng J, Wan H, Sun Y, Fu S, Liu S, He B, Cai G, Cao Y, Huang H, Li Q, Ma Y, Chen S, Wang F, Jiang H (2023) A mitochondrial SCF-FBXL4 ubiquitin E3 ligase complex degrades BNIP3 and NIX to restrain mitophagy and prevent mitochondrial disease. EMBO J 42:e113033. https://doi.org/10.15252/embj.2022113033
Catanzaro MP, Weiner A, Kaminaris A, Li C, Cai F, Zhao F, Kobayashi S, Kobayashi T, Huang Y, Sesaki H, Liang Q (2019) Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J 33:11096–11108. https://doi.org/10.1096/fj.201802663R
Chakrabarti R, Ji W-K, Stan RV, de Juan SJ, Ryan TA, Higgs HN (2018) INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol 217:251–268. https://doi.org/10.1083/jcb.201709111
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, Duan L, Wang X, Liu L, Liu X, Shen Y, Zhu Y, Chen Q (2014) A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54:362–377. https://doi.org/10.1016/j.molcel.2014.02.034
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189–200. https://doi.org/10.1083/jcb.200211046
Chen W, Ma M, Song Y, Hua Y, Jia H, Liu J, Wang Y (2023) Exercise attenuates myocardial ischemia-reperfusion injury by regulating endoplasmic reticulum stress and mitophagy through M(2) acetylcholine receptor. Antioxid Redox Signal. https://doi.org/10.1089/ars.2022.0168
Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, Wang Y, Sehgal SA, Siraj S, Wang X, Wang J, Zhu Y, Chen Q (2017) Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep 18:495–509. https://doi.org/10.15252/embr.201643309
Cho B, Cho HM, Jo Y, Kim HD, Song M, Moon C, Kim H, Kim K, Sesaki H, Rhyu IJ, Kim H, Sun W (2017) Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat Commun 8:15754. https://doi.org/10.1038/ncomms15754
Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu C-H, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515:431–435. https://doi.org/10.1038/nature13909
Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Wang KZQ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE (2013) Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15:1197–1205. https://doi.org/10.1038/ncb2837
Cianfanelli V, De Zio D, Di Bartolomeo S, Nazio F, Strappazzon F, Cecconi F (2015) Ambra1 at a glance. J Cell Sci 128:2003–2008. https://doi.org/10.1242/jcs.168153
Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, Zeviani M (2015) Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 21:845–854. https://doi.org/10.1016/j.cmet.2015.04.016
Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, Vandenberghe W (2014) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23:5227–5242. https://doi.org/10.1093/hmg/ddu244
Coronado M, Fajardo G, Nguyen K, Zhao M, Kooiker K, Jung G, Hu DQ, Reddy S, Sandoval E, Stotland A, Gottlieb RA, Bernstein D (2018) Physiological mitochondrial fragmentation is a normal cardiac adaptation to increased energy demand. Circ Res 122:282–295. https://doi.org/10.1161/CIRCRESAHA.117.310725
D’Souza K, Nzirorera C, Kienesberger PC (2016) Lipid metabolism and signaling in cardiac lipotoxicity. Biochim Biophys Acta 1861:1513–1524. https://doi.org/10.1016/j.bbalip.2016.02.016
Dagar N, Kale A, Steiger S, Anders H-J, Gaikwad AB (2022) Receptor-mediated mitophagy: an emerging therapeutic target in acute kidney injury. Mitochondrion 66:82–91. https://doi.org/10.1016/j.mito.2022.08.004
Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26:207–210. https://doi.org/10.1038/79936
Dhingra A, Jayas R, Afshar P, Guberman M, Maddaford G, Gerstein J, Lieberman B, Nepon H, Margulets V, Dhingra R, Kirshenbaum LA (2017) Ellagic acid antagonizes Bnip3-mediated mitochondrial injury and necrotic cell death of cardiac myocytes. Free Radic Biol Med 112:411–422. https://doi.org/10.1016/j.freeradbiomed.2017.08.010
Di Rita A, Peschiaroli A, Acunzo DP, Strobbe D, Hu Z, Gruber J, Nygaard M, Lambrughi M, Melino G, Papaleo E, Dengjel J, El Alaoui S, Campanella M, Dötsch V, Rogov VV, Strappazzon F, Cecconi F (2018) HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat Commun 9:3755. https://doi.org/10.1038/s41467-018-05722-3
Ding W-X, Ni H-M, Li M, Liao Y, Chen X, Stolz DB, Dorn GW, Yin X-M (2010) Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and parkin-ubiquitin-p62-mediated mitochondrial priming*. J Biol Chem 285:27879–27890. https://doi.org/10.1074/jbc.M110.119537
Ding X, Jiang X, Tian R, Zhao P, Li L, Wang X, Chen S, Zhu Y, Mei M, Bao S, Liu W, Tang Z, Sun Q (2019) RAB2 regulates the formation of autophagosome and autolysosome in mammalian cells. Autophagy 15:1774–1786. https://doi.org/10.1080/15548627.2019.1596478
Drew BG, Ribas V, Le JA, Henstridge DC, Phun J, Zhou Z, Soleymani T, Daraei P, Sitz D, Vergnes L, Wanagat J, Reue K, Febbraio MA, Hevener AL (2014) HSP72 is a mitochondrial stress sensor critical for Parkin action, oxidative metabolism, and insulin sensitivity in skeletal muscle. Diabetes 63:1488–1505. https://doi.org/10.2337/db13-0665
Duan C, Kuang L, Xiang X, Zhang J, Zhu Y, Wu Y, Yan Q, Liu L, Li T (2020) Drp1 regulates mitochondrial dysfunction and dysregulated metabolism in ischemic injury via Clec16a-, BAX-, and GSH- pathways. Cell Death Dis 11:251. https://doi.org/10.1038/s41419-020-2461-9
Durcan TM, Tang MY, Pérusse JR, Dashti EA, Aguileta MA, McLelland G-L, Gros P, Shaler TA, Faubert D, Coulombe B, Fon EA (2014) USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J 33:2473–2491. https://doi.org/10.15252/embj.201489729
Egan DF, Chun MGH, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang C-C, Sheffler DJ, Teriete P, Asara JM, Turk BE, Cosford NDP, Shaw RJ (2015) Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 59:285–297. https://doi.org/10.1016/j.molcel.2015.05.031
Epstein PN, Overbeek PA, Means AR (1989) Calmodulin-induced early-onset diabetes in transgenic mice. Cell 58:1067–1073. https://doi.org/10.1016/0092-8674(89)90505-9
Estaquier J, Vallette F, Vayssiere J-L, Mignotte B (2012) The mitochondrial pathways of apoptosis. Adv Exp Med Biol 942:157–183. https://doi.org/10.1007/978-94-007-2869-1_7
