
Abstract
Soon after birth, the regenerative capacity of the mammalian heart is lost, cardiomyocytes withdraw from the cell cycle and demonstrate a minimal proliferation rate. Despite improved treatment and reperfusion strategies, the uncompensated cardiomyocyte loss during injury and disease results in cardiac remodeling and subsequent heart failure. The promising field of regenerative medicine aims to restore both the structure and function of damaged tissue through modulation of cellular processes and regulatory mechanisms involved in cardiac cell cycle arrest to boost cardiomyocyte proliferation. Non-coding RNAs (ncRNAs), such as microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) are functional RNA molecules with no protein-coding function that have been reported to engage in cardiac regeneration and repair. In this review, we summarize the current understanding of both the biological functions and molecular mechanisms of ncRNAs involved in cardiomyocyte proliferation. Furthermore, we discuss their impact on the structure and contractile function of the heart in health and disease and their application for therapeutic interventions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heart failure is a burgeoning health problem worldwide. Often a consequence of cardiovascular events, heart failure has become a major cause of morbidity and mortality and its incidence is expected to grow further in the future [137]. Upon cardiac injury, the adult mammalian heart does not regenerate sufficiently to compensate for the lost myocardium, resulting in a cardiomyocyte deficiency, and hence a lack of functional recovery of the infarcted or failing heart. Therefore, there is an unmet need to device effective regenerative strategies to induce cardiomyocyte renewal and replace the post-infarct fibrotic tissue with contractile muscle cells.
For many years, the human heart has been considered a post-mitotic and fully differentiated organ; this paradigm, however, has been challenged since various studies have reported a residual capacity of cardiomyocytes to proliferate after birth [8, 120]. This process has been shown to occur at a low rate and it progressively and continuously declines with age so much so that in a 60 years old human heart it does not exceed 0.5% [8]. On the contrary, other species such as zebrafish and newt can completely regenerate their hearts after myocardial injury by activating cardiomyocyte proliferation [4, 71]. Similarly, neonatal mammals such as rodents and pigs show some degree of regenerative capacity after injury which is lost during the first week after birth [54, 112, 166]. A human case report has recently suggested that a similar capability could be present in humans and demonstrated complete recovery of a 1-day-old newborn following a severe cardiac injury caused by myocardial infarction (MI) [55]. Taken together, these examples highlight the fact that cardiomyocytes harbor endogenous proliferative capabilities, suggesting that these mechanisms could be reactivated to boost proliferation in adult human cardiac tissue, thus providing new possibilities to be exploited for novel therapeutic strategies.
Non-coding RNA (ncRNA) account for the vast majority of the transcribed genome [28]. Unlike messenger RNA (mRNA), these sequences do not encode proteins, but rather act as regulators in the epigenetic, post-transcriptional, and translational coordination of gene expression [50]. In recent years, the reduced cost of next-generation sequencing technologies has led to an explosion of newly identified ncRNAs, and accumulating evidence has provided a more comprehensive knowledge of their biological functions [35]. Interestingly, a large number of ncRNAs seem to be cardiac specific [102], and different classes of ncRNAs have been linked to cardiac regeneration [108]. Gain- and loss-of-function approaches demonstrated an essential role of various ncRNAs in regulating the proliferation of cardiomyocytes in both in vivo and in vitro models. These modulations were sufficient to promote cardiac regenerative and reparative responses, which were observed at the functional, structural, and cellular levels [41, 132]. Among the different classes of ncRNA, microRNAs (miRNA) were the first to be identified as early as 1993 [79], thus they had a head start in regenerative research. So far, numerous miRNAs have been established for their role and mechanism in cardiac regeneration and their capability to promote cardiomyocyte proliferation10. On the contrary, long non-coding RNA (lncRNA) and circular RNA (circRNA) have only recently emerged as potential targets to boost cardiomyocyte proliferation. The aim of this review is to provide an overview of the current understanding of the roles that miRNAs, lncRNAs, and circRNAs play in the process of cardiomyocyte proliferation, as a means of cardiac regeneration. We discuss their potential to induce division of cardiomyocytes both in vitro and in vivo, and to attenuate tissue remodeling in terms of function and structure following cardiac injury. Finally, current limitations and future directions for successful translational research are discussed.
Cardiomyocyte proliferation and heart regeneration
The mammalian heart responds to myocardial infarction and consecutive cell death by the progressive replacement of the injured myocardium with a meshwork of extracellular matrix and proliferating cells that forms a scar. This wound healing process involves an inflammatory response [48] and the simultaneous activation of local stromal cells [13]. The fibrotic scar has been shown to be essential to safeguard the cardiac wall and prevent the fatal rupture of the dead myocardium [45]. Yet, the progressive accumulation of extracellular matrix and associated tissue remodeling have profound effects on the cardiac function and structure [13]. The remaining cardiomyocytes experience abnormal mechanical stress and develop compensatory hypertrophy [77] and reorganization of their sarcomere structure [38]. Additionally, cardiomyocyte remodeling results in impaired intracellular calcium handling and altered contraction and relaxation [60]. These major alterations in cardiac function and structure have led to the emergence of novel regenerative strategies that aim to attenuate the progression of myocardial remodeling and repopulate the fibrotic scar with cardiomyocytes which are mechanically and electrically integrated with the surrounding tissue. Embryonic stem cells (ESC) and induced pluripotent stem cells (iPSC) have been extensively studied for cell therapy as an exogenous origin of new cardiomyocytes. Either delivered by direct injection or cardiac patch, pluripotent stem cell-derived cardiomyocytes were shown to engraft in infarcted hearts and promote beneficial effects on regional and global cardiac function [21, 25, 93, 123]. Nevertheless, challenges like the immature phenotype and poor survival of cardiomyocytes [99], risk for tumorgenicity [100], and increased propensity to develop arrhythmic events [158] need to be overcome before their translation to clinical use. An alternative is direct reprogramming which aims to convert resident cardiac fibroblasts into cardiomyocytes in situ. Several small molecules, transcription factors, and ncRNAs have been identified and used to mediate the direct trans-differentiation [32, 65]. This evidence, however, is still not sufficiently robust to determine whether this approach can generate the large number of cardiomyocytes needed to replenish the lost myocardium. Finally, cardiomyocyte cell cycle re-activation and stimulation of their proliferative capacity seem to be the most direct approach. Accumulating data are offering various strategies to trigger this process and generate new cells. Despite some challenges which will be discussed later in the text, this is an exciting avenue with great translational potential.
Cardiomyocyte proliferation in the steady-state
During the embryonic development, cardiomyocytes are generated by two main mechanisms: (1) the commitment and differentiation of cardiogenic precursor cells derived from the mesoderm cell layer, and (2) the proliferation of existing cardiomyocytes. The former is dominant throughout the formation and expansion of the early heart tubes [14], while the development of the heart tubes into the four-chambered heart shape is driven mainly by the proliferation of differentiated myocytes [126]. Later phases of the heart development, such as trabeculation and wall maturation, are also dependent predominantly on cardiomyocyte division [51, 52]. After birth, cardiac growth in mammals shifts abruptly from hyperplasia to hypertrophy, and eventually, an almost complete mitotic arrest occurs around postnatal day 4 (P4) in mice [82]. In this brief window of time, mammalian newborns retain a transient capacity for cardiomyocyte renewal. Surgical resection of the cardiac apex in P1 mice was shown to trigger a robust upregulation of myocyte proliferation and near-complete regeneration of the injury site by day 21 [112]. The cell cycle withdrawal after birth was shown to be mediated by a complex of regulatory factors which can be extrinsic, e.g., oxygen level, mechanical forces, and macrophage-associated factors, or intrinsic, e.g., Hippo/Yap pathway, Neuregulin/ErbB2 pathway, Meis1, E2F and miR-15 [113, 140]. The potential contribution of these factors to cardiac regeneration, however, is still not fully determined and currently they are the subject of intensive research.
Non-mammalian species, like teleost fish and amphibians, have a pronounced regenerative capacity which has also been exploited for translational research. Studies on zebrafish have identified many factors such as JAK/STAT pathway, retinoic acid, Notch, and non-coding RNAs such as miR-133, miR-99/100 and Let-7a/c which are involved in cytokinesis [1, 36, 74, 155, 164]. The modulation of miR-99/100 and Let-7a/c and their target genes SMARCA5 and FTNB was shown to induce cardiomyocyte dedifferentiation and proliferation in mammals suggesting that the molecular machinery responsible for zebrafish heart regeneration can be effectively utilized on mammalian hearts [1]. Although these data are promising, other molecular mechanisms have failed to achieve the same goal [114], indicating the need for further investigations to better understand the applicability and limitations of this approach.
Current approaches to induce cardiomyocyte proliferation and pathways involved
Several physiological conditions, such as oxygen level and mechanical load, have been reported to push cardiomyocytes into mitosis. By measuring 15N-thymidine labeling of DNA synthesis, Vujic et al. [139] have reported increased cardiomyocyte proliferation in both healthy and MI adult mice which had to perform exercise for 8 weeks [139], suggesting that exercise promotes cell cycle re-entry [139]. In addition to exercise, oxygen levels are also known to play an important role in cell cycle withdrawal. One of the major changes that newborns experience after birth is the abrupt increase in oxygen levels. An increase in reactive oxygen species (ROS) production and consequent activation of DNA damage responses have a pivotal role in cardiomyocyte cell cycle arrest [115]. On the contrary, exposure to severe systemic hypoxemia of 7% O2 for 2 weeks in adult mice was shown to reduce ROS generation and reactivate the cell cycle in cardiomyocytes. Additionally, when hypoxia was applied to post-MI mice, it was able to induce cardiomyocyte proliferation accompanied by improved cardiac function, vascular expansion, and decreased fibrosis [97]. Mechanical unloading of the left ventricle has also been reported to induce cardiomyocyte proliferation. Increased mechanical stress induced in volume overload models activates mitochondrial biogenesis, which increases ROS production; thus, similar to hyperoxemia it results in the activation of DNA damage responses [109]. Canseco et al. [19] examined the effects of human ventricular unloading after implantation of left ventricular assist devices (LVADs) by comparing left ventricular samples collected at the time of LVAD implantation (pre-LVAD) and at the time of explantation (post-LVAD). Their findings demonstrated that post-LVAD hearts had increased cardiomyocyte proliferation, suggesting that mechanical unloading promotes the cell cycle progression.
The modulation of several coordinated signal transduction pathways is known to reactivate the cell cycle in adults [37]. The Hippo/YAP transduction pathway is an evolutionarily conserved regulator of organ size in different tissues [159]. In the heart, it was shown to be a key regulator of cardiomyocyte proliferation during heart development and cardiac regeneration in the early postnatal stage [138, 152]. Following the activation of the cascade, the transcriptional co-activators Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) become phosphorylated and inactive, which results in their dissociation from transcription factors that normally drive cell division [161]. Blocking the Hippo/YAP pathway by targeting its components has been shown as an effective approach to boost cardiomyocyte proliferation accompanied by improvement in total cardiac function [56, 78, 152]. Meis1 is another transcription factor that was linked to cardiac myocyte proliferation during development and in the early postnatal regenerative phases [87, 127]. Meis1 facilitates the cell cycle withdrawal by activating the expression of the cyclin-dependent kinase (CDK) inhibitors p15, p16, and p21 [87]. The transcription factor TBx20 has also direct interaction with Meis1 [150]. The downregulation of Meis1 was shown to reactivate cardiomyocyte mitosis in the adult heart with no deleterious effect on cardiac function [87]; however, whether this ameliorates the cardiac structure and function in the post-MI setting still needs to be determined. Several paracrine factors have been reported to regulate cardiomyocyte proliferation. Neuregulin-1 (NRG-1) is secreted from microvascular endothelial cells and acts on cardiomyocytes through its receptors ErbB2 and ErbB4 [40]. Activation of NRG-ErbB pathway either by overexpression of ErbB4 or by using a recombinant NRG-1 was reported to improve cardiac function and decrease post-MI fibrosis [10]. Moreover, clinical trials reported a favorable effect of recombinant NRG-1 on the cardiac function in heart failure patients [44, 67]. These findings however were not confirmed in a mouse study performed by Reuter et al. [117]. Experimental evidence indicated that NRG-1 did not activate cardiomyocyte DNA synthesis in either healthy or infarcted mice, thus suggesting that improved cardiac structure and function likely occurred independently of apparent myocardial regeneration. More work will be required to better understand NRG-1 cardiac reparative effects and its role in cardiomyocyte proliferation.
