
Abstract
A new developed tunable diode laser spectrometer for the measurement of ammonia (NH3) mole fractions in exhaust gas matrices with strong CO2 and H2O background at temperatures up to 800 K is presented. In situ diagnostics in harsh exhaust environments during SCR after treatment are enabled by the use of ammonia transitions in the ν2 + ν3 near-infrared band around 2300 nm. Therefore, three lines have been selected, coinciding near 2200.5 nm (4544.5 cm−1) with rather weak temperature dependency and minimal interference with CO2 and H2O. A fiber-coupled 2.2-μm distributed feedback laser diode was used and attached to the hot gas flow utilizing adjustable gas tight high-temperature fiber ports. The spectrometer spans four coplanar optical channels across the measurement plane and simultaneously detects the direct absorption signal via a fiber-coupled detector unit. An exhaust simulation test rig was used to characterize the spectrometer’s performance in ammonia-doped hot gas environments. We achieved a temporal resolution of 13 Hz and temperature-dependent precisions of NH3 mole fraction ranging from 50 to 70 ppmV. There the spectrometer achieved normalized ammonia detection limits of 7–10 \({\text{ppm}}_{\text{V}}\cdot{\text{m}}\) and 2–3 \({\text{ppm}}_{\text{V}}\cdot{\text{m}}/\sqrt{\text{Hz}}\).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Modern regulations of motor vehicle exhaust pollutants require further decrease in emissions, particularly unburned hydrocarbons (HC), carbon monoxide (CO), nitrogen oxides (NOx) and particulate matter (PM) or number (PN) [1, 2]. To meet upcoming European regulation standards for diesel engines and comparable regulations worldwide, like US Environmental Protection Agency’s Tier 3 [3] for example, an improved exhaust aftertreatment approach is necessary. The combination of diesel oxidation catalysts (DOC), diesel particulate filters (DPF) and selective catalytic reduction (SCR) systems is a state of the art and promising exhaust gas treatment concept already in use on heavy-duty vehicles as well as passenger cars. The SCR technology uses ammonia (NH3) as the reactant with NOx directly in the exhaust. Due to the risks and restrictions when handling gaseous ammonia, a non-toxic aqueous urea solution consisting of 32.5 % urea (AUS32/AdBlue) [4] is commonly used to provide a liquid pre-stage of ammonia.
A modern diesel exhaust system is quite complex and equipped with several sensors for air–fuel ratio λ, NOx, temperature and pressure drop. For SCR, AUS32 is injected downstream of the DOC and DPF, forming H2O, CO2 and NH3. The latter reduces the NOx to water vapor and molecular nitrogen in a subsequent catalyst [5]. With urea-SCR and an optimal NO/NO2 ratio of 1, NOx conversion rates up to 100 % can be achieved [6].
The drawbacks of a SCR system are evidently the increased complexity, higher weight and the need to refill an extra operating fluid. However, unlike the concurrent denox technology lean NOx trap (LNT), the SCR technology does not impede engine optimization concerning performance and efficiency. Thus, up to 10 % lower fuel consumption compared to vehicles using LNT can be achieved and even operational costs decrease [7], which is why SCR is the only exhaust aftertreatment system used by European heavy-duty vehicle manufacturers and also applied in mid-size and bigger passenger cars [5].
In current applications, the amount of AUS32 injected is always less than the stoichiometric ratio in order to avoid ammonia downstream of the catalyst, also known as ammonia slip. This is contradictory to maximum catalyst performance. In particular, in vehicle applications, the control of this technology is based on an engine performance map and for correct dosing all possible operational states of an engine must be investigated and characterized during the design process. Since NOx is evenly distributed in the exhaust gas, the injection system needs to deliver ammonia homogeneously to prevent local under- or overdosing.