Fan Q, Maejima Y, Wei L, Nakagama S, Shiheido-Watanabe Y, Sasano T (2022) The pathophysiological significance of “Mitochondrial Ejection” from cells. Biomolecules. https://doi.org/10.3390/biom12121770
Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW (2016) Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540:74–79. https://doi.org/10.1038/nature20156
Fu M, St-Pierre P, Shankar J, Wang PTC, Joshi B, Nabi IR (2013) Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 24:1153–1162. https://doi.org/10.1091/mbc.E12-08-0607
Fukuda T, Furukawa K, Maruyama T, Yamashita S-i, Noshiro D, Song C, Ogasawara Y, Okuyama K, Alam JM, Hayatsu M, Saigusa T, Inoue K, Ikeda K, Takai A, Chen L, Lahiri V, Okada Y, Shibata S, Murata K, Klionsky DJ, Noda NN, Kanki T (2023) The mitochondrial intermembrane space protein mitofissin drives mitochondrial fission required for mitophagy. Mol Cell 83:2045-2058.e2049. https://doi.org/10.1016/j.molcel.2023.04.022
Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, Wang G (2015) The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet 24:2528–2538. https://doi.org/10.1093/hmg/ddv017
Gedik N, Thielmann M, Kottenberg E, Peters J, Jakob H, Heusch G, Kleinbongard P (2014) No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PLoS ONE 9:e96567. https://doi.org/10.1371/journal.pone.0096567
Gegg ME, Cooper JM, Chau K-Y, Rojo M, Schapira AHV, Taanman J-W (2010) Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19:4861–4870. https://doi.org/10.1093/hmg/ddq419
Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131. https://doi.org/10.1038/ncb2012
Gersch M, Gladkova C, Schubert AF, Michel MA, Maslen S, Komander D (2017) Mechanism and regulation of the Lys6-selective deubiquitinase USP30. Nat Struct Mol Biol 24:920–930. https://doi.org/10.1038/nsmb.3475
Ghavami S, Cunnington RH, Yeganeh B, Davies JJ, Rattan SG, Bathe K, Kavosh M, Los MJ, Freed DH, Klonisch T, Pierce GN, Halayko AJ, Dixon IM (2012) Autophagy regulates trans fatty acid-mediated apoptosis in primary cardiac myofibroblasts. Biochim Biophys Acta 1823:2274–2286. https://doi.org/10.1016/j.bbamcr.2012.09.008
Gomes LC, Benedetto GD, Scorrano L (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13:589–598. https://doi.org/10.1038/ncb2220
Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW 2nd (2015) Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350:aad2459. https://doi.org/10.1126/science.aad2459
Guerroué FL, Bunker EN, Rosencrans WM, Nguyen JT, Basar MA, Werner A, Chou T-F, Wang C, Youle RJ (2023) TNIP1 inhibits selective autophagy via bipartite interaction with LC3/GABARAP and TAX1BP1. Mol Cell 83:927-941.e928. https://doi.org/10.1016/j.molcel.2023.02.023
Halestrap AP, Richardson AP (2015) The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78:129–141. https://doi.org/10.1016/j.yjmcc.2014.08.018
Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, Li H, Kitsis RN, Dorn GW II, Sadoshima J, Ellisman MH, Gustafsson AB (2017) A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 8:14050. https://doi.org/10.1038/ncomms14050
Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB (2012) Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy *. J Biol Chem 287:19094–19104. https://doi.org/10.1074/jbc.M111.322933
Harper JW, Ordureau A, Heo J-M (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19:93–108. https://doi.org/10.1038/nrm.2017.129
Harris MP, Zhang QJ, Cochran CT, Ponce J, Alexander S, Kronemberger A, Fuqua JD, Zhang Y, Fattal R, Harper T, Murry ML, Grueter CE, Abel ED, Lira VA (2022) Perinatal versus adult loss of ULK1 and ULK2 distinctly influences cardiac autophagy and function. Autophagy 18:2161–2177. https://doi.org/10.1080/15548627.2021.2022289
Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, Martin SE, Youle RJ (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504:291–295. https://doi.org/10.1038/nature12748
Heusch G (2020) Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol 17:773–789. https://doi.org/10.1038/s41569-020-0403-y
Hollville E, Carroll RG, Cullen SP, Martin SJ (2014) Bcl-2 family proteins participate in mitochondrial quality control by regulating Parkin/PINK1-dependent mitophagy. Mol Cell 55:451–466. https://doi.org/10.1016/j.molcel.2014.06.001
Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S (2013) Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun 4:2308. https://doi.org/10.1038/ncomms3308
Hoshino A, Wang WJ, Wada S, McDermott-Roe C, Evans CS, Gosis B, Morley MP, Rathi KS, Li J, Li K, Yang S, McManus MJ, Bowman C, Potluri P, Levin M, Damrauer S, Wallace DC, Holzbaur ELF, Arany Z (2019) The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 575:375–379. https://doi.org/10.1038/s41586-019-1667-4
Huang W, Xie W, Zhong H, Cai S, Huang Q, Liu Y, Zeng Z, Liu Y (2022) Cytosolic p53 inhibits parkin-mediated mitophagy and promotes acute liver injury induced by heat stroke. Front Immunol 13:859231. https://doi.org/10.3389/fimmu.2022.859231
Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K (2013) Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta 1833:503–510. https://doi.org/10.1016/j.bbamcr.2012.11.018
Ikeda G, Santoso MR, Tada Y, Li AM, Vaskova E, Jung JH, O’Brien C, Egan E, Ye J, Yang PC (2021) Mitochondria-rich extracellular vesicles from autologous stem cell-derived cardiomyocytes restore energetics of ischemic myocardium. J Am Coll Cardiol 77:1073–1088. https://doi.org/10.1016/j.jacc.2020.12.060
Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J (2015) Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116:264–278. https://doi.org/10.1161/CIRCRESAHA.116.303356
Jahani-Asl A, Huang E, Irrcher I, Rashidian J, Ishihara N, Lagace DC, Slack RS, Park DS (2015) CDK5 phosphorylates DRP1 and drives mitochondrial defects in NMDA-induced neuronal death. Hum Mol Genet 24:4573–4583. https://doi.org/10.1093/hmg/ddv188