Finally, ncRNAs are also emerging as common targets to modulate cardiomyocyte proliferation. These molecules form a complex regulatory network with other molecular elements such as mRNAs and proteins which regulate cardiomyocyte cycle progression. Here we discuss miRNAs, lncRNAs, and circRNAs with such proliferative capacity and present their underlying mechanism.
MicroRNAs as targets for cardiomyocyte proliferation
MicroRNAs are small (≈22 peptides), single-stranded, noncoding RNAs that regulate gene expression at the post-transcription stage. These molecules repress protein expression by interacting with completely or partially complementary sequences of the 3′-untranslated regions (3′-UTR) of mRNAs. This binding leads to degradation or translational repression of the targeted mRNA [6] (Fig. 1). A single miRNA can target several to hundreds of genes, whereas various miRNAs can collectively target one mRNA [124]. These combinatorial interactions allow for a complex fine-tuning of several regulatory processes. It is not surprising that disruption of miRNA biogenesis and function contributes to many human diseases, including cancer, cardiovascular diseases, and neurological disorders [125, 130, 163].

miRNA mode of action. In most cases, miRNAs bind to a specific sequence at the 3′ UTR of their target mRNAs. This binding results in either translational repression or degradation of the respective mRNA target
Since the initial discovery of miRNAs in 1993 in the nematode Caenorhabditis elegans [80], thousands of miRNA genes have been identified in various species, among these more than 1500 were identified in humans [73]. It is estimated that two-thirds of the human protein-coding genes have miRNA target sites in their 3′ UTR; thus, they are potentially regulated by these molecules in both health and disease [39]. Numerous miRNAs have been identified in the cardiovascular system and were shown to control a wide range of biological processes, including cardiac repair, lineage commitment, proliferation, and cardiomyocyte survival [131]. miRNAs have been studied in the field of cardiac regeneration and have been found to tightly control cell cycle re-entry in cardiomyocytes. Here, we summarize a group of recently discovered miRNAs in the field and we outline their mechanism of action and potential gene targets (Table 1).
In 2012, Eulalio et al. identified a large series of human miRNAs reported to induce cardiomyocyte proliferation in vitro [34]. The same group has recently shown that the ten most effective miRNAs converge in the regulation of the Hippo pathway [134]. This pathway is a highly conserved signal transduction cascade that was first identified in Drosophila [53, 129]. It comprises a wide network of components that integrate diverse signals to eventually regulate cell proliferation and control organ size [161]. Activation of the Hippo pathway results in the phosphorylation of the master transcriptional cofactor YAP, thus blocking its activity. On the contrary, when YAP is dephosphorylated, it localizes to the nucleus and associates with the transcriptional enhanced associate domain (TEAD) 1–4 transcription factors to drive gene expression and stimulate cell proliferation [162] (Fig. 2a). Consistently, YAP is an essential factor in early heart development [138] and it is currently one of the most important targets for cardiac regeneration [152]. The miRNAs that were investigated include human miR-590-3p, miR-199a-3p, members of the miR-302 family (miR-302d, miR-302c, and miR-373), miR-1825, miR-1248, miR-18a, miR-33b, and miR-30e, all of which were shown to significantly increase the dephosphorylated YAP levels in the nucleus and improve TEAD activity in vitro. These effects were prevented by the knockdown of YAP, suggesting it is an essential requisite to mediate the pro-proliferative outcome of the investigated miRNAs [134]. These findings were confirmed by a study performed on hiPSC-derived cardiomyocytes, which found that 84 out of 96 miRNAs that promote proliferation upon overexpression induced nuclear translocation of YAP, and most of these miRNAs (67/84) required YAP for their proliferative activity [27]. These miRNAs act through different pathways to induce YAP activation. Some were found to directly target components of the Hippo pathway, such as the kinases MST1/2 and LATS1/2, while others regulate YAP via other mechanisms. For example, an intriguing interplay between YAP activation and the cytoskeletal arrangement was reported [134]. In particular, miR-199a-3p, miR-1825, miR-302d, miR-373, and miR-33b were found to downregulate the protein cofilin 2 and, except for miR-33b, this was achieved by directly binding to the 3′ UTR of cofilin 2 mRNA. Cofilin 2 is an actin-regulatory protein that binds actin monomers and filaments, causing their depolymerization and preventing their re-assembly [47], thus suggesting that proliferation was induced by the modulation of the actin cytoskeleton network (Fig. 2b).

Hippo pathway mediates the activity of miRNAs inducing cardiomyocyte proliferation. a The active dephosphorylated form if YAP/TAZ localizes to the nucleus and associates with TEAD transcription factors to drive cell proliferation genes expression. b miR-199a-3p, miR-1825, miR-302d, miR-373 and miR-33b downregulate cofilin 2, which disassembles actin filaments. The resulting cytoskeletal rearrangement leads to YAP activation and nuclear localization. c When Hippo signaling is on, MST1/2 activate LATS1/2 kinases, which in turn phosphorylate and inactivate the downstream effectors YAP and TAZ. miR-302/367 complex directly targets the expression of MST1, LATS2 and MOB1B, thereby blocking the Hippo signaling. miR-1825 activates miR-199a-3p, resulting in the downregulation of its target genes TAOK1 and β-TrCP, eventually leading to Hippo pathway repression and prevention of YAP degradation. miR-31a-5p downregulates RhoBTB1 and results in Hippo deactivation
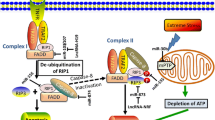
Among the investigated miRNAs, miR-199a-3p is one of the most potent promoters of cardiomyocyte proliferation and one of the first miRNAs shown to activate, rather than suppress the cardiac cell cycle [34]. Cardiomyocyte treatment with miR-199a-3p demonstrates downregulation of both the TAO kinase1 (TAOK1) and the E3 ubiquitin-ligase β-transducing repeat-containing protein (β-TrCP) [134] (Fig. 2c). The former phosphorylates and activates the MST and LATS kinases [11, 111], whereas the latter catalyzes YAP ubiquitination, resulting in its degradation [160]. Additionally, miR-199a-3p, along with miR-590a-3p, are known to target the genes HOMER1, which encodes a protein that modulates calcium signaling in the heart, and HOP homeobox (HOPX), which encodes a homeodomain protein that inhibits cardiomyocyte proliferation [34]. Both miR-199a-3p and miR-590a-3p are undetectable in the postnatal state, but when administered to isolated adult cardiomyocytes they are effective inducer of cell cycle re-entry [34]. These miRNAs were also tested using different delivery methods in vivo and have shown promising effects in both small and large mammals. Intracardiac delivery and vector-based overexpression of miR-199a-3p and miR-590a-3p in neonatal rodents resulted in ventricular wall thickening and increased cardiac myocyte proliferation with no signs of cardiomyocyte hypertrophy. In a post-MI adult mouse model, they also increased cardiomyocyte proliferation markers, decreased the infarct size and improved cardiac function [34]. Even a single intracardiac injection in mice, immediately after MI, led to persistent recovery of cardiac function [80]. Consistent with these experiments on small mammals, a study performed on pigs showed that expression of human miR-199a in infarcted porcine hearts can boost cardiac repair. This was determined by the marked increase in cardiomyocyte proliferation markers, improvements in both global and regional contractility, increased muscle mass, and reduced scar size. Of note, persistent and uncontrolled expression of this miRNA resulted in sudden arrhythmic death of most of the treated pigs, suggesting that the dosage of this therapy needs to be tightly controlled [41]. Taken together, these data suggest that both miR-199a-3p and miR-590a-3p are powerful regulators and valid candidates for future clinical trials.
miR-1825 is also one of the miRNAs that target cofilin 2 and induce YAP activation [134]. In addition, a recent study has demonstrated a second mechanism that involves miR-199a. The authors showed that miR-199a has significantly increased in adult cardiomyocytes post-miR-1825 transfection, indicating that miR-199a acts downstream of miR-1825 [104]. While it is well established that miRNAs act by binding to the 3′ UTR of mRNAs, there have been very few reports on miRNA targeting other miRNAs [18, 88, 89]. Through miR-199a, miR-1825 treatment downregulated the cell cycle inhibitory genes Meis2, Rb1, and P16 [104]. Consequently, the overexpression of miR-1825 in adult rat cardiomyocytes increased both their number and their proliferation markers. Furthermore, a reduction in mitochondrial number and a decrease in ROS and DNA damage were observed. In vivo, miR-1825 administration to P1 neonatal mice resulted in enlarged hearts and increased cardiomyocyte numbers. Prolonged delivery of miR-1825 following MI increased cardiomyocyte proliferation improved heart function and decreased scar size in adult mice [104].
Another important regulator of the Hippo pathway is the miRNA cluster miR302-367. This cluster is made of five miRNAs that are expressed in the heart during the early embryonic stage. Tian et al. [132] have shown that when this cluster is suppressed in mouse embryos, proliferation markers and expression of the cell cycle proliferation gene Ccnd1 were downregulated, as well as other genes involved in cell differentiation such as Nkx2.5, Gata4, Myh6, and Myh7. When overexpressed, miR302-367 increased cardiomyocyte proliferation and induced profound cardiac enlargement and cardiomegaly. Interestingly, although prolonged overexpression of miR302-367 in post-MI mice reduced fibrosis and improved cardiomyocyte proliferation, the treatment resulted in an overall altered cardiac function. On the contrary, a transient administration (over 1 week) resulted in a significant improvement in cardiac function. Experimental evidence suggested that miR302-367 can promote a proliferative state, but it also induced cardiomyocyte dedifferentiation. This dedifferentiation, however, can be avoided by short-term treatments. In addition to the actin-regulatory function of miR-302d, which is mediated by the interaction with cofilin 2 [134], miR302-367 inhibits the kinases Mst1, Lats2, and Mob1b. These are core components of the Hippo signaling cascade, whose inactivation eventually results in the active dephosphorylated form of YAP and, as a result, in a proliferative state [132].
Normally, miRNAs that function as positive regulators of cardiomyocyte proliferation are upregulated in the embryonic and fetal stages and downregulated afterward. On the contrary, miR-31a-5p was found to be significantly upregulated in P10 cardiomyocytes compared to P0, yet it was shown to activate the cardiomyocyte cell cycle [151]. This unusual pattern of differential expression was considered as a compensatory response. miR-31a-5p overexpression in neonatal rat ventricular myocytes (NRVM) and in rat neonates was sufficient to induce cardiomyocyte proliferation, assessed by immunostaining and the expression levels of proliferating cell nuclear antigen (PCNA) [151]. Bioinformatics analysis identified RhoBTB1 as the target gene of miR-31a-5p. RhoBTB1 is a tumor suppressor gene involved in different types of cancer [7, 90], but its role in cardiac pathologies is still unclear. Interestingly, a recent study also suggested a regulatory role of RhoBTB on the Hippo signaling [98]. This role was reported in Drosophila and human (HEK293T) cells and was shown to be mediated by ubiquitination-dependent regulation of LKB1 kinase [151]. LKB1 controls the Hippo pathway through phosphorylation of MARK kinases and thereby modulation of YAP activity [96].