For current and future developments of SCR systems, measurement methods for exhaust diagnostics must be able to measure ammonia mole fractions directly in the exhaust pipe with low cross-sensitivity toward other exhaust species and deliver information about the spatial distribution. A high temporal resolution is essential to extend diagnostic possibilities to unsteady or transitional operational states. Ammonia concentration in exhaust applications is commonly detected with mass spectrometers [6, 8] or Fourier transform infrared spectrometers [9], which both have temporal resolutions in the range of seconds. Other ammonia sensors make use of absorption spectroscopy in various spectral regions with multipass cells [10–12] or other laboratory setups with absorption paths in the range of meters [13–15] that make low detection limits possible. However, most commercially available systems work with a gas sample that has to be extracted from the exhaust and routed to the system [e.g., AVL AMA i60, Fraunhofer IPM DEGAS IV (1 ppmV, 10 ms)]. The extractive methods are invasive, can change the gas composition and have a fixed region of extraction. Spatial resolution can thus only be obtained by sequentially scanning different measurement points, which can be quite cumbersome and time-consuming. In addition to that, these devices need controlled environments and are quite complex to set up for complete engine measurements. One in situ TDLAS measurement system suitable for analyzing hot gas is the SIEMENS LDS6 (0.5 ppmV, 1.25 m, 3 s) employing rather big sensor lances (163 mm outer diameter, 765 mm length). With its dimensions and a mandatory absorption length of at least 1 m, its usability in exhaust applications is limited to a single path parallel to the flow direction. Exhaust diagnosis directly in front of or behind the SCR catalyst and spatial analysis would therefore not be possible with this system.
Our approach is a fiber-coupled TDLAS-based spectrometer with multiple optical channels integrated into the exhaust system in planar alignment perpendicular to the flow. Measuring absolute mole fractions without the need for calibration paired with high temporal resolution can result in more reliable data about NH3 inhomogeneity at the catalyst surface.
2 Direct TDL absorption spectroscopy
2.1 Measurement principle
For the in situ measurements of absolute mole fractions, we used direct absorption spectroscopy (DAS) [16–21]. This method utilizes the resonant photon absorption of molecules by tuning a diode laser in the wavelength range of one or more molecule specific absorption lines. The TDLAS principle therefore measures the wavelength-dependent extinction of laser light having passed the sample volume. The correlation between detected intensity and number density of the gas species is described by the Beer–Lambert law [22]:
Herein, I 0(ν) and I(ν) define the incident and the attenuated laser light intensity, respectively. Furthermore, the absorption coefficient includes the temperature-dependent line strength S(T), the number density N, the absorption path length L and the normalized line shape function g(ν − ν 0 ) at the central frequency ν 0. It is essentially determined by Doppler and collision broadening. Due to broadband absorption, particle scattering and beam steering, a significant part of the light transmission can be strongly attenuated, which is considered by the term Tr(t). Furthermore, an additional amount of background radiation (i.e., thermal radiation) will be incorporated by the emission correction E(t).
Rearranging Eq. (1) with respect to N and integration of the line shape function over the spectral tuning range yield [22]:
With knowledge of the absorption length L, the laser tuning coefficient ∂ν/∂t and the line strength, the number density can be calculated. The laser tuning coefficient is a characteristic of the diode laser, while the line strength can be extracted from spectroscopic databases (e.g., HITRAN2012, [23]) or measured in prior laboratory experiments. The transmission and emission corrections (Tr(t), E(t)) can be determined from the detected signal itself. The absorption line area is extracted by using a recursive Levenberg–Marquardt algorithm to fit Voigt line shapes [24] to the detected line profile. Together with the measurement of temperature T and pressure p, the ideal gas law can be applied to calculate the absolute mole fraction of the gas species. Due to the fact that all parameters are known or can be measured simultaneously with the laser signal, this method does not require any additional in situ calibration.
The TDLAS principle offers the possibility to measure absolute gas species concentration in harsh environments (see [16, 25]), just like NH3 in hot exhaust gases. In situ application and fast response times allow for direct measurement of NH3 even in non-steady engine states. As a line-of-sight measurement technique, a single TDLAS absorption path is sufficient for spatially integrated concentration measurements, whereas multiple beams can deliver an instant image of the concentration distribution, e.g., by tomography [26, 27].