Jahania SM, Sengstock D, Vaitkevicius P, Andres A, Ito BR, Gottlieb RA, Mentzer RM Jr (2013) Activation of the homeostatic intracellular repair response during cardiac surgery. J Am Coll Surg 216:719–726. https://doi.org/10.1016/j.jamcollsurg.2012.12.034. (discussion 726–719)
Javed R, Jain A, Duque T, Hendrix E, Paddar MA, Khan S, Claude-Taupin A, Jia J, Allers L, Wang F, Mudd M, Timmins G, Lidke K, Rusten TE, Akepati PR, He Y, Reggiori F, Eskelinen E-L, Deretic V (2023) Mammalian ATG8 proteins maintain autophagosomal membrane integrity through ESCRTs. EMBO J 42:e112845. https://doi.org/10.15252/embj.2022112845
Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Höke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H (2014) Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J 33:2798–2813. https://doi.org/10.15252/embj.201488658
Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, Geng J, Mao K, Yang Z, Yen WL, Klionsky DJ (2009) A genomic screen for yeast mutants defective in selective mitochondria autophagy. Mol Biol Cell 20:4730–4738. https://doi.org/10.1091/mbc.E09-03-0225
Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF (2015) Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell 57:537–551. https://doi.org/10.1016/j.molcel.2015.01.002
Kataura T, Otten EG, Rabanal-Ruiz Y, Adriaenssens E, Urselli F, Scialo F, Fan L, Smith GR, Dawson WM, Chen X, Yue WW, Bronowska AK, Carroll B, Martens S, Lazarou M, Korolchuk VI (2023) NDP52 acts as a redox sensor in PINK1/Parkin-mediated mitophagy. EMBO J 42:e111372. https://doi.org/10.15252/embj.2022111372
Khaminets A, Behl C, Dikic I (2016) Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol 26:6–16. https://doi.org/10.1016/j.tcb.2015.08.010
Kim J-a, Wei Y, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102:401–414. https://doi.org/10.1161/CIRCRESAHA.107.165472
Kim J, Kundu M, Viollet B, Guan K-L (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13:132–141. https://doi.org/10.1038/ncb2152
Kobayashi S, Xu X, Chen K, Liang Q (2012) Suppression of autophagy is protective in high glucose-induced cardiomyocyte injury. Autophagy 8:577–592. https://doi.org/10.4161/auto.18980
Koncha RR, Ramachandran G, Sepuri NBV, Ramaiah KVA (2021) CCCP-induced mitochondrial dysfunction—characterization and analysis of integrated stress response to cellular signaling and homeostasis. FEBS J 288:5737–5754. https://doi.org/10.1111/febs.15868
Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC (2004) Structural basis of mitochondrial tethering by mitofusin complexes. Science 305:858–862. https://doi.org/10.1126/science.1099793
Kubli DA, Cortez MQ, Moyzis AG, Najor RH, Lee Y, Gustafsson ÅB (2015) PINK1 is dispensable for mitochondrial recruitment of parkin and activation of mitophagy in cardiac myocytes. PLoS ONE 10:e0130707. https://doi.org/10.1371/journal.pone.0130707
Kubli DA, Quinsay MN, Huang C, Lee Y, Gustafsson ÅB (2008) Bnip3 functions as a mitochondrial sensor of oxidative stress during myocardial ischemia and reperfusion. Am J Physiol Heart Circul Physiol 295:H2025–H2031. https://doi.org/10.1152/ajpheart.00552.2008
Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson ÅB (2013) Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction♦. J Biol Chem 288:915–926. https://doi.org/10.1074/jbc.M112.411363
Kuma A, Komatsu M, Mizushima N (2017) Autophagy-monitoring and autophagy-deficient mice. Autophagy 13:1619–1628. https://doi.org/10.1080/15548627.2017.1343770
Kundu M, Lindsten T, Yang C-Y, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB (2008) Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112:1493–1502. https://doi.org/10.1182/blood-2008-02-137398
Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, Kim T, Sussman MA, Gustafsson AB (2019) BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy 15:1182–1198. https://doi.org/10.1080/15548627.2019.1580095
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524:309–314. https://doi.org/10.1038/nature14893
Lee J-Y, Koga H, Kawaguchi Y, Tang W, Wong E, Gao Y-S, Pandey UB, Kaushik S, Tresse E, Lu J, Taylor JP, Cuervo AM, Yao T-P (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 29:969–980. https://doi.org/10.1038/emboj.2009.405
Lee J-Y, Nagano Y, Taylor JP, Lim KL, Yao T-P (2010) Disease-causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J Cell Biol 189:671–679. https://doi.org/10.1083/jcb.201001039
Lee Y, Lee H-Y, Hanna RA, Gustafsson ÅB (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301:H1924-1931. https://doi.org/10.1152/ajpheart.00368.2011
Lei Y, VanPortfliet JJ, Chen YF, Bryant JD, Li Y, Fails D, Torres-Odio S, Ragan KB, Deng J, Mohan A, Wang B, Brahms ON, Yates SD, Spencer M, Tong CW, Bosenberg MW, West LC, Shadel GS, Shutt TE, Upton JW, Li P, West AP (2023) Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell 186(3013–3032):e3022. https://doi.org/10.1016/j.cell.2023.05.039
Lemasters JJ (2014) Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2:749–754. https://doi.org/10.1016/j.redox.2014.06.004
Li J, Qi W, Chen G, Feng D, Liu J, Ma B, Zhou C, Mu C, Zhang W, Chen Q, Zhu Y (2015) Mitochondrial outer-membrane E3 ligase MUL1 ubiquitinates ULK1 and regulates selenite-induced mitophagy. Autophagy 11:1216–1229. https://doi.org/10.1080/15548627.2015.1017180
Li Q, Liu Y, Huang Q, Yi X, Qin F, Zhong Z, Lin L, Yang H, Gong G, Wu W (2022) Hypoxia acclimation protects against heart failure postacute myocardial infarction via Fundc1-mediated mitophagy. Oxid Med Cell Longev 2022:8192552. https://doi.org/10.1155/2022/8192552
Liao X, Zhang R, Lu Y, Prosdocimo DA, Sangwung P, Zhang L, Zhou G, Anand P, Lai L, Leone TC, Fujioka H, Ye F, Rosca MG, Hoppel CL, Schulze PC, Abel ED, Stamler JS, Kelly DP, Jain MK (2015) Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J Clin Invest 125:3461–3476. https://doi.org/10.1172/JCI79964
Lim GG, Lim K-L (2017) Parkin-independent mitophagy-FKBP8 takes the stage. EMBO Rep 18:864–865. https://doi.org/10.15252/embr.201744313
Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14:177–185. https://doi.org/10.1038/ncb2422