Despite the prominence of the Hippo transduction pathway in miRNA-mediated cardiomyocyte proliferation, other pathways are also involved. The cyclin family is a key factor in the decision as to whether somatic cells become quiescent or resume active proliferation [42]. This mechanism is also highly regulated by miRNAs. For example, miRNAs miR-1 [43], miR-499 [84] and miR-34a [154] control cardiac myocyte proliferation through the regulation of cyclin D1, while miR-29a [20], miR-133a [86] and miR let-7i-5p [61] target cyclin D2. Moreover, miR-294 [12] and miR-128 [64] regulate cyclin B1 and cyclin E, respectively. Except for miR-499 and miR-29a, all these miRNAs have been reported to stimulate cardiomyocyte proliferation in vivo, indicating a high potential for successful translational applications. In addition to the cyclin proteins, phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that can be the target for miRNAs promoting cardiac cell cycle progression. The miR-17–92 cluster, which directly targets PTEN, is known to be required and sufficient to induce cardiomyocyte proliferation. The overexpression of this cluster in adult animal hearts attenuated myocardial infarction-induced injury [23]. miR-29b-3p [153] and miR-34a [9] have been described to target the Notch signaling pathway which also plays an important role in the differentiation and proliferation of cardiomyocytes [107]. Of note, Notch is known to act downstream of Hippo, as suppression of Hippo is a potent activator of Notch signaling [66]. miR-29b-3p inactivates the NOTCH2 gene and its inhibition promoted cardiomyocyte proliferation in vitro and in vivo [153]. miR-34a, however, targets NOTCH1 and its regulator enzyme Pofut1 [9]. Inhibition of miR-34a reduced the scar size and improved cardiac function post-MI [154].
The data presented here propose a great potential of miRNAs as therapeutic targets in regenerative medicine. It is not surprising that miRNAs are gaining a lot of interest among pharmaceutical companies, which are dedicating significant efforts to develop efficient, safe, and easy-to-deliver miRNA products. Nevertheless, several obstacles remain to be overcome, the most important of which is the off-target biological effects which are complicated by the miRNA pleiotropic nature. Another important aspect is the fundamental genetic difference between humans and the animal models used for testing which further complicates the translatability of results, ultimately delaying the human clinical application. Furthermore, some miRNAs were shown to effectively stimulate the proliferation of adult cardiomyocytes by controlling the expression of target molecules, while the modulation of the respective targets was not sufficient to produce the same outcome. The overexpression of cyclin D1 in mice, for example, resulted in DNA replication and multinucleation of cardiomyocytes, but these cells did not proceed to cytokinesis and complete cell division [125]. These results suggest that miRNAs that target cyclin D1 also target other genes to successfully complete cell division. Moreover, activation of Notch signaling in zebrafish hearts unexpectedly suppressed cardiomyocyte proliferation and heart regeneration. This could be attributed to the over-stimulation of Notch signaling in the endocardium and/or epicardium which might lead to the production of paracrine signal that controls myocardial cell division [164]. These, however are speculations and a deeper understanding of miRNAs molecular targets and mechanisms are required to develop more efficient, effective, and safe therapeutic strategies.
Long non-coding RNAs as targets for cardiomyocyte proliferation
lncRNAs are defined as RNA transcripts of > 200 nucleotides with no evidence for a protein-coding function [94]. Initially termed ‘mRNA-like noncoding RNA’ after its first discovery in 1999 [33], this subset of ncRNA has quickly gained popularity among researchers, and according to NONCODE database, 548,640 lncRNAs have already been annotated from 17 species [35]. From the human genome alone, 96,308 lncRNA transcripts are known, by far exceeding the number of protein-coding genes by about fivefold [118]. Unlike protein-coding genes and miRNAs, the majority of lncRNAs have poor interspecies sequence conservation [105], although it is speculated that conservation of their secondary structure might exist [69].
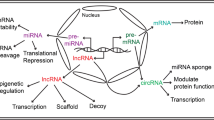
Based on genomic location and orientation to protein-coding genes, there are six major classes of lncRNAs: (1) sense—spanning multiple introns or exons within a gene, (2) sense intronic—located within one intron, (3) antisense—transcribed from the opposite strand of a gene, (4) bidirectional—located on the antisense strand within 1 kb of a promoter on the sense strand, (5) enhancer—located in an enhancer region and (6) intergenic—located between two genes [26]. Another type of classification relies on the cellular function of lncRNAs which are divided to signal, decoy, guide, scaffold, enhancer, or sponge lncRNA (Fig. 3) [5].

lncRNA modes of action. (1) signal—acts in response to stimuli, (2) decoy—sequesters transcription factors/protein complex, (3) guide—guides transcription factors/protein complex to a specific target site, (4) scaffold—brings together multi-protein complexes, (5) enhancer—induces chromosomal looping to increase association between enhancer and promoter regions, and (6) sponge—acts as a competing endogenous RNA (ceRNA) and sponge miRNAs
Accumulating evidence shows that lncRNAs play key roles in a large number of key biological processes, such as the circuitry controlling pluripotency and differentiation, immune responses, disease etiology, and chromosome dynamics [35]. In the cardiovascular system, lncRNAs are expressed in an abundance and it has been suggested that they compose a complex regulatory network governing the physiology (and pathology) of the heart. They are indeed involved in the regulation of various biological functions: for example, CARMEN [101], H19 [116] and Braveheart [76] play crucial roles in cardiomyocyte differentiation, Meg3 [149] and CARL [144] are essentially involved in apoptosis, and Chast [136] and Chaer [147] are associated with cardiac hypertrophy. Here, we highlight and summarize some of the recent developments and experimental findings related to the roles that different lncRNAs play in controlling cardiomyocyte proliferation and their implication in cardiac regenerative medicine (Table 2).
Similarly to miRNAs, lncRNAs can regulate cardiomyocyte proliferation either by promoting (positive regulators) or halting (negative regulators) cell cycle progression. Positive regulating lncRNAs are expressed in abundance during heart development to allow for effective cytokinesis and heart growth; however, their expression is undetectable in adulthood. Endogenous cardiac regeneration-associated regulator (ECRAR) was shown to be upregulated in human fetal hearts and downregulated during adulthood [24]. ECRAR exhibits a pro-proliferative effect on adult rat cardiomyocytes both in vitro and in vivo. ECRAR overexpression in both P7 and adult post-MI rats resulted in elevated cardiomyocyte proliferation markers and higher capillary density both in the infarct and peri-infarct areas. Additionally, reduced fibrosis and enhanced cardiac function were also observed. In contrast, the knockdown of ECRAR in P1 post-MI rats revealed decreased proliferation and increased fibrosis markers [24]. ECRAR transcription is regulated by E2F transcription factor 1 (E2F1). Acting as a signal lncRNA, ECRAR binds directly to extracellular signal-regulated kinases 1 and 2 (ERK1/2) and accelerates their phosphorylation and translocation to the nucleus where they boost the transcription of several genes involved in cell proliferation. These genes act as activators of cyclin D1 and cyclin E, and increase the expression of E2F1, creating a positive feedback loop [24]. Another positive regulator is NR_045363, which is a mouse homolog of the human LOC101927497. NR_045363 is known to be conserved among various species, including other mammals and birds, suggesting the functional importance of this lncRNA through evolution [142]. This lncRNA is highly expressed in the hearts of embryonic mice and diminishes during adulthood. In vitro, the downregulation of NR_045363 in primary embryonic mouse cardiomyocytes inactivates cell proliferation, and its re-expression in postnatal cardiomyocytes is sufficient to reactivate cardiomyocyte cell cycle progression [142]. Moreover, in in vivo mouse MI models, it was demonstrated that overexpression of NR_045363 resulted in a smaller scar, improved heart function, and enhanced cardiomyocyte proliferation [142]. Mechanistic analyses reveal that NR_045363 acts through direct interaction with miR-216a via a sponge effect. miR-216a is presumed to block the activity of JAK2 [59], a member of the receptor-associated Janus kinase family, which phosphorylates and activates the transcription factor signal transducer and activator of transcription 3 (STAT3) [70]. Through the miR-216a/JAK2-STAT3 pathway, lncRNA NR_045363 seems to act as a potent regulator of cardiac cell proliferation [142].
Antisense (AS) lncRNA is a class of lncRNA that is transcribed from the opposite strand of its sense protein-coding gene [26]. An example is silent information regulator factor 2 related enzyme 1 (Sirt1) AS which has been reported to induce cell proliferation in various cell types, including myoblasts and endothelial progenitor cells [95, 141]. Recently, a similar effect of Sirt1 AS lncRNA on cardiomyocytes was also reported [81]. Sirt1 AS lncRNA expression was found to be high in mouse embryonic hearts and to decrease after birth. When overexpressed in vitro, Sirt1 AS lncRNA induced mitosis, upregulation of dedifferentiation markers, and a reduction in cell size. When silenced, the opposite results were obtained [81]. Depletion of Sirt1 AS lncRNA in vivo in neonatal mice significantly decreased cardiomyocyte proliferation, while its overexpression in the adult mice induced cell cycle re-activation. When myocardial infarction was induced, Sirt1 AS lncRNA was able to improve survival and cardiac function, decreased scar size, and enhanced proliferation markers [81]. Interestingly, Sirt1 AS lncRNA was found to act by direct binding to Sirt1 mRNA, thus enhancing its stability and increasing the levels of Sirt1 at both the mRNA and protein levels. Moreover, co-transfection with Sirt1 AS lncRNA and small interfering-Sirt1 (which targets Sirt1 mRNA) revealed decreased extent of cardiomyocyte proliferation compared to Sirt1 AS lncRNA alone, suggesting that Sirt1 is involved in Sirt1 AS lncRNA-induced cardiomyocyte proliferation [81].
In contrast to ECRAR, NR_045363, and Sirt1 AS lncRNA, negative regulating lncRNAs are minimally expressed during embryogenesis and development and upregulated in adulthood. Cardiac regeneration-related lncRNA (CAREL) is a 748-nucleotide lncRNA that was found to be progressively upregulated in postnatal mice [15]. In the early neonatal stage, cardiomyocyte-specific CAREL transgenic mice and CAREL-adenovirus-transfected mice in which apical resection surgery was performed, exhibited a loss of cardiac regenerative capability, characterized by decreased proliferative potential and hypertrophied cardiomyocytes, increased fibrosis, and altered cardiac function [15]. On the contrary, when CAREL was silenced in adult mice, myocardial infarction resulted in smaller infarct size and improved contractile function [15]. CAREL effects were also investigated in human IPS cell-derived cardiomyocytes. Overexpression of the human conserved sequence of CAREL reduced cell proliferation markers, whereas its knockdown had the opposite effect [15]. Mechanistically, CAREL enhances cardiac regeneration and repair by acting as ceRNA and sponging miRNAs, thus influencing the expression of their target genes. CAREL has a sponge effect on miR-296 which has the ability to induce mitosis and boost cell proliferation by targeting Trp53inp1 and Itm2a whose specific role and function, however, is still unclear [15]. High-throughput sequencing of fetal and adult human cardiac tissue reported differential expression of another lncRNA, NONHSAG007671, proposing it as a negative regulator of cardiomyocyte regeneration [22]. Furthermore, species conservation analysis showed that the exons of NONHSAG007671 are highly conserved across humans, chimps, gorilla, mice, and rats. Designated as cardiomyocyte regeneration-related lncRNA (CRRL), this lncRNA was shown to suppress cardiomyocyte proliferation [22]. In vitro CRRL knockdown studies showed that it facilitated cell proliferation in P1 and P7 rat cardiomyocytes, without changing the cell size [22]. CRRL knockdown in vivo also induced a significant increase in the rate of cardiomyocyte proliferation and resulted in a marked increase in heart weight of both P1 and P7 neonatal rats. In post-MI neonatal and adult rats, blocking CRRL promoted cardiomyocyte proliferation in the peri-infarct area, improved cardiac function, and attenuated post-infarction cardiac remodeling, resulting in a smaller scar area and reduced fibrosis [22]. Conversely, overexpression of CRRL in P1 rats inhibited the post-MI regenerative response. CRRL inhibits cardiomyocyte proliferation by acting as a sponge and absorbing miR-199a-3p. One of the target genes of miR-199a-3p is HOPX [22], which is a critical factor that participates with the enzyme histone deacetylase 2 (Hdac2) in mediating the deacetylation of Gata4 to regulate cardiomyocyte proliferation [135]. These results suggest CRRL as a new potential therapeutic target for heart failure.