The detection limit Δx molec which we discuss in this paper is defined as:
where A peak(ν 0 ) describes the peak absorption at the line center, ΔA residual the standard deviation 1σ of the fit residual over the complete tuning range and x molec the measured absolute mole fraction of the molecule. This way, the detection limit is found at a signal-to-noise ratio of 1. In the context of the mole fraction measurements shown here, the unit ppmV (parts per million) is used to describe the detection limit for the given boundary conditions (path length, acquisition rate). To compare the minimal detectable mole fraction with other configurations, the unit \({\text{ppm}}_{\text{V}}\cdot{\text{m}}\) gives the normalization regarding the path length and \({\text{ppm}}_{\text{V}}\cdot{\text{m}}/\sqrt{\text{Hz}}\) the additional normalization regarding the integration time. In a noise-dominated signal, the detection limit (ppmV) can be improved with an increasing path length, which can be expressed by the path normalization (\({\text{ppm}}_{\text{V}}\cdot{\text{m}}\)). In contrast, a spectral background from interfering species will also increase in conjunction with the increasing path length and will therefore not affect the detection limit. This has to be considered during line selection, spectrometer design and discussion of the results.
2.2 Absorption line selection
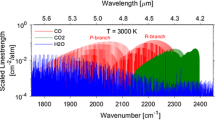
Ammonia absorption measurements are commonly conducted in the 1.5-µm band [10, 13, 28–31] of the near-infrared region (NIR) or the stronger absorption band in the mid IR (MIR > 3 µm [32–34]). Due to the higher line strength, the MIR bands are used for trace gas ammonia. Since the use of quantum cascade lasers (QCL), necessary MIR optics and MIR detection components is possible [35–38], but not yet convenient for industrial application, multiple studies have been done in the very weak NIR bands. Well-developed diode lasers, detection systems, optics as well as fiber delivery modules are commercially available in these bands. However, both bands are masked with at least one of the water or carbon dioxide absorption bands. In particular, measurements in exhaust gas matrices with harsh boundary conditions like strong H2O and CO2 backgrounds at elevated temperatures (up to 800 K) require detailed line selection which should also include hot band structures of the interfering species. Based on new spectral ammonia data in HITRAN2012 [23], we selected the ν2 + ν3 band of 14NH3 [39, 40], which shows sufficient absorption in the 2.3 µm region where H2O and CO2 have only weak absorption lines (Fig. 1).

Line strength spectrum in the NIR combination bands of NH3 and the interfering species H2O and CO2. The simulation is based on HITRAN2012 [23] and was calculated for room temperature (296 K). The commonly used spectral region around 1.5 µm is a combination of seven different ammonia absorption bands [41], while the absorptions at 2 and 2.3 µm are combinations of only two bands each [39]
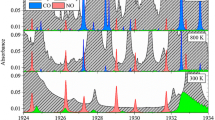
For the selection of suitable ammonia absorption lines, we simulated spectra for typical exhaust gas conditions (0.8–1.5 bar, 400–800 K, 15 %Vol H2O and CO2) at low NH3 mole fractions in combination with further species that could possibly be found in exhaust gas, namely CO, N2O, NO, NO2, CH4, C2H2, C2H4, C2H6 and SO2. A promising line triple was found at 2200.5 nm (4544.5 cm−1) (Table 1), which shows sufficient self- and foreign line separation, favorable temperature behavior and enough combined line strength for our application conditions. Only water, carbon dioxide and methane (in concentrations >10 ppmV) show significant absorption (absorbance > 10−5 after 10 cm) nearby. Because methane absorption would interfere with ammonia absorption, the influence has been investigated. If not compensated, a rather high methane concentration for exhaust systems of 5 ppmV would lead to an overestimation in ammonia concentration of 1 % at temperatures of 400 K and even less at higher temperatures. The use of DOC makes the existence of methane in single-digit ppmV quantities improbable, which is why its influence is neglected during further considerations.
The three ammonia lines are within 0.01 cm−1 and coincide due to pressure broadening, which is why they cannot be made out separately in Fig. 2. Due to the strong uncertainties of the line strength and positions as well as incomplete foreign broadening parameters in HITRAN2012 [23], it is necessary to revise the line data for improved accuracy. Because line data are not within the focus of this paper, this issue will be discussed in future publications.

Exemplary simulated spectrum for worst case exhaust conditions with only 100 ppmV NH3, 15 %Vol H2O and 15 %Vol CO2 at 1 bar and various temperatures at absorption path length of 10 cm
One of the major issues of a line-of-sight method like TDLAS is the nonlinear integration of the signal along an inhomogeneous absorption path. Therefore, the influence of strong fluctuations of temperature in the measurement volume has to be minimized. Due to differences in temperature dependency of the line strengths, the combined absorption of this line triple (Fig. 2) shows only small connection to temperature variation (