Losón OC, Song Z, Chen H, Chan DC (2013) Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell 24:659–667. https://doi.org/10.1091/mbc.E12-10-0721
Lu W, Sun J, Yoon JS, Zhang Y, Zheng L, Murphy E, Mattson MP, Lenardo MJ (2016) Mitochondrial protein PGAM5 regulates mitophagic protection against cell necroptosis. PLoS ONE 11:e0147792. https://doi.org/10.1371/journal.pone.0147792
Ma X, Rawnsley DR, Kovacs A, Islam M, Murphy JT, Zhao C, Kumari M, Foroughi L, Liu H, Qi K, Diwan A, Hyrc K, Evans S, Satoh T, French BA, Margulies KB, Javaheri A, Razani B, Mann DL, Mani K, Diwan A (2022) TRAF2, an innate immune sensor, reciprocally regulates mitophagy and inflammation to maintain cardiac myocyte homeostasis. JACC Basic Transl Sci 7:223–243. https://doi.org/10.1016/j.jacbts.2021.12.002
MacVicar T, Langer T (2016) OPA1 processing in cell death and disease—the long and short of it. J Cell Sci 129:2297–2306. https://doi.org/10.1242/jcs.159186
Mao K, Wang K, Liu X, Klionsky DJ (2013) The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell 26:9–18. https://doi.org/10.1016/j.devcel.2013.05.024
Marinković M, Šprung M, Novak I (2021) Dimerization of mitophagy receptor BNIP3L/NIX is essential for recruitment of autophagic machinery. Autophagy 17:1232–1243. https://doi.org/10.1080/15548627.2020.1755120
Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, Sugita Y, Nomura N, Iwata S, Ohsumi Y, Fujimoto T, Nakatogawa H, Kikkawa M, Noda NN (2020) Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol 27:1185–1193. https://doi.org/10.1038/s41594-020-00518-w
McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57:39–54. https://doi.org/10.1016/j.molcel.2014.11.006
McLelland G-L, Soubannier V, Chen CX, McBride HM, Fon EA (2014) Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33:282–295. https://doi.org/10.1002/embj.201385902
Mellor KM, Reichelt ME, Delbridge LM (2011) Autophagy anomalies in the diabetic myocardium. Autophagy 7:1263–1267. https://doi.org/10.4161/auto.7.10.17148
Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR, Rossignol R, Bénard G (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17:719–730. https://doi.org/10.1016/j.cmet.2013.03.014
Merline R, Rödig H, Zeng-Brouwers J, Poluzzi C, Tascher G, Michaelis J, Lopez-Mosqueda J, Rhiner A, Huber LS, Diehl V, Dikic I, Kögel D, Münch C, Wygrecka M, Schaefer L (2023) A20 binding and inhibitor of nuclear factor kappa B (NF-κB)-1 (ABIN-1): a novel modulator of mitochondrial autophagy. Am J Physiol Cell Physiol 324:C339–C352. https://doi.org/10.1152/ajpcell.00493.2022
Migneault F, Hebert MJ (2021) Autophagy, tissue repair, and fibrosis: a delicate balance. Matrix Biol 100–101:182–196. https://doi.org/10.1016/j.matbio.2021.01.003
Misaka T, Murakawa T, Nishida K, Omori Y, Taneike M, Omiya S, Molenaar C, Uno Y, Yamaguchi O, Takeda J, Shah AM, Otsu K (2018) FKBP8 protects the heart from hemodynamic stress by preventing the accumulation of misfolded proteins and endoplasmic reticulum-associated apoptosis in mice. J Mol Cell Cardiol 114:93–104. https://doi.org/10.1016/j.yjmcc.2017.11.004
Miyamoto S, Brown JH (2016) Drp1 and mitochondrial autophagy lend a helping hand in adaptation to pressure overload. Circulation 133:1225–1227. https://doi.org/10.1161/CIRCULATIONAHA.116.021796
Moore AS, Holzbaur ELF (2016) Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci 113:E3349–E3358. https://doi.org/10.1073/pnas.1523810113
Morales PE, Arias-Durán C, Ávalos-Guajardo Y, Aedo G, Verdejo HE, Parra V, Lavandero S (2020) Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med 71:100822. https://doi.org/10.1016/j.mam.2019.09.006
Moyzis AG, Lally NS, Liang W, Najor RH, Gustafsson AB (2022) Mcl-1 differentially regulates autophagy in response to changes in energy status and mitochondrial damage. Cells. https://doi.org/10.3390/cells11091469
Mozdy AD, McCaffery JM, Shaw JM (2000) Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J Cell Biol 151:367–380. https://doi.org/10.1083/jcb.151.2.367
Müller AJ, Proikas-Cezanne T (2015) Function of human WIPI proteins in autophagosomal rejuvenation of endomembranes? FEBS Lett 589:1546–1551. https://doi.org/10.1016/j.febslet.2015.05.008
Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, Taneike M, Misaka T, Omiya S, Shah AM, Yamamoto A, Nishida K, Ohsumi Y, Okamoto K, Sakata Y, Otsu K (2015) Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6:7527. https://doi.org/10.1038/ncomms8527
Murphy E, Steenbergen C (2008) Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev 88:581–609. https://doi.org/10.1152/physrev.00024.2007
N T, N I, A J, T O, K M (2007) Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. https://doi.org/10.1074/jbc.M607279200
Nah J, Miyamoto S, Sadoshima J (2017) Mitophagy as a protective mechanism against myocardial stress. Compr Physiol 7:1407–1424. https://doi.org/10.1002/cphy.c170005
Nah J, Shirakabe A, Mukai R, Zhai P, Sung EA, Ivessa A, Mizushima W, Nakada Y, Saito T, Hu C, Jung YK, Sadoshima J (2022) Ulk1-dependent alternative mitophagy plays a protective role during pressure overload in the heart. Cardiovasc Res 118:2638–2651. https://doi.org/10.1093/cvr/cvac003
Nah J, Zhai P, Huang CY, Fernandez AF, Mareedu S, Levine B, Sadoshima J (2020) Upregulation of Rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest 130:2978–2991. https://doi.org/10.1172/JCI132366
Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13:619–624. https://doi.org/10.1038/nm1574
Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ (2010) p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6:1090–1106. https://doi.org/10.4161/auto.6.8.13426
Nguyen TN, Padman BS, Usher J, Oorschot V, Ramm G, Lazarou M (2016) Atg8 family LC3/GABARAP proteins are crucial for autophagosome-lysosome fusion but not autophagosome formation during PINK1/Parkin mitophagy and starvation. J Cell Biol 215:857–874. https://doi.org/10.1083/jcb.201607039
Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, Rubio-Ponce A, Li JL, Balachander A, Quintana JA, Martinez-de-Mena R, Castejon-Vega B, Pun-Garcia A, Traves PG, Bonzon-Kulichenko E, Garcia-Marques F, Cusso L, Noelia AG, Gonzalez-Guerra A, Roche-Molina M, Martin-Salamanca S, Crainiciuc G, Guzman G, Larrazabal J, Herrero-Galan E, Alegre-Cebollada J, Lemke G, Rothlin CV, Jimenez-Borreguero LJ, Reyes G, Castrillo A, Desco M, Munoz-Canoves P, Ibanez B, Torres M, Ng LG, Priori SG, Bueno H, Vazquez J, Cordero MD, Bernal JA, Enriquez JA, Hidalgo A (2020) A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183:94-109e123. https://doi.org/10.1016/j.cell.2020.08.031
Nicolas-Avila JA, Pena-Couso L, Munoz-Canoves P, Hidalgo A (2022) Macrophages, metabolism and heterophagy in the heart. Circ Res 130:418–431. https://doi.org/10.1161/CIRCRESAHA.121.319812
Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S (2009) Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461:654–658. https://doi.org/10.1038/nature08455
Nishimura T, Tooze SA (2020) Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov 6:32. https://doi.org/10.1038/s41421-020-0161-3
Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I (2010) Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11:45–51. https://doi.org/10.1038/embor.2009.256
Ohsumi Y (2001) Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2:211–216. https://doi.org/10.1038/35056522
Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, Akira S, Yamamoto A, Komuro I, Otsu K (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485:251–255. https://doi.org/10.1038/nature10992
Onishi M, Yamano K, Sato M, Matsuda N, Okamoto K (2021) Molecular mechanisms and physiological functions of mitophagy. EMBO J 40:e104705. https://doi.org/10.15252/embj.2020104705
Ordureau A, Sarraf SA, Duda DM, Heo J-M, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW (2014) Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56:360–375. https://doi.org/10.1016/j.molcel.2014.09.007
Orvedahl A, Sumpter R, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby-Phelps K, Xavier RJ, Xie Y, Levine B (2011) Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480:113–117. https://doi.org/10.1038/nature10546
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20:1013–1022. https://doi.org/10.1038/s41556-018-0176-2
Park S, Choi S-G, Yoo S-M, Son JH, Jung Y-K (2014) Choline dehydrogenase interacts with SQSTM1/p62 to recruit LC3 and stimulate mitophagy. Autophagy 10:1906–1920. https://doi.org/10.4161/auto.32177
Parzych KR, Klionsky DJ (2014) An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal 20:460–473. https://doi.org/10.1089/ars.2013.5371
Peng SZ, Chen XH, Chen SJ, Zhang J, Wang CY, Liu WR, Zhang D, Su Y, Zhang XK (2021) Phase separation of Nur77 mediates celastrol-induced mitophagy by promoting the liquidity of p62/SQSTM1 condensates. Nat Commun. https://doi.org/10.1038/s41467-021-26295-8
Pickles S, Vigié P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28:R170–R185. https://doi.org/10.1016/j.cub.2018.01.004
Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F (2013) Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol 4:102. https://doi.org/10.3389/fphys.2013.00102
Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L (2010) The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 5:e10054. https://doi.org/10.1371/journal.pone.0010054
Praefcke GJK, McMahon HT (2004) The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol 5:133–147. https://doi.org/10.1038/nrm1313
Queliconi BB, Kowaltowski AJ, Gottlieb RA (2016) Bicarbonate increases ischemia–reperfusion damage by inhibiting mitophagy. PLoS ONE 11:e0167678. https://doi.org/10.1371/journal.pone.0167678
Rabinovich-Nikitin I, Dhingra R, Kirshenbaum LA (2019) Activation of mitophagy in high-fat diet-induced diabetic cardiomyopathy. Circ Res 124:1288–1290. https://doi.org/10.1161/CIRCRESAHA.119.314967
Rabinovich-Nikitin I, Rasouli M, Reitz CJ, Posen I, Margulets V, Dhingra R, Khatua TN, Thliveris JA, Martino TA, Kirshenbaum LA (2021) Mitochondrial autophagy and cell survival is regulated by the circadian Clock gene in cardiac myocytes during ischemic stress. Autophagy 17:3794–3812. https://doi.org/10.1080/15548627.2021.1938913
Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH (2000) BNIP3 Heterodimerizes with Bcl-2/Bcl-XL and induces cell death independent of a Bcl-2 Homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites*. J Biol Chem 275:1439–1448. https://doi.org/10.1074/jbc.275.2.1439
Rogov VV, Suzuki H, Marinković M, Lang V, Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S, Hamacher-Brady A, Dötsch V, Dikic I, Brady NR, Novak I (2017) Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep 7:1131. https://doi.org/10.1038/s41598-017-01258-6
Rosdah AA, Abbott BM, Langendorf CG, Deng Y, Truong JQ, Waddell HMM, Ling NXY, Smiles WJ, Delbridge LMD, Liu GS, Oakhill JS, Lim SY, Holien JK (2022) A novel small molecule inhibitor of human Drp1. Sci Rep 12:21531. https://doi.org/10.1038/s41598-022-25464-z
Rothermel BA, Hill JA (2008) Autophagy in load-induced heart disease. Circ Res 103:1363–1369. https://doi.org/10.1161/CIRCRESAHA.108.186551
Sadoshima J (2022) TRAF2 mediates physiological mitophagy. JACC Basic Transl Sci 7:244–246. https://doi.org/10.1016/j.jacbts.2021.11.012
Sagar S, Faizan MI, Chaudhary N, Singh V, Singh P, Gheware A, Sharma K, Azmi I, Singh VP, Kharya G, Mabalirajan U, Agrawal A, Ahmad T, Sinha Roy S (2023) Obesity impairs cardiolipin-dependent mitophagy and therapeutic intercellular mitochondrial transfer ability of mesenchymal stem cells. Cell Death Dis 14:1–19. https://doi.org/10.1038/s41419-023-05810-3
Saito T, Nah J, Oka S-i, Mukai R, Monden Y, Maejima Y, Ikeda Y, Sciarretta S, Liu T, Li H, Baljinnyam E, Fraidenraich D, Fritzky L, Zhai P, Ichinose S, Isobe M, Hsu C-P, Kundu M, Sadoshima J (2019) An alternative mitophagy pathway mediated by Rab9 protects the heart against ischemia. J Clin Invest 129:802–819. https://doi.org/10.1172/JCI122035
Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM Jr, Gottlieb RA, Przyklenk K (2010) Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia–reperfusion injury. Circulation 122:S179-184. https://doi.org/10.1161/CIRCULATIONAHA.109.928242
Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454:232–235. https://doi.org/10.1038/nature07006
Sciarretta S, Maejima Y, Zablocki D, Sadoshima J (2018) The role of autophagy in the heart. Annu Rev Physiol 80:1–26. https://doi.org/10.1146/annurev-physiol-021317-121427
Sekine S, Youle RJ (2018) PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 16:2. https://doi.org/10.1186/s12915-017-0470-7
Shang L, Chen S, Du F, Li S, Zhao L, Wang X (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc Natl Acad Sci 108:4788–4793. https://doi.org/10.1073/pnas.1100844108
Shen Z, Li Y, Gasparski AN, Abeliovich H, Greenberg ML (2017) Cardiolipin regulates mitophagy through the protein kinase C pathway. J Biol Chem 292:2916–2923. https://doi.org/10.1074/jbc.M116.753574
Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu C-P, Nomura M, Egashira K, Levine B, Sadoshima J (2016) Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation 133:1249–1263. https://doi.org/10.1161/CIRCULATIONAHA.115.020502
Shoji-Kawata S, Sumpter R, Leveno M, Campbell GR, Zou Z, Kinch L, Wilkins AD, Sun Q, Pallauf K, MacDuff D, Huerta C, Virgin HW, Helms JB, Eerland R, Tooze SA, Xavier R, Lenschow DJ, Yamamoto A, King D, Lichtarge O, Grishin NV, Spector SA, Kaloyanova DV, Levine B (2013) Identification of a candidate therapeutic autophagy-inducing peptide. Nature 494:201–206. https://doi.org/10.1038/nature11866
Shu L, Hu C, Xu M, Yu J, He H, Lin J, Sha H, Lu B, Engelender S, Guan M, Song Z (2021) ATAD3B is a mitophagy receptor mediating clearance of oxidative stress-induced damaged mitochondrial DNA. EMBO J 40:e106283. https://doi.org/10.15252/embj.2020106283
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 87:1157–1180. https://doi.org/10.1007/s00204-013-1034-4
Soh JEC, Shimizu A, Molla MR, Zankov DP, Nguyen LKC, Khan MR, Tesega WW, Chen S, Tojo M, Ito Y, Sato A, Hitosugi M, Miyagawa S, Ogita H (2023) RhoA rescues cardiac senescence by regulating Parkin-mediated mitophagy. J Biol Chem. https://doi.org/10.1016/j.jbc.2023.102993
Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd (2015) Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21:273–286. https://doi.org/10.1016/j.cmet.2014.12.011
Soubannier V, McLelland G-L, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM (2012) A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22:135–141. https://doi.org/10.1016/j.cub.2011.11.057
Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, Cecconi F (2015) AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 22:419–432. https://doi.org/10.1038/cdd.2014.139
Sui Z, Wang MM, Xing Y, Qi J, Wang W (2022) Targeting MCOLN1/TRPML1 channels to protect against ischemia-reperfusion injury by restoring the inhibited autophagic flux in cardiomyocytes. Autophagy 18:3053–3055. https://doi.org/10.1080/15548627.2022.2072657
Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, Xie Y, Carlson D, Rothermel BA, Sun Y, Levine B, Hill JA, Wolf SE, Minei JP, Zang QS (2018) Beclin-1-dependent autophagy protects the heart during sepsis. Circulation 138:2247–2262. https://doi.org/10.1161/CIRCULATIONAHA.117.032821
Szargel R, Shani V, Abd Elghani F, Mekies LN, Liani E, Rott R, Engelender S (2016) The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet 25:3476–3490. https://doi.org/10.1093/hmg/ddw189
Szulik MW, Valdez S, Walsh M, Davis K, Bia R, Horiuchi E, O’Very S, Laxman AK, Sandaklie-Nicolova L, Eberhardt DR, Durrant JR, Sheikh H, Hickenlooper S, Creed M, Brady C, Miller M, Wang L, Garcia-Llana J, Tracy C, Drakos SG, Funai K, Chaudhuri D, Boudina S, Franklin S (2023) SMYD1a protects the heart from ischemic injury by regulating OPA1-mediated cristae remodeling and supercomplex formation. Basic Res Cardiol. https://doi.org/10.1007/s00395-023-00991-6
Tahrir FG, Knezevic T, Gupta MK, Gordon J, Cheung JY, Feldman AM, Khalili K (2017) Evidence for the role of BAG3 in mitochondrial quality control in cardiomyocytes. J Cell Physiol 232:797–805. https://doi.org/10.1002/jcp.25476
Tan HWS, Lu G, Dong H, Cho YL, Natalia A, Wang L, Chan C, Kappei D, Taneja R, Ling SC, Shao H, Tsai SY, Ding WX, Shen HM (2022) A degradative to secretory autophagy switch mediates mitochondria clearance in the absence of the mATG8-conjugation machinery. Nat Commun 13:3720. https://doi.org/10.1038/s41467-022-31213-7
Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L (2020) Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat Rev Cardiol 17:585–607. https://doi.org/10.1038/s41569-020-0339-2
Tanaka A, Cleland MM, Xu S, Narendra DP, Suen D-F, Karbowski M, Youle RJ (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191:1367–1380. https://doi.org/10.1083/jcb.201007013
Tang X, Zhao S, Liu J, Liu X, Sha X, Huang C, Hu L, Sun S, Gao Y, Chen H, Zhang Z, Wang D, Gu Y, Chen S, Wang L, Gu A, Chen F, Pu J, Chen X, Yu B, Xie L, Huang Z, Han Y, Ji Y (2023) Mitochondrial GSNOR alleviates cardiac dysfunction via ANT1 denitrosylation. Circ Res 133:220–236. https://doi.org/10.1161/CIRCRESAHA.123.322654
Tanida I, Ueno T, Kominami E (2004) LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol 36:2503–2518. https://doi.org/10.1016/j.biocel.2004.05.009
Tian X, Teng J, Chen J (2021) New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy 17:2680–2688. https://doi.org/10.1080/15548627.2020.1823124
Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pépin G, Germain M (2021) Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun 12:1971. https://doi.org/10.1038/s41467-021-21984-w
Tong M, Mukai R, Mareedu S, Zhai P, Oka S-i, Huang C-Y, Hsu C-P, Yousufzai FAK, Fritzky L, Mizushima W, Babu GJ, Sadoshima J (2023) Distinct roles of DRP1 in conventional and alternative mitophagy in obesity cardiomyopathy. Circ Res 133:6–21. https://doi.org/10.1161/CIRCRESAHA.123.322512