Ponnusamy et al. [110] observed that the expression levels of lncRNA AK080084, designated as Cardiomyocyte Proliferation Regulator (CPR), were also low in the embryonic mouse cardiomyocytes, and dramatically increased in postnatal and adult hearts, suggesting that it may play a role in cell cycle arrest in the adult mammalian heart. Cell proliferation assays have shown that silencing of lncRNA CPR in neonatal and adult mouse cardiomyocytes resulted in increased proliferation levels in vivo. On the contrary, CPR overexpression inhibited cardiomyocyte proliferation in neonatal mice and decreased their regenerative capacity after myocardial injury, leading to a larger fibrotic scar. Functionally, the contractile functions of CPR-overexpressed neonatal hearts were significantly depressed after myocardial infarction, while CPR deletion in adult mice resulted in improved left ventricular wall thickness, ejection fraction, and fractional shortening values, with a smaller scar size [110]. Transcriptomic analysis of CPR knockout hearts revealed that 1780 genes were differentially expressed. Among the most upregulated genes was minichromosome maintenance 3 (MCM3), an important factor in initiating eukaryotic genome replication [2]. The authors demonstrated that CPR acts as a guide lncRNA by direct interaction with DNA methyltransferase 3A (DNMT3A) to induce MCM3 promoter methylation, thus blocking the cell cycle progression [110]. Another negative regulating lncRNA identified by Cai et al. is lncRNA dachshund homolog 1 (lncDACH1). This 2085-nucleotide, highly conserved lncRNA was previously shown to be involved in the development of heart failure [17], and more recently a prominent role in the regulation of cardiomyocyte proliferation and cardiac regeneration was also reported [16]. When overexpressed in transgenic mice, lncDACH1 reduced proliferation markers expression and increased cardiomyocyte cross-sectional area, suggesting a blockage of mitosis and proliferation mechanisms [16]. Moreover, when apical resection was performed to neonatal transgenic mice, lncDACH1 treatment decreased their heart regenerative capacity. On the contrary, silencing of lncDACH1 enhanced the proliferative potential of cardiomyocytes after ischemic injury in adult mice, and resulted in smaller infarct size, improved cardiac function, and increased proliferation markers [16]. To study the effects of lncDACH1 on human iPSC-derived cardiomyocytes, a conserved sequence of the lncRNA was tested with gain and loss-of-function approaches. Overexpression of lncDACH1 resulted in reduced cardiomyocyte proliferation, while its inhibition promoted cell cycle re-entry [16]. The molecular mechanism underlying the effects of lncDACH1 on cardiac repair was reported to involve a decoy activity by direct interaction with protein phosphatase 1 alpha (PP1A) and a reduction in its activity. PP1A enzyme dephosphorylates the transcriptional cofactor YAP1, thereby activating the Hippo transduction pathway which is, as already stated, an essential regulator of cardiomyocyte proliferation [138]. These findings highlight the importance of lncDACH1 in the regulation of the Hippo/YAP pathway, suggesting that, similarly to miRNAs, several lncRNAs can also modulate it.
Finally, AZIN2-sv is a splice variant of the AZIN2 gene that was found to be highly expressed in adult human hearts compared to the fetal stage thus suggesting a negative regulatory role [83]. In vitro, AZIN2-sv was shown to suppress the proliferation genes in cardiomyocytes, while silencing AZIN2-sv resulted in their increase. Overexpression of AZIN2-sv in neonatal rats at P1 and P7 revealed decreased levels of cardiomyocyte proliferation, whereas its knockdown markedly increased these markers and elevated their hearts’ size and weight while the cardiomyocyte cross-sectional area remained unchanged. In post-MI adult rats, reduction in AZIN2-sv resulted in better cardiac function, smaller scars, and less tissue fibrosis [84]. Together, these findings propose a pivotal role of AZIN2-sv in blocking the cell cycle and stop cardiomyocyte proliferation. As to its mechanism of action, AZIN2-sv directly binds PTEN and improves its stability. Additionally, it also sponges miR-214 which has inhibitory effects on PTEN. The overall effect is a significant increase in PTEN levels which results in lower levels of phosphorylated Akt and cyclin D and, consequently, block of the cell cycle [84].
In summary, these studies support the potential of lncRNA-based strategies to boost cardiomyocyte proliferation and cardiac regeneration. The various and intricate regulatory mechanisms are still under intense investigation; however, it is unquestionable that our knowledge of lncRNAs not only adds a new dimension to the molecular architecture of human disease, but also opens up a whole new range of opportunities for future treatments.
Circular RNAs as targets for cardiomyocyte proliferation
Circular RNA is a special subclass of ncRNA characterized by linking the 3′ and 5′ ends to form a covalently closed loop structure. The circular structure maintains the stability of these ncRNAs, reduces the exonuclease susceptibility, and ultimately results in a longer half-life compared to linear RNA molecules [92]. Recently, circRNAs were shown to be extensively expressed in eukaryotes [145] and to exhibit some degree of evolutionary conservation among mammals [68]. They are dynamically expressed in both tissue development and pathophysiological conditions and they follow tissue- and age-dependent patterns [119, 148]. circRNAs expression is a finely tuned and controlled process involved in various biological roles and for this reason, recent years have witnessed a growing interest in the scientific community.
Different models to describe the modes of action of circRNAs have been proposed. Some circRNAs were found to act as ceRNAs and bind miRNAs [165], whereas others sequester RNA-binding proteins (RBPs) and compete with their mRNA targets [3]. Other proposed modes of action involve competing with the splicing of mRNA [3], interacting directly with regulatory proteins [30], or small nuclear RNA (snRNA) [85] to regulate gene expression, and even being translated to proteins [103].
With the advancement of RNA sequencing technologies, many circRNAs have been confirmed to show abnormal expression in various pathological conditions including cancer [156], neurological [121], and metabolic disorders [146]. circRNAs are also involved in several biological pathological conditions of the cardiovascular system such as cardiomyocyte hypertrophy [143], senescence [29], and apoptosis [157]. Although circRNA investigation in the field of cardiac regeneration and repair has only just started, some encouraging data have already been collected. Circ-Amotl1 is a circular RNA that can boost cardiomyocyte survival following in vitro exposure to H2O2. It was also shown to attenuate doxorubicin-induced cardiomyopathy in mice by binding phosphoinositide-dependent kinase 1 (PDK1) and AKT1, resulting in AKT1 phosphorylation and nuclear translocation [157]. Another example is circFndc3b which harbor cardioprotective effects by reducing cardiomyocyte apoptosis following in vitro stress conditions such as exposure to H2O2 serum deprivation and hypoxic stress. In vivo data also demonstrated a reduction in cardiomyocyte apoptosis and improved neovascularization and left ventricular functions post-MI in circFndc3b treated animals [46].
Very limited scientific evidence is currently available to robustly support the contribution of circRNAs in the regulation of cardiomyocyte cell cycle. So far, only one circRNA was demonstrated to regulate cardiac myocyte proliferation. CircRNA Nfix is a super enhancer-associated circRNA that is highly conserved in humans, rats, and mice, and was found to be enriched in adult cardiomyocytes. When overexpressed in neonatal mice, circNfix reduced both proliferation and angiogenesis. Overexpression in MI neonatal mice decreased cardiomyocyte proliferation rate, ultimately resulting in impaired cardiac function and larger infarct size. On the contrary, the loss of circNfix stimulated cardiomyocyte proliferation in vitro and resulted in improved cell survival after exposure to H2O2 treatment. Moreover, circNfix silencing in adult mice induced enhanced cardiomyocytes number and proliferation capacity, together with increased arteriolar density and decreased apoptosis, ultimately resulting in improved cardiac function and reduced infarct area and cardiac fibrosis [63]. CircNfix was found to be regulated by Meis1, a key transcription factor controlling cardiomyocyte cell cycle arrest, which binds and activates the super-enhancer region of circNfix. CircNfix was suggested to act by binding to the transcription factor Ybx1 while recruiting the E3 ubiquitin ligase Nedd4l, thereby mediating the arrest of Ybx1 in the cytoplasm and its degradation through ubiquitination–proteasome pathways. Nuclear Ybx1 is known to bind to the promoters of CCNA2 (cyclin A2) and CCNB1 (cyclin B1) and activate their transcription, facilitating cell cycle entry [72]. In parallel, circNfix’s proangiogenic activity is mediated by sponging of miR-214, thus inhibiting Gsk3β expression. CircNfix/miR-214/Gsk3β pathway regulates VEGF release from cardiomyocytes and regulates the formation of blood vessels.
As demonstrated by the limited literature, circRNAs research particularly in the regenerative medicine field is still in its infancy, and because of its potential gaining increasing interest from the scientific community. The deeper understanding of circRNAs mechanisms and development of new technologies will further contribute to this field providing new insights and therapeutic possibilities for the treatment of heart failure.
Current limitations and future insights
The field of cardiac regeneration has made enormous progress and various regulatory networks that control cardiomyocyte proliferation have been unravelled. Several methodological and technological limitations still remain. For a long time, the field has been enfeebled by the improper use of techniques that claim to indicate cardiomyocyte proliferation. Ki-67, phosphohistone H3 (pH3), Aurora B, and nucleotide analogs are examples of immunohistochemical markers that are frequently used to demonstrate cell cycle activity [15, 17, 24, 34, 81, 84, 132]. These methods, however, have crucial limitations which, if not considered, can result in misleading data interpretation. Nucleotide analogs are incorporated into the newly synthesized DNA and can be easily detected by immunofluorescent staining [49]. Cardiomyocytes, however, undergo various processes such as endoreduplication, endomitosis, and multinucleation, which imply DNA synthesis but do not result in complete cell division. pH3 is also a popular marker which labels phosphorylated histone H3 that is believed to be crucial for chromosome condensation and cell cycle progression during mitosis [58]. Histone H3 phosphorylation occurs from late G2 phase to early telophase [57]. Similarly to nucleotide analogs, this assay is not sufficient to indicate cell proliferation, as cells undergoing endomitosis or multinucleation will also be labeled [31, 91]. Ki67 is a protein required for the formation of the perichromosomal layer and plays a key role in preventing the aggregation of mitotic chromosomes [128]. It is clearly required for cell proliferation, but it is also re-expressed in cardiomyocytes undergoing hypertrophy, endoreduplication, and endomitosis [91]. Taken together, these considerations suggest that a composite approach which includes the use of various markers, that distinguish different parts of the cell cycle, in combination with other approaches such as cell proliferation assays, cell count, and clonal analysis, can provide a more reliable readout.
Another challenge that still needs to be addressed is the development of strategies that can modulate cardiomyocyte proliferation rate as a mean of increase but also limit this process. Uncontrolled proliferation can indeed result in pathological conditions such as uncontrolled hypertrophy and enlarged myocardium. Several studies on both rodents and pigs have reported adverse effects in response to prolonged treatment with various miRNAs; these included cardiomegaly, impaired cardiac function, arrhythmic events and sudden death [41, 132]. An additional aspect that remains to be verified is the actual impact that enhanced cardiomyocyte proliferation can play in the context of the whole heart. Several studies have reported significant improvements in cardiac function and structure which were attributed to this process. However, in various cases a minor increase in cardiomyocyte cell cycle activity was demonstrated, which was not sufficient to justify the overall beneficial effects [16, 122, 142]. Other factors such as enhanced angiogenesis, hypertrophy, reduced apoptosis, or altered metabolic activity may also contribute and, thus, should be considered.
Furthermore, humans have both genetic and physiological features that are different from other species, and which are only partially recapitulated by research models. These aspects are certainly confounding factors and they contribute to complicating the translation of findings into human applications. A clear example is lncRNAs, as they are poorly conserved among species; therefore, humanized models or organoid cultures are often required for translatability of finding to human biology [81]. Moreover, accumulating evidence seems to indicate the existence of a specific subpopulation of mononuclear diploid cardiomyocytes in the murine hearts which contribute widely to new cardiomyocyte formation [75, 106]. Thus, it appears important to determine whether this subpopulation of cardiomyocytes exists in humans and other mammals and how it could be targeted and used to boost regenerative processes. Finally, it should be considered that the myocardium consists of several cell types that communicate and interact in a variety of ways [133]. Thus, it seems essential to elucidate the associations between these multicellular interactions and regenerative processes. A better understanding of this area could be exploited to control and boost cardiomyocyte proliferation.