Tong M, Saito T, Zhai P, Oka S-i, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J (2021) Alternative mitophagy protects the heart against obesity-associated cardiomyopathy. Circ Res 129:1105–1121. https://doi.org/10.1161/CIRCRESAHA.121.319377
Tong M, Saito T, Zhai P, Oka S-i, Mizushima W, Nakamura M, Ikeda S, Shirakabe A, Sadoshima J (2019) Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res 124:1360–1371. https://doi.org/10.1161/CIRCRESAHA.118.314607
Tong M, Zablocki D, Sadoshima J (2020) The role of Drp1 in mitophagy and cell death in the heart. J Mol Cell Cardiol 142:138–145. https://doi.org/10.1016/j.yjmcc.2020.04.015
Torii S, Yamaguchi H, Nakanishi A, Arakawa S, Honda S, Moriwaki K, Nakano H, Shimizu S (2020) Identification of a phosphorylation site on Ulk1 required for genotoxic stress-induced alternative autophagy. Nat Commun 11:1754. https://doi.org/10.1038/s41467-020-15577-2
Turco E, Witt M, Abert C, Bock-Bierbaum T, Su MY, Trapannone R, Sztacho M, Danieli A, Shi X, Zaffagnini G, Gamper A, Schuschnig M, Fracchiolla D, Bernklau D, Romanov J, Hartl M, Hurley JH, Daumke O, Martens S (2019) FIP200 claw domain binding to p62 promotes autophagosome formation at ubiquitin condensates. Mol Cell 74(330–346):e311. https://doi.org/10.1016/j.molcel.2019.01.035
Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (2008) Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27:433–446. https://doi.org/10.1038/sj.emboj.7601963
Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, Menabo R, Costa V, Civiletto G, Pesce P, Viscomi C, Zeviani M, Di Lisa F, Mongillo M, Sandri M, Scorrano L (2015) The OPA1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab 21:834–844. https://doi.org/10.1016/j.cmet.2015.05.007
Villa E, Proïcs E, Rubio-Patiño C, Obba S, Zunino B, Bossowski JP, Rozier RM, Chiche J, Mondragón L, Riley JS, Marchetti S, Verhoeyen E, Tait SWG, Ricci J-E (2017) Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep 20:2846–2859. https://doi.org/10.1016/j.celrep.2017.08.087
Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, Rupérez FJ, Barbas C, Ibañez B, Langer T (2015) Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science 350:aad0116. https://doi.org/10.1126/science.aad0116
Wai T, Langer T (2016) Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab 27:105–117. https://doi.org/10.1016/j.tem.2015.12.001
Wang B, Nie J, Wu L, Hu Y, Wen Z, Dong L, Zou MH, Chen C, Wang DW (2018) AMPKalpha2 protects against the development of heart failure by enhancing mitophagy via PINK1 phosphorylation. Circ Res 122:712–729. https://doi.org/10.1161/CIRCRESAHA.117.312317
Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V, Kirshenbaum LA (2013) p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 62:70–77. https://doi.org/10.1161/HYPERTENSIONAHA.113.01028
Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL (2011) PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147:893–906. https://doi.org/10.1016/j.cell.2011.10.018
Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT, Kim PK, McQuibban GA (2015) Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11:595–606. https://doi.org/10.1080/15548627.2015.1034408
Wei Y, Chiang W-C, Sumpter R, Mishra P, Levine B (2017) Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell 168:224-238.e210. https://doi.org/10.1016/j.cell.2016.11.042
Wen X, Klionsky DJ (2020) Phosphorylation of ULK1 serine 746 dictates ATG5-independent autophagy. Autophagy 16:1557–1558. https://doi.org/10.1080/15548627.2020.1780844
Westermann B (2010) Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol 11:872–884. https://doi.org/10.1038/nrm3013
Wu H, Xue D, Chen G, Han Z, Huang L, Zhu C, Wang X, Jin H, Wang J, Zhu Y, Liu L, Chen Q (2014) The BCL2L1 and PGAM5 axis defines hypoxia-induced receptor-mediated mitophagy. Autophagy 10:1712–1725. https://doi.org/10.4161/auto.29568
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhao B, Feng D (2014) ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15:566–575. https://doi.org/10.1002/embr.201438501
X W, W J, Y Y, T G, J H, Z T, R Z (2014) RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat Immunol 15. https://doi.org/10.1038/ni.3015.
Xiao Y, Zhou Y, Lu Y, Zhou K, Cai W (2018) PHB2 interacts with LC3 and SQSTM1 is required for bile acids-induced mitophagy in cholestatic liver. Cell Death Dis 9:160. https://doi.org/10.1038/s41419-017-0228-8
Xie Z, Lau K, Eby B, Lozano P, He C, Pennington B, Li H, Rathi S, Dong Y, Tian R, Kem D, Zou M-H (2011) Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes 60:1770–1778. https://doi.org/10.2337/db10-0351
Xing Y, Sui Z, Liu Y, Wang MM, Wei X, Lu Q, Wang X, Liu N, Lu C, Chen R, Wu M, Wang Y, Zhao YH, Guo F, Cao JL, Qi J, Wang W (2022) Blunting TRPML1 channels protects myocardial ischemia/reperfusion injury by restoring impaired cardiomyocyte autophagy. Basic Res Cardiol 117:20. https://doi.org/10.1007/s00395-022-00930-x
Xu Q, Liu S, Gong Q, Zhu R, Liu J, Wu Q, Zhou X (2022) Notch1 protects against ischemic–reperfusion injury by suppressing PTEN-Pink1-mediated mitochondrial dysfunction and mitophagy. Cells. https://doi.org/10.3390/cells12010137
Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, Gulick J, Yue Z, Robbins J, Epstein PN, Liang Q (2013) Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem 288:18077–18092. https://doi.org/10.1074/jbc.M113.474650
Xu Y, Shen J, Ran Z (2020) Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 16:3–17. https://doi.org/10.1080/15548627.2019.1603547
Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, Kato T, Araki Y, Huganir RL, Dawson TM, Yanagawa T, Okamoto K, Iijima M, Sesaki H (2018) Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab 28:588-604.e585. https://doi.org/10.1016/j.cmet.2018.06.014
Yamaguchi H, Honda S, Torii S, Shimizu K, Katoh K, Miyake K, Miyake N, Fujikake N, Sakurai HT, Arakawa S, Shimizu S (2020) Wipi3 is essential for alternative autophagy and its loss causes neurodegeneration. Nat Commun 11:5311. https://doi.org/10.1038/s41467-020-18892-w
Yamashita SI, Jin X, Furukawa K, Hamasaki M, Nezu A, Otera H, Saigusa T, Yoshimori T, Sakai Y, Mihara K, Kanki T (2016) Mitochondrial division occurs concurrently with autophagosome formation but independently of Drp1 during mitophagy. J Cell Biol 215:649–665. https://doi.org/10.1083/jcb.201605093
Yogalingam G, Hwang S, Ferreira JCB, Mochly-Rosen D (2013) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cδ (PKCδ) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem 288:18947–18960. https://doi.org/10.1074/jbc.M113.466870
Yoon Y, Krueger EW, Oswald BJ, McNiven MA (2003) The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol Cell Biol 23:5409–5420. https://doi.org/10.1128/MCB.23.15.5409-5420.2003
Youle RJ, van der Bliek AM (2012) Mitochondrial fission, fusion, and stress. Science 337:1062–1065. https://doi.org/10.1126/science.1219855
Yu R, Jin S-B, Lendahl U, Nistér M, Zhao J (2019) Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J 38:e99748. https://doi.org/10.15252/embj.201899748
Yu S, Du M, Yin A, Mai Z, Wang Y, Zhao M, Wang X, Chen T (2020) Bcl-xL inhibits PINK1/Parkin-dependent mitophagy by preventing mitochondrial Parkin accumulation. Int J Biochem Cell Biol 122:105720. https://doi.org/10.1016/j.biocel.2020.105720
Yu W, Xu M, Zhang T, Zhang Q, Zou C (2019) Mst1 promotes cardiac ischemia-reperfusion injury by inhibiting the ERK-CREB pathway and repressing FUNDC1-mediated mitophagy. J Physiol Sci 69:113–127. https://doi.org/10.1007/s12576-018-0627-3
Yun J, Puri R, Yang H, Lizzio MA, Wu C, Sheng Z-H, Guo M (2014) MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. Elife 3:e01958. https://doi.org/10.7554/eLife.01958
Zachari M, Ganley Ian G (2017) The mammalian ULK1 complex and autophagy initiation. Essays Biochem 61:585–596. https://doi.org/10.1042/EBC20170021
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283:10892–10903. https://doi.org/10.1074/jbc.M800102200
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia*. J Biol Chem 283:10892–10903. https://doi.org/10.1074/jbc.M800102200
Zhang H, Wang P, Bisetto S, Yoon Y, Chen Q, Sheu SS, Wang W (2017) A novel fission-independent role of dynamin-related protein 1 in cardiac mitochondrial respiration. Cardiovasc Res 113:160–170. https://doi.org/10.1093/cvr/cvw212
Zhang H, Yang N, He H, Chai J, Cheng X, Zhao H, Zhou D, Teng T, Kong X, Yang Q, Xu Z (2021) The zinc transporter ZIP7 (Slc39a7) controls myocardial reperfusion injury by regulating mitophagy. Basic Res Cardiol 116:54. https://doi.org/10.1007/s00395-021-00894-4
Zhang J, Zeng W, Han Y, Lee WR, Liou J, Jiang Y (2023) Lysosomal LAMP proteins regulate lysosomal pH by direct inhibition of the TMEM175 channel. Mol Cell. https://doi.org/10.1016/j.molcel.2023.06.004
Zhang N, Wang S, Li Y, Che L, Zhao Q (2013) A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci Lett 535:104–109. https://doi.org/10.1016/j.neulet.2012.12.049
Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, Billiar TR, Tao WA, Zhang Z (2016) BNIP3 protein suppresses PINK1 Kinase proteolytic cleavage to promote mitophagy*. J Biol Chem 291:21616–21629. https://doi.org/10.1074/jbc.M116.733410
Zhang W, Ren H, Xu C, Zhu C, Wu H, Liu D, Wang J, Liu L, Li W, Ma Q, Du L, Zheng M, Zhang C, Liu J, Chen Q (2016) Hypoxic mitophagy regulates mitochondrial quality and platelet activation and determines severity of I/R heart injury. Elife 5:e21407. https://doi.org/10.7554/eLife.21407
Zhang Y, Wang Z, Lan D, Zhao J, Wang L, Shao X, Wang D, Wu K, Sun M, Huang X, Yan M, Liang H, Rong X, Diao H, Guo J (2022) MicroRNA-24-3p alleviates cardiac fibrosis by suppressing cardiac fibroblasts mitophagy via downregulating PHB2. Pharmacol Res 177:106124. https://doi.org/10.1016/j.phrs.2022.106124
Zheng J, Cao Y, Yang J, Jiang H (2022) UBXD8 mediates mitochondria-associated degradation to restrain apoptosis and mitophagy. EMBO Rep 23:e54859. https://doi.org/10.15252/embr.202254859
Zhi FM, Zhang Q, Liu L, Chang X, Xu HT (2023) Novel insights into the role of mitochondria in diabetic cardiomyopathy: molecular mechanisms and potential treatments. Cell Stress Chaperon. https://doi.org/10.1007/s12192-023-01361-w
Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y (2018) Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ 25:1080–1093. https://doi.org/10.1038/s41418-018-0086-7
Zhou J, Liu H, Zhang T, Wang Z, Zhang J, Lu Y, Li Z, Kong W, Zhao J (2023) MORN4 protects cardiomyocytes against ischemic injury via MFN2-mediated mitochondrial dynamics and mitophagy. Free Radic Biol Med 196:156–170. https://doi.org/10.1016/j.freeradbiomed.2023.01.016
Zhu P, Wan K, Yin M, Hu P, Que Y, Zhou X, Zhang L, Li T, Du Y, Xu G, Fang X (2021) RIPK3 induces cardiomyocyte necroptosis via inhibition of AMPK-parkin-mitophagy in cardiac remodelling after myocardial infarction. Oxid Med Cell Longev 2021:6635955. https://doi.org/10.1155/2021/6635955
Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A, Brady NR (2013) Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis*. J Biol Chem 288:1099–1113. https://doi.org/10.1074/jbc.M112.399345
Acknowledgements
We thank Jihoon Nah (Chungbuk National University) for critical reading. This study was supported in part by U.S. Public Health Service grants HL67724, HL91469, HL102738, HL112330, HL138720, HL144626, HL150881, and AG23039 (J.S.). This work was also supported by an American Heart Association Predoctoral Fellowship Award 915784 (E-A.S.), Merit Award 20 Merit 35120374 (J.S.), and by the Foundation Leducq Transatlantic Network of Excellence 15CVD04 (J.S.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no coflict of interests.
Additional information
This article is part of the topical collection “Mitochondria at the heart of cardioprotection”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Titus, A.S., Sung, EA., Zablocki, D. et al. Mitophagy for cardioprotection. Basic Res Cardiol 118, 42 (2023). https://doi.org/10.1007/s00395-023-01009-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-023-01009-x