Although several challenges remain, the field of cardiomyocyte proliferation has made enormous steps forward and ncRNAs hold tremendous promise for regenerative medicine. A recent translational study, in a large animal model, indicated improved contractility, increased muscle mass, and reduced post-MI scar size in response to miRNA treatment [41]. To date, no clinical trials have been held to investigate the proliferative potential of ncRNAs in the heart; however, preclinical development of an inhibitor for the pro-hypertrophic miR-132 has shown favorable pharmacokinetics, safety, and tolerability of the drug, now leading to a first phase 1b clinical trial in heart failure patients [62]. These findings suggest the enormous clinical therapeutic potential for miRNA mimics and inhibitors and more preclinical and clinical studies can be expected in the near future.
Conclusions
The field of cardiac regeneration has been exploring numerous avenues to achieve effective cardiac repair, and being able to restore (and control) the proliferative capacity of adult cardiomyocytes is possibly the biggest challenge in regenerative medicine. The information collected and summarized in this review indicates that cardiomyocytes have an intrinsic capacity to proliferate which, in the steady-state, is suppressed to various extents by either extra- or intracellular factors. The nature and importance of such stimuli and how they impact the cardiomyocyte cell cycle are still under intensive investigation. The understanding of the regenerative capacity of the heart along evolution has been essential to improve our knowledge of the cellular mechanisms which are often shared between lower vertebrates and neonatal mammals. This holds promise as the cardiomyogenesis regulation mechanisms identified in these animal models seem to act in a similar manner in humans. Both short and long ncRNAs are primary orchestrators of crucial gene regulatory networks controlling various cellular processes. miRNAs experimentation is leading the research and it is ahead in comparison to other ncRNAs. lncRNAs remain an immense, poorly explored, and inadequately understood source for new regenerative targets. Recent advancements in sequencing have allowed large-scale target identification and the new gene therapy technologies provide new tools for their precise in vivo modulation. These are crucial requirements to elucidate the regulatory networks implicated in cell cycle activation and arrest. Micro, circular, and lncRNAs were shown to have extremely versatile modes of action and to be expressed abundantly in the heart tissue, suggesting that they are an appealing target for future cardiac regenerative research. The mechanisms that underlie the activity of some of the described long non-coding and circular RNAs seem to largely overlap with pathways that were shown being essential in microRNA regulation, suggesting that a more refined and possibly combined approach might lead to more effective strategies. Understanding and taking advantage of the mechanisms will eventually help to remove and control the breaks on myocardial regeneration.
References
Aguirre A, Montserrat N, Zacchigna S, Nivet E, Hishida T, Krause MN, Kurian L, Ocampo A, Vazquez-Ferrer E, Rodriguez-Esteban C, Kumar S, Moresco JJ, Yates JR, Campistol JM, Sancho-Martinez I, Giacca M, Izpisua Belmonte JC (2014) In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell Stem Cell 15:589–604. https://doi.org/10.1016/j.stem.2014.10.003
Alvarez S, Díaz M, Flach J, Rodriguez-Acebes S, López-Contreras AJ, Martínez D, Cañamero M, Fernández-Capetillo O, Isern J, Passegué E, Méndez J (2015) Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat Commun 6:8548. https://doi.org/10.1038/ncomms9548
Ashwal-Fluss R, Meyer M, Pamudurti NR, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, Kadener S (2014) CircRNA biogenesis competes with Pre-mRNA splicing. Mol Cell 56:55–66. https://doi.org/10.1016/j.molcel.2014.08.019
Bader D, Oberpriller JO (1978) Repair and reorganization of minced cardiac muscle in the adult newt (Notophthalmus viridescens). J Morphol 155:349–357. https://doi.org/10.1002/jmor.1051550307
Bär C, Chatterjee S, Thum T (2016) Long noncoding RNAs in cardiovascular pathology, diagnosis, and therapy. Circulation 134:1484–1499. https://doi.org/10.1161/CIRCULATIONAHA.116.023686
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Beder LB, Gunduz M, Ouchida M, Gunduz E, Sakai A, Fukushima K, Nagatsuka H, Ito S, Honjo N, Nishizaki K, Shimizu K (2006) Identification of a candidate tumor suppressor gene RHOBTB1 located at a novel allelic loss region 10q21 in head and neck cancer. J Cancer Res Clin Oncol 132:19–27. https://doi.org/10.1007/s00432-005-0033-0
Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisén J (2009) Evidence for cardiomyocyte renewal in humans. Science 324:98–102. https://doi.org/10.1126/science.1164680
Bernardo BC, Gao XM, Winbanks CE, Boey EJH, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, Lin RCY, McMullen JR (2012) Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A 109:17615–17620. https://doi.org/10.1073/pnas.1206432109
Bersell K, Arab S, Haring B, Kühn B (2009) Neuregulin1/ErbB4 signaling induces cardiomyocyte proliferation and repair of heart injury. Cell 138:257–270. https://doi.org/10.1016/j.cell.2009.04.060
Boggiano JC, Vanderzalm PJ, Fehon RG (2011) Tao-1 phosphorylates hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Dev Cell 21:888–895. https://doi.org/10.1016/j.devcel.2011.08.028
Borden A, Kurian J, Nickoloff E, Yang Y, Troupes CD, Ibetti J, Lucchese AM, Gao E, Mohsin S, Koch WJ, Houser SR, Kishore R, Khan M (2019) Transient Introduction of miR-294 in the heart promotes cardiomyocyte cell cycle reentry after injury. Circ Res 125:14–25. https://doi.org/10.1161/CIRCRESAHA.118.314223
Van Den Borne SWM, Diez J, Blankesteijn WM, Verjans J, Hofstra L, Narula J (2010) Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol 7:30–37
Buckingham M, Meilhac S, Zaffran S (2005) Building the mammalian heart from two sources of myocardial cells. Nat Rev Genet 6:826–835
Cai B, Ma W, Ding F, Zhang L, Huang Q, Wang X, Hua B, Xu J, Li J, Bi C, Guo S, Yang F, Han Z, Li Y, Yan G, Yu Y, Bao Z, Yu M, Li F, Tian Y, Pan Z, Yang B (2018) The long noncoding RNA CAREL controls cardiac regeneration. J Am Coll Cardiol 72:534–550. https://doi.org/10.1016/j.jacc.2018.04.085
Cai B, Ma W, Wang X, Sukhareva N, Hua B, Zhang L, Xu J, Li X, Li S, Liu S, Yu M, Xu Y, Song R, Xu B, Yang F, Han Z, Ding F, Huang Q, Yu Y, Zhao Y, Wang J, Bamba D, Zagidullin N, Li F, Tian Y, Pan Z, Yang B (2020) Targeting LncDACH1 promotes cardiac repair and regeneration after myocardium infarction. Cell Death Differ. https://doi.org/10.1038/s41418-020-0492-5
Cai B, Zhang Y, Zhao Y, Wang J, Li T, Zhang Y, Jiang Y, Jin X, Xue G, Li P, Sun Y, Huang Q, Zhang X, Su W, Yang Y, Sun Y, Shi L, Li X, Lu Y, Yang B, Pan Z (2019) Long noncoding RNA-DACH1 (Dachshund Homolog 1) regulates cardiac function by inhibiting SERCA2a (Sarcoplasmic Reticulum Calcium ATPase 2a). Hypertens (Dallas, Tex 1979) 74:833–842. https://doi.org/10.1161/HYPERTENSIONAHA.119.12998
Cannell IG, Kong YW, Bushell M (2008) How do microRNAs regulate gene expression? Biochem Soc Trans 36:1224–1231
Canseco DC, Kimura W, Garg S, Mukherjee S, Bhattacharya S, Abdisalaam S, Das S, Asaithamby A, Mammen PPA, Sadek HA (2015) Human ventricular unloading induces cardiomyocyte proliferation. J Am Coll Cardiol 65:892–900. https://doi.org/10.1016/j.jacc.2014.12.027
Cao X, Wang J, Wang Z, Du J, Yuan X, Huang W, Meng J, Gu H, Nie Y, Ji B, Hu S, Zheng Z (2013) MicroRNA profiling during rat ventricular maturation: a role for miR-29a in regulating cardiomyocyte cell cycle re-entry. FEBS Lett 587:1548–1555. https://doi.org/10.1016/j.febslet.2013.01.075
Caspi O, Huber I, Kehat I, Habib M, Arbel G, Gepstein A, Yankelson L, Aronson D, Beyar R, Gepstein L (2007) Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J Am Coll Cardiol 50:1884–1893. https://doi.org/10.1016/j.jacc.2007.07.054
Chen G, Li H, Li X, Li B, Zhong L, Huang S, Zheng H, Li M, Jin G, Liao W, Liao Y, Chen Y, Bin J (2018) Loss of long non-coding RNA CRRL promotes cardiomyocyte regeneration and improves cardiac repair by functioning as a competing endogenous RNA. J Mol Cell Cardiol 122:152–164. https://doi.org/10.1016/j.yjmcc.2018.08.013
Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang DZ (2013) Mir-17-92 induce cardiomyocyte proliferation in postnatal and adult hearts, 2013. Circ Res 112:1557–1566. https://doi.org/10.1161/CIRCRESAHA.112.300658
Chen Y, Li X, Li B, Wang H, Li M, Huang S, Sun Y, Chen G, Si X, Huang C, Liao W, Liao Y, Bin J (2019) Long non-coding RNA ECRAR triggers post-natal myocardial regeneration by activating ERK1/2 signaling. Mol Ther 27:29–45. https://doi.org/10.1016/j.ymthe.2018.10.021
Chow A, Stuckey DJ, Kidher E, Rocco M, Jabbour RJ, Mansfield CA, Darzi A, Harding SE, Stevens MM, Athanasiou T (2017) Human induced pluripotent stem cell-derived cardiomyocyte encapsulating bioactive hydrogels improve rat heart function post myocardial infarction. Stem Cell Reports 9:1415–1422. https://doi.org/10.1016/j.stemcr.2017.09.003
Devaux Y, Zangrando J, Schroen B, Creemers EE, Pedrazzini T, Chang CP, Dorn GW, Thum T, Heymans S (2015) Long noncoding RNAs in cardiac development and ageing. Nat Rev Cardiol 12:415–425
Diez-Cuñado M, Wei K, Bushway PJ, Maurya MR, Perera R, Subramaniam S, Ruiz-Lozano P, Mercola M (2018) miRNAs that induce human cardiomyocyte proliferation converge on the hippo pathway. Cell Rep 23:2168–2174. https://doi.org/10.1016/j.celrep.2018.04.049
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guig R, Gingeras TR (2012) Landscape of transcription in human cells. Nature 489:101–108. https://doi.org/10.1038/nature11233
Du WW, Yang W, Chen Y, Wu Z-K, Foster FS, Yang Z, Li X, Yang BB (2017) Foxo3 circular RNA promotes cardiac senescence by modulating multiple factors associated with stress and senescence responses. Eur Heart J 38:1402–1412. https://doi.org/10.1093/eurheartj/ehw001
Du WW, Yang W, Liu E, Yang Z, Dhaliwal P, Yang BB (2016) Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res 44:2846–2858. https://doi.org/10.1093/nar/gkw027
Engel FB, Schebesta M, Keating MT (2006) Anillin localization defect in cardiomyocyte binucleation. J Mol Cell Cardiol 41:601–612. https://doi.org/10.1016/j.yjmcc.2006.06.012
Engel JL, Ardehali R (2018) Direct cardiac reprogramming: progress and promise. Stem Cells Int. https://doi.org/10.1155/2018/1435746
Erdmann VA, Szymanski M, Hochberg A, de Groot N, Barciszewski J (1999) Collection of mRNA-like non-coding RNAs. Nucleic Acids Res 27:192–195. https://doi.org/10.1093/nar/27.1.192
Eulalio A, Mano M, Ferro MD, Zentilin L, Sinagra G, Zacchigna S, Giacca M (2012) Functional screening identifies miRNAs inducing cardiac regeneration. Nature 492:376–381. https://doi.org/10.1038/nature11739
Fang S, Zhang L, Guo J, Niu Y, Wu Y, Li H, Zhao L, Li X, Teng X, Sun X, Sun L, Zhang MQ, Chen R, Zhao Y (2018) NONCODEV5: a comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res 46:D308–D314. https://doi.org/10.1093/nar/gkx1107
Fang Y, Gupta V, Karra R, Holdway JE, Kikuchi K, Poss KD (2013) Translational profiling of cardiomyocytes identifies an early Jak1/Stat3 injury response required for zebrafish heart regeneration. Proc Natl Acad Sci USA 110:13416–13421. https://doi.org/10.1073/pnas.1309810110
Foglia MJ, Poss KD (2016) Building and re-building the heart by cardiomyocyte proliferation. Dev 143:729–740
Frey N, Olson EN (2003) Cardiac Hypertrophy: The Good, the Bad, and the Ugly. Annu Rev Physiol 65:45–79. https://doi.org/10.1146/annurev.physiol.65.092101.142243
Friedman RC, Farh KKH, Burge CB, Bartel DP (2009) Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19:92–105. https://doi.org/10.1101/gr.082701.108
Fuller SJ, Sivarajah K, Sugden PH (2008) ErbB receptors, their ligands, and the consequences of their activation and inhibition in the myocardium. J Mol Cell Cardiol 44:831–854. https://doi.org/10.1016/j.yjmcc.2008.02.278
Gabisonia K, Prosdocimo G, Aquaro GD, Carlucci L, Zentilin L, Secco I, Ali H, Braga L, Gorgodze N, Bernini F, Burchielli S, Collesi C, Zandonà L, Sinagra G, Piacenti M, Zacchigna S, Bussani R, Recchia FA, Giacca M (2019) MicroRNA therapy stimulates uncontrolled cardiac repair after myocardial infarction in pigs. Nature 569:418–422
Galderisi U, Jori FP, Giordano A (2003) Cell cycle regulation and neural differentiation. Oncogene 22:5208–5219
Gan J, Tang FMK, Su X, Lu G, Xu J, Lee HSS, Lee KKH (2019) microRNA-1 inhibits cardiomyocyte proliferation in mouse neonatal hearts by repressing CCND1 expression. Ann Transl Med 7:455–455. https://doi.org/10.21037/atm.2019.08.68
Gao R, Zhang J, Cheng L, Wu X, Dong W, Yang X, Li T, Liu X, Xu Y, Li X, Zhou M (2010) A phase II, randomized, double-blind, multicenter, based on standard therapy, placebo-controlled study of the efficacy and safety of recombinant human neuregulin-1 in patients with chronic heart failure. J Am Coll Cardiol 55:1907–1914. https://doi.org/10.1016/j.jacc.2009.12.044
Gao XM, White DA, Dart AM, Du XJ (2012) Post-infarct cardiac rupture: Recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther 134:156–179
Garikipati VNS, Verma SK, Cheng Z, Liang D, Truongcao MM, Cimini M, Yue Y, Huang G, Wang C, Benedict C, Tang Y, Mallaredy V, Ibetti J, Grisanti L, Schumacher SM, Gao E, Rajan S, Wilusz JE, Goukassian D, Houser SR, Koch WJ, Kishore R (2019) Circular RNA CircFndc3b modulates cardiac repair after myocardial infarction via FUS/VEGF-A axis. Nat Commun 10:1–14. https://doi.org/10.1038/s41467-019-11777-7
Ghosh M, Song X, Mouneimne G, Sidani M, Lawrence DS, Condeelis JS (2004) Cofilin promotes actin polymerization and defines the direction of cell motility. Science (80- ) 304:743–746. https://doi.org/10.1126/science.1094561
Godwin JW, Pinto AR, Rosenthal NA (2017) Chasing the recipe for a pro-regenerative immune system. Semin Cell Dev Biol 61:71–79. https://doi.org/10.1016/j.semcdb.2016.08.008
Gratzner HG (1982) Monoclonal antibody to 5-bromo- and 5-iododeoxyuridine: a new reagent for detection of DNA replication. Science (80- ) 218:474–475. https://doi.org/10.1126/science.7123245
Greco CM, Condorelli G (2015) Epigenetic modifications and noncoding RNAs in cardiac hypertrophy and failure. Nat Rev Cardiol 12:488–497
Grego-Bessa J, Luna-Zurita L, del Monte G, Bolós V, Melgar P, Arandilla A, Garratt AN, Zang H, Suke MY, Chen H, Shou W, Ballestar E, Esteller M, Rojas A, Pérez-Pomares JM, de la Pompa JL (2007) Notch signaling is essential for ventricular chamber development. Dev Cell 12:415–429. https://doi.org/10.1016/j.devcel.2006.12.011
Gupta V, Poss KD (2012) Clonally dominant cardiomyocytes direct heart morphogenesis. Nature 484:479–484. https://doi.org/10.1038/nature11045
Harvey KF, Pfleger CM, Hariharan IK (2003) The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 114:457–467. https://doi.org/10.1016/S0092-8674(03)00557-9
Haubner BJ, Adamowicz-Brice M, Khadayate S, Tiefenthaler V, Metzler B, Aitman T, Penninger JM (2012) Complete cardiac regeneration in a mouse model of myocardial infarction. Aging (Albany NY) 4:966–977. https://doi.org/10.18632/aging.100526
Haubner BJ, Schneider J, Schweigmann U, Schuetz T, Dichtl W, Velik-Salchner C, Stein JI, Penninger JM (2016) Functional recovery of a human neonatal heart after severe myocardial infarction. Circ Res 118:216–221. https://doi.org/10.1161/CIRCRESAHA.115.307017
Heallen T, Morikawa Y, Leach J, Tao G, Willerson JT, Johnson RL, Martin JF (2013) Hippo signaling impedes adult heart regeneration. Dev 140:4683–4690. https://doi.org/10.1242/dev.102798
Hendzel MJ, Wei Y, Mancini MA, Van Hooser A, Ranalli T, Brinkley BR, Bazett-Jones DP, Allis CD (1997) Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma 106:348–360. https://doi.org/10.1007/s004120050256
Hirata A, Inada KI, Tsukamoto T, Sakai H, Mizoshita T, Yanai T, Masegi T, Goto H, Inagaki M, Tatematsu M (2004) Characterization of a monoclonal antibody, HTA28, recognizing a histone H3 phosphorylation site as a useful marker of M-phase cells. J Histochem Cytochem 52:1503–1509. https://doi.org/10.1369/jhc.4A6285.2004
Hou B, Jian Z, Cui P, Li S, Tian R, Ou J (2015) miR-216a may inhibit pancreatic tumor growth by targeting JAK2. FEBS Lett 589:2224–2232. https://doi.org/10.1016/j.febslet.2015.06.036
Høydal MA, Kirkeby-Garstad I, Karevold A, Wiseth R, Haaverstad R, Wahba A, Stølen TL, Contu R, Condorelli G, Ellingsen Ø, Smith GL, Kemi OJ, Wisløff U (2018) Human cardiomyocyte calcium handling and transverse tubules in mid-stage of post-myocardial-infarction heart failure. ESC Hear Fail 5:332–342. https://doi.org/10.1002/ehf2.12271
Hu Y, Jin G, Li B, Chen Y, Zhong L, Chen G, Chen X, Zhong J, Liao W, Liao Y, Wang Y, Bin J (2019) Suppression of miRNA let-7i-5p promotes cardiomyocyte proliferation and repairs heart function post injury by targetting CCND2 and E2F2. Clin Sci 133:425–441. https://doi.org/10.1042/CS20181002
Huang CK, Kafert-Kasting S, Thum T (2020) Preclinical and clinical development of noncoding RNA therapeutics for cardiovascular disease. Circ Res 126:663–678
Huang S, Li X, Zheng H, Si X, Li B, Wei G, Li C, Chen Y, Chen Y, Liao W, Liao Y, Bin J (2019) Loss of super-enhancer-regulated circRNA Nfix Induces cardiac regeneration after myocardial infarction in adult mice. Circulation 139:2857–2876. https://doi.org/10.1161/CIRCULATIONAHA.118.038361
Huang W, Feng Y, Liang J, Yu H, Wang C, Wang B, Wang M, Jiang L, Meng W, Cai W, Medvedovic M, Chen J, Paul C, Davidson WS, Sadayappan S, Stambrook PJ, Yu XY, Wang Y (2018) Loss of microRNA-128 promotes cardiomyocyte proliferation and heart regeneration. Nat Commun 9. doi: 10.1038/s41467-018-03019-z
Ieda M, Fu JD, Delgado-Olguin P, Vedantham V, Hayashi Y, Bruneau BG, Srivastava D (2010) Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142:375–386. https://doi.org/10.1016/j.cell.2010.07.002
Irvine KD (2012) Integration of intercellular signaling through the Hippo pathway. Semin Cell Dev Biol 23:812–817
Jabbour A, Hayward CS, Keogh AM, Kotlyar E, McCrohon JA, England JF, Amor R, Liu X, Li XY, Zhou MD, Graham RM, Macdonald PS (2011) Parenteral administration of recombinant human neuregulin-1 to patients with stable chronic heart failure produces favourable acute and chronic haemodynamic responses. Eur J Heart Fail 13:83–92. https://doi.org/10.1093/eurjhf/hfq152
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE (2013) Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 19:141–157. https://doi.org/10.1261/rna.035667.112
Johnsson P, Lipovich L, Grandér D, Morris KV (2014) Evolutionary conservation of long non-coding RNAs; Sequence, structure, function. Biochim Biophys Acta - Gen Subj 1840:1063–1071
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud H-R, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 6:844–851. https://doi.org/10.1038/ni1229
Jopling C, Sleep E, Raya M, Martí M, Raya A, Belmonte JCI (2010) Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 464:606–609. https://doi.org/10.1038/nature08899
Jurchott K, Bergmann S, Stein U, Walther W, Janz M, Manni I, Piaggio G, Fietze E, Dietel M, Royer H-D (2003) YB-1 as a cell cycle-regulated transcription factor facilitating cyclin A and cyclin B1 gene expression. J Biol Chem 278:27988–27996. https://doi.org/10.1074/jbc.M212966200
Kern F, Fehlmann T, Solomon J, Schwed L, Grammes N, Backes C, Van Keuren-Jensen K, Craig DW, Meese E, Keller A (2020) miEAA 2.0: integrating multi-species microRNA enrichment analysis and workflow management systems. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa309
Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, Poss KD (2011) Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev Cell 20:397–404. https://doi.org/10.1016/j.devcel.2011.01.010
Kimura W, Xiao F, Canseco DC, Muralidhar S, Thet S, Zhang HM, Abderrahman Y, Chen R, Garcia JA, Shelton JM, Richardson JA, Ashour AM, Asaithamby A, Liang H, Xing C, Lu Z, Zhang CC, Sadek HA (2015) Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 523:226–230. https://doi.org/10.1038/nature14582
Klattenhoff CA, Scheuermann JC, Surface LE, Bradley RK, Fields PA, Steinhauser ML, Ding H, Butty VL, Torrey L, Haas S, Abo R, Tabebordbar M, Lee RT, Burge CB, Boyer LA (2013) Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 152:570–583. https://doi.org/10.1016/j.cell.2013.01.003
Kramer CM, Rogers WJ, Park CS, Seibel PS, Shaffer A, Theobald TM, Reichek N, Onodera T, Gerdes AM (1998) Regional myocyte hypertrophy parallels regional myocardial dysfunction during post-infarct remodeling. J Mol Cell Cardiol 30:1773–1778. https://doi.org/10.1006/jmcc.1998.0741
Leach JP, Heallen T, Zhang M, Rahmani M, Morikawa Y, Hill MC, Segura A, Willerson JT, Martin JF (2017) Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 550:260–264. https://doi.org/10.1038/nature24045
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75:843–854. https://doi.org/10.1016/0092-8674(93)90529-Y
Lesizza P, Prosdocimo G, Martinelli V, Sinagra G, Zacchigna S, Giacca M (2017) Single-dose intracardiac injection of pro-regenerative microRNAs improves cardiac function after myocardial infarction. Circ Res 120:1298–1304. https://doi.org/10.1161/CIRCRESAHA.116.309589
Li B, Hu Y, Li X, Jin G, Chen X, Chen G, Chen Y, Huang S, Liao W, Liao Y, Teng Z, Bin J (2018) Sirt1 antisense long noncoding RNA promotes cardiomyocyte proliferation by enhancing the stability of sirt1. J Am Heart Assoc 7:e009700. https://doi.org/10.1161/JAHA.118.009700
Li F, Wang X, Capasso JM, Gerdes AM (1996) Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28:1737–1746. https://doi.org/10.1006/jmcc.1996.0163
Li X, He X, Wang H, Li M, Huang S, Chen G, Jing Y, Wang S, Chen Y, Liao W, Liao Y, Bin J (2018) Loss of AZIN2 splice variant facilitates endogenous cardiac regeneration. Cardiovasc Res 114:1642–1655. https://doi.org/10.1093/cvr/cvy075
Li X, Wang J, Jia Z, Cui Q, Zhang C, Wang W, Chen P, Ma K, Zhou C (2013) MiR-499 regulates cell proliferation and apoptosis during late-stage cardiac differentiation via Sox6 and cyclin D1. PLoS One 8. doi: 10.1371/journal.pone.0074504
Li Z, Huang C, Bao C, Chen L, Lin M, Wang X, Zhong G, Yu B, Hu W, Dai L, Zhu P, Chang Z, Wu Q, Zhao Y, Jia Y, Xu P, Liu H, Shan G (2015) Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol 22:256–264. https://doi.org/10.1038/nsmb.2959
Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN (2008) microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev 22:3242–3254. https://doi.org/10.1101/gad.1738708
Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA (2013) Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 497:249–253. https://doi.org/10.1038/nature12054
Martinez-Sanchez A, Murphy CL (2013) MicroRNA target identification-experimental approaches. Biology (Basel) 2:189–205
Matkovich SJ, Hu Y, Dorn GW (2013) Regulation of cardiac microRNAs by cardiac microRNAs. Circ Res 113:62–71. https://doi.org/10.1161/CIRCRESAHA.113.300975
McKinnon CM, Mellor H (2017) The tumor suppressor RhoBTB1 controls Golgi integrity and breast cancer cell invasion through METTL7B. BMC Cancer 17. doi: 10.1186/s12885-017-3138-3
Meckert PC, Rivello HG, Vigliano C, González P, Favaloro R, Laguens R (2005) Endomitosis and polyploidization of myocardial cells in the periphery of human acute myocardial infarction. Cardiovasc Res 67:116–123. https://doi.org/10.1016/j.cardiores.2005.02.017
Memczak S, Jens M, Elefsinioti A, Torti F, Krueger J, Rybak A, Maier L, Mackowiak SD, Gregersen LH, Munschauer M, Loewer A, Ziebold U, Landthaler M, Kocks C, Le Noble F, Rajewsky N (2013) Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495:333–338. https://doi.org/10.1038/nature11928
Menasché P, Vanneaux V, Hagège A, Bel A, Cholley B, Cacciapuoti I, Parouchev A, Benhamouda N, Tachdjian G, Tosca L, Trouvin JH, Fabreguettes JR, Bellamy V, Guillemain R, Suberbielle Boissel C, Tartour E, Desnos M, Larghero J (2015) Human embryonic stem cell-derived cardiac progenitors for severe heart failure treatment: First clinical case report. Eur Heart J 36:2011–2017. https://doi.org/10.1093/eurheartj/ehv189
Mercer TR, Dinger ME, Mattick JS (2009) Long non-coding RNAs: Insights into functions. Nat Rev Genet 10:155–159
Ming G-F, Wu K, Hu K, Chen Y, Xiao J (2016) NAMPT regulates senescence, proliferation, and migration of endothelial progenitor cells through the SIRT1 AS lncRNA/miR-22/SIRT1 pathway. Biochem Biophys Res Commun 478:1382–1388. https://doi.org/10.1016/j.bbrc.2016.08.133
Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, Wei C, Frazier M, Samson O, Wong KK, Kim C, Camargo FD (2014) A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol 16:108–117. https://doi.org/10.1038/ncb2884
Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah AM, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Hu Z, Deberardinis RJ, Schiattarella G, Hill JA, Oz O, Lu Z, Zhang CC, Kimura W, Sadek HA (2017) Hypoxia induces heart regeneration in adult mice. Nature 541:222–227. https://doi.org/10.1038/nature20173
Nguyen TH, Ralbovska A, Kugler JM (2020) RhoBTB proteins regulate the hippo pathway by antagonizing ubiquitination of LKB1. G3 (Bethesda) 10:1319–1325. doi: 10.1534/g3.120.401038
Nunes SS, Miklas JW, Liu J, Aschar-Sobbi R, Xiao Y, Zhang B, Jiang J, Massé S, Gagliardi M, Hsieh A, Thavandiran N, Laflamme MA, Nanthakumar K, Gross GJ, Backx PH, Keller G, Radisic M (2013) Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat Methods 10:781–787. https://doi.org/10.1038/nmeth.2524
Nussbaum J, Minami E, Laflamme MA, Virag JAI, Ware CB, Masino A, Muskheli V, Pabon L, Reinecke H, Murry CE (2007) Transplantation of undifferentiated murine embryonic stem cells in the heart: teratoma formation and immune response. FASEB J 21:1345–1357. https://doi.org/10.1096/fj.06-6769com
Ounzain S, Micheletti R, Arnan C, Plaisance I, Cecchi D, Schroen B, Reverter F, Alexanian M, Gonzales C, Ng SY, Bussotti G, Pezzuto I, Notredame C, Heymans S, Guigó R, Johnson R, Pedrazzini T (2015) CARMEN, a human super enhancer-associated long noncoding RNA controlling cardiac specification, differentiation and homeostasis. J Mol Cell Cardiol 89:98–112. https://doi.org/10.1016/j.yjmcc.2015.09.016
Ounzain S, Micheletti R, Beckmann T, Schroen B, Alexanian M, Pezzuto I, Crippa S, Nemir M, Sarre A, Johnson R, Dauvillier J, Burdet F, Ibberson M, Guigó R, Xenarios I, Heymans S, Pedrazzini T (2015) Genome-wide profiling of the cardiac transcriptome after myocardial infarction identifies novel heart-specific long non-coding RNAs. Eur Heart J 36:353–368. https://doi.org/10.1093/eurheartj/ehu180
Pamudurti NR, Bartok O, Jens M, Ashwal-Fluss R, Stottmeister C, Ruhe L, Hanan M, Wyler E, Perez-Hernandez D, Ramberger E, Shenzis S, Samson M, Dittmar G, Landthaler M, Chekulaeva M, Rajewsky N, Kadener S (2017) Translation of CircRNAs. Mol Cell 66:9–21.e7. https://doi.org/10.1016/j.molcel.2017.02.021
Pandey R, Velasquez S, Durrani S, Jiang M, Neiman M, Crocker JS, Benoit JB, Rubinstein J, Paul A, Ahmed RPH (2017) MicroRNA-1825 induces proliferation of adult cardiomyocytes and promotes cardiac regeneration post ischemic injury. Am J Transl Res 9:3120–3137
Pang KC, Frith MC, Mattick JS (2006) Rapid evolution of noncoding RNAs: Lack of conservation does not mean lack of function. Trends Genet 22:1–5
Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, Shen H, Allayee H, Crump JG, Force TI, Lien CL, Makita T, Lusis AJ, Kumar SR, Sucov HM (2017) Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet 49:1346–1353. https://doi.org/10.1038/ng.3929
Pedrazzini T (2007) Control of cardiogenesis by the notch pathway. Trends Cardiovasc Med 17:83–90
Piccoli M-T, Gupta SK, Thum T (2015) Noncoding RNAs as regulators of cardiomyocyte proliferation and death. J Mol Cell Cardiol 89:59–67. https://doi.org/10.1016/j.yjmcc.2015.02.002
Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F (2013) Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front Physiol 4 MAY. doi: 10.3389/fphys.2013.00102
Ponnusamy M, Liu F, Zhang Y-H, Li R-B, Zhai M, Liu F, Zhou L-Y, Liu C-Y, Yan K-W, Dong Y-H, Wang M, Qian L-L, Shan C, Xu S, Wang Q, Zhang Y-H, Li P-F, Zhang J, Wang K (2019) Long noncoding RNA CPR (Cardiomyocyte Proliferation Regulator) regulates cardiomyocyte proliferation and cardiac repair. Circulation 139:2668–2684. https://doi.org/10.1161/CIRCULATIONAHA.118.035832
Poon CLC, Lin JI, Zhang X, Harvey KF (2011) The sterile 20-like kinase Tao-1 controls tissue growth by regulating the Salvador-Warts-Hippo pathway. Dev Cell 21:896–906. https://doi.org/10.1016/j.devcel.2011.09.012
Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, Sadek HA (2011) Transient regenerative potential of the neonatal mouse heart. Science (80- ) 331:1078–1080. https://doi.org/10.1126/science.1200708
Porrello ER, Olson EN (2014) A neonatal blueprint for cardiac regeneration. Stem Cell Res 13:556–570
Poss KD (2010) Advances in understanding tissue regenerative capacity and mechanisms in animals. Nat Rev Genet 11:710–722
Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, Grinsfelder D, Rothermel BA, Chen R, Garcia JA, Santos CX, Thet S, Mori E, Kinter MT, Rindler PM, Zacchigna S, Mukherjee S, Chen DJ, Mahmoud AI, Giacca M, Rabinovitch PS, Aroumougame A, Shah AM, Szweda LI, Sadek HA (2014) The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell 157:565–579. https://doi.org/10.1016/j.cell.2014.03.032
Ragina NP, Schlosser K, Knott JG, Senagore PK, Swiatek PJ, Chang EA, Fakhouri WD, Schutte BC, Kiupel M, Cibelli JB (2012) Downregulation of H19 improves the differentiation potential of mouse parthenogenetic embryonic stem cells. Stem Cells Dev 21:1134–1144. https://doi.org/10.1089/scd.2011.0152
Reuter S, Soonpaa MH, Firulli AB, Chang AN, Field LJ (2014) Recombinant neuregulin 1 does not activate cardiomyocyte DNA synthesis in normal or infarcted adult mice. PLoS One 9. doi: 10.1371/journal.pone.0115871
Salzberg SL (2018) Open questions: How many genes do we have? BMC Biol 16:94
Salzman J, Chen RE, Olsen MN, Wang PL, Brown PO (2013) Cell-Type Specific Features of Circular RNA Expression. PLoS Genet 9. doi: 10.1371/journal.pgen.1003777
Senyo SE, Steinhauser ML, Pizzimenti CL, Yang VK, Cai L, Wang M, Wu T-D, Guerquin-Kern J-L, Lechene CP, RTL (2017) Mammalian heart renewal by preexisting cardiomyocytes. Physiol Behav 176:139–148. https://doi.org/10.1016/j.physbeh.2017.03.040
Shao Y, Chen Y (2016) Roles of circular RNAs in neurologic disease. Front Mol Neurosci 9:25. https://doi.org/10.3389/fnmol.2016.00025
Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T, Wang H, Xuan Q, Chen P, Xu J, Che L, Liu H, Zhong J, Sluijter JPG, Li X, Rosenzweig A, Xiao J (2017) miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial ischemia-reperfusion injury. Theranostics 7:664–676. https://doi.org/10.7150/thno.15162
Shiba Y, Gomibuchi T, Seto T, Wada Y, Ichimura H, Tanaka Y, Ogasawara T, Okada K, Shiba N, Sakamoto K, Ido D, Shiina T, Ohkura M, Nakai J, Uno N, Kazuki Y, Oshimura M, Minami I, Ikeda U (2016) Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 538:388–391. https://doi.org/10.1038/nature19815
Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469:336–342
Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, Kim KK, Field LJ (1997) Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest 99:2644–2654. https://doi.org/10.1172/JCI119453
Soufan AT, van den Berg G, Ruijter JM, de Boer PAJ, van den Hoff MJB, Moorman AFM (2006) Regionalized sequence of myocardial cell growth and proliferation characterizes early chamber formation. Circ Res 99:545–552. https://doi.org/10.1161/01.RES.0000239407.45137.97
Stankunas K, Shang C, Twu KY, Kao S-C, Jenkins NA, Copeland NG, Sanyal M, Selleri L, Cleary ML, Chang C-P (2008) Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ Res 103:702–709. https://doi.org/10.1161/CIRCRESAHA.108.175489
Sun X, Kaufman PD (2018) Ki-67: more than a proliferation marker. Chromosoma 127:175–186
Tapon N, Harvey KF, Bell DW, Wahrer DCR, Schiripo TA, Haber DA, Hariharan IK (2002) salvador promotes both cell cycle exit and apoptosis in Drosophila and is mutated in human cancer cell lines. Cell 110:467–478. https://doi.org/10.1016/S0092-8674(02)00824-3
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM (2006) Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 20:2202–2207. https://doi.org/10.1101/gad.1444406
Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J (2007) MicroRNAs in the human heart. Circulation 116:258–267. https://doi.org/10.1161/CIRCULATIONAHA.107.687947
Tian Y, Liu Y, Wang T, Zhou N, Kong J, Chen L, Snitow M, Morley M, Li D, Petrenko N, Zhou S, Lu M, Gao E, Koch WJ, Stewart KM, Morrisey EE (2015) A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci Transl Med 7:279ra38. doi: 10.1126/scitranslmed.3010841
Tirziu D, Giordano FJ, Simons M (2010) Cell communications in the heart. Circulation 122:928–937. https://doi.org/10.1161/CIRCULATIONAHA.108.847731
Torrini C, Cubero RJ, Dirkx E, Braga L, Ali H, Prosdocimo G, Gutierrez MI, Collesi C, Licastro D, Zentilin L, Mano M, Zacchigna S, Vendruscolo M, Marsili M, Samal A, Giacca M (2019) Common regulatory pathways mediate activity of microRNAs inducing cardiomyocyte proliferation. Cell Rep 27:2759–2771.e5. https://doi.org/10.1016/j.celrep.2019.05.005
Trivedi CM, Zhu W, Wang Q, Jia C, Kee HJ, Li L, Hannenhalli S, Epstein JA (2010) Hopx and Hdac2 interact to modulate Gata4 acetylation and embryonic cardiac myocyte proliferation. Dev Cell 19:450–459. https://doi.org/10.1016/j.devcel.2010.08.012
Viereck J, Kumarswamy R, Foinquinos A, Xiao K, Avramopoulos P, Kunz M, Dittrich M, Maetzig T, Zimmer K, Remke J, Just A, Fendrich J, Scherf K, Bolesani E, Schambach A, Weidemann F, Zweigerdt R, de Windt LJ, Engelhardt S, Dandekar T, Batkai S, Thum T (2016) Long noncoding RNA Chast promotes cardiac remodeling. Sci Transl Med 8:326ra22. https://doi.org/10.1126/scitranslmed.aaf1475
Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Shay CM, Spartano NL, Stokes A, Tirschwell DL, VanWagner LB, Tsao CW (2020) Heart disease and stroke statistics-2020 update: a report from the american heart association. Circulation 141:e139–e596. https://doi.org/10.1161/CIR.0000000000000757
von Gise A, Lin Z, Schlegelmilch K, Honor LB, Pan GM, Buck JN, Ma Q, Ishiwata T, Zhou B, Camargo FD, Pu WT (2012) YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc Natl Acad Sci USA 109:2394–2399. https://doi.org/10.1073/pnas.1116136109
Vujic A, Lerchenmüller C, Wu T Di, Guillermier C, Rabolli CP, Gonzalez E, Senyo SE, Liu X, Guerquin-Kern JL, Steinhauser ML, Lee RT, Rosenzweig A (2018) Exercise induces new cardiomyocyte generation in the adult mammalian heart. Nat Commun 9. doi: 10.1038/s41467-018-04083-1
Vujic A, Natarajan N, Lee RT (2020) Molecular mechanisms of heart regeneration. Semin Cell Dev Biol 100:20–28
Wang GQ, Wang Y, Xiong Y, Chen XC, Ma ML, Cai R, Gao Y, Sun YM, Yang GS, Pang WJ (2016) Sirt1 AS lncRNA interacts with its mRNA to inhibit muscle formation by attenuating function of miR-34a. Sci Rep 6:1–13. https://doi.org/10.1038/srep21865
Wang J, Chen X, Shen D, Ge D, Chen J, Pei J, Li Y, Yue Z, Feng J, Chu M, Nie Y (2019) A long noncoding RNA NR_045363 controls cardiomyocyte proliferation and cardiac repair. J Mol Cell Cardiol 127:105–114. https://doi.org/10.1016/j.yjmcc.2018.12.005
Wang K, Long B, Liu F, Wang J-X, Liu C-Y, Zhao B, Zhou L-Y, Sun T, Wang M, Yu T, Gong Y, Liu J, Dong Y-H, Li N, Li P-F (2016) A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J 37:2602–2611. https://doi.org/10.1093/eurheartj/ehv713
Wang K, Long B, Zhou L-Y, Liu F, Zhou Q-Y, Liu C-Y, Fan Y-Y, Li P-F (2014) CARL lncRNA inhibits anoxia-induced mitochondrial fission and apoptosis in cardiomyocytes by impairing miR-539-dependent PHB2 downregulation. Nat Commun 5:3596. https://doi.org/10.1038/ncomms4596
Wang PL, Bao Y, Yee M-C, Barrett SP, Hogan GJ, Olsen MN, Dinneny JR, Brown PO, Salzman J (2014) Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 9:e90859. https://doi.org/10.1371/journal.pone.0090859
Wang T, Pan W, Hu J, Zhang Z, Li G, Liang Y (2018) Circular RNAs in metabolic diseases. Advances in experimental medicine and biology. Springer, New York LLC, pp 275–285
Wang Z, Zhang X-J, Ji Y-X, Zhang P, Deng K-Q, Gong J, Ren S, Wang X, Chen I, Wang H, Gao C, Yokota T, Ang YS, Li S, Cass A, Vondriska TM, Li G, Deb A, Srivastava D, Yang H-T, Xiao X, Li H, Wang Y (2016) The long noncoding RNA Chaer defines an epigenetic checkpoint in cardiac hypertrophy. Nat Med 22:1131–1139. https://doi.org/10.1038/nm.4179
Westholm JO, Miura P, Olson S, Shenker S, Joseph B, Sanfilippo P, Celniker SE, Graveley BR, Lai EC (2014) Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 9:1966–1980. https://doi.org/10.1016/j.celrep.2014.10.062
Wu H, Zhao Z-A, Liu J, Hao K, Yu Y, Han X, Li J, Wang Y, Lei W, Dong N, Shen Z, Hu S (2018) Long noncoding RNA Meg3 regulates cardiomyocyte apoptosis in myocardial infarction. Gene Ther 25:511–523. https://doi.org/10.1038/s41434-018-0045-4
Xiang FL, Guo M, Yutzey KE (2016) Overexpression of Tbx20 in adult cardiomyocytes promotes proliferation and improves cardiac function after myocardial infarction. Circulation 133:1081–1092. https://doi.org/10.1161/CIRCULATIONAHA.115.019357
Xiao J, Liu H, Cretoiu D, Toader DO, Suciu N, Shi J, Shen S, Bei Y, Sluijter JPG, Das S, Kong X, Li X (2017) Mir-31a-5p promotes postnatal cardiomyocyte proliferation by targeting RhoBTB1. Exp Mol Med 49:386. https://doi.org/10.1038/emm.2017.150
Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, Richardson JA, Sadek HA, Bassel-Duby R, Olson EN (2013) Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci USA 110:13839–13844. https://doi.org/10.1073/pnas.1313192110
Yang Q, Wu F, Mi Y, Wang F, Cai K, Yang X, Zhang R, Liu L, Zhang Y, Wang Y, Wang X, Xu M, Gui Y, Li Q (2020) Aberrant expression of miR-29b-3p influences heart development and cardiomyocyte proliferation by targeting NOTCH2. Cell Prolif 53:e12764. https://doi.org/10.1111/cpr.12764
Yang Y, Cheng HW, Qiu Y, Dupee D, Noonan M, Lin YD, Fisch S, Unno K, Sereti KI, Liao R (2015) MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circ Res 117:450–459. https://doi.org/10.1161/CIRCRESAHA.117.305962
Yin VP, Lepilina A, Smith A, Poss KD (2012) Regulation of zebrafish heart regeneration by miR-133. Dev Biol 365:319–327. https://doi.org/10.1016/j.ydbio.2012.02.018
Yin Y, Long J, He Q, Li Y, Liao Y, He P, Zhu W (2019) Emerging roles of circRNA in formation and progression of cancer. J Cancer 10:5015–5021
Zeng Y, Du WW, Wu Y, Yang Z, Awan FM, Li X, Yang W, Zhang C, Yang Q, Yee A, Chen Y, Yang F, Sun H, Huang R, Yee AJ, Li RK, Wu Z, Backx PH, Yang BB (2017) A circular RNA binds to and activates AKT phosphorylation and nuclear localization reducing apoptosis and enhancing cardiac repair. Theranostics 7:3842–3855. https://doi.org/10.7150/thno.19764
Zhang YM, Hartzell C, Narlow M, Dudley SC (2002) Stem cell-derived cardiomyocytes demonstrate arrhythmic potential. Circulation 106:1294–1299. https://doi.org/10.1161/01.cir.0000027585.05868.67
Zhao B, Lei Q-Y, Guan K-L (2008) The Hippo-YAP pathway: new connections between regulation of organ size and cancer. Curr Opin Cell Biol 20:638–646. https://doi.org/10.1016/j.ceb.2008.10.001
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCFβ-TRCP. Genes Dev 24:72–85. https://doi.org/10.1101/gad.1843810
Zhao B, Tumaneng K, Guan KL (2011) The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat Cell Biol 13:877–883
Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, Lai ZC, Guan KL (2008) TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22:1962–1971. https://doi.org/10.1101/gad.1664408
Zhao C, Sun G, Li S, Shi Y (2009) A feedback regulatory loop involving microRNA-9 and nuclear receptor TLX in neural stem cell fate determination. Nat Struct Mol Biol 16:365–371. https://doi.org/10.1038/nsmb.1576
Zhao L, Borikova AL, Ben-Yair R, Guner-Ataman B, MacRae CA, Lee RT, Geoffrey Burns C, Burns CE (2014) Notch signaling regulates cardiomyocyte proliferation during zebrafish heart regeneration. Proc Natl Acad Sci USA 111:1403–1408. https://doi.org/10.1073/pnas.1311705111
Zheng Q, Bao C, Guo W, Li S, Chen J, Chen B, Luo Y, Lyu D, Li Y, Shi G, Liang L, Gu J, He X, Huang S (2016) Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Commun 7:1–13. https://doi.org/10.1038/ncomms11215
Zhu W, Zhang E, Zhao M, Chong Z, Fan C, Tang Y, Hunter JD, Borovjagin AV, Walcott GP, Chen JY, Qin G, Zhang J (2018) Regenerative potential of neonatal porcine hearts. Circulation 138:2809–2816. https://doi.org/10.1161/CIRCULATIONAHA.118.034886
Acknowledgements
Open Access funding provided by Projekt DEAL. Prof. Thum was supported by grants from ERC Longheart and ERANET CVD Expert and KFO311.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
TT is founder and shareholder of Cardior Pharmaceuticals GmbH. All the other authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Abbas, N., Perbellini, F. & Thum, T. Non-coding RNAs: emerging players in cardiomyocyte proliferation and cardiac regeneration. Basic Res Cardiol 115, 52 (2020). https://doi.org/10.1007/s00395-020-0816-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-020-0816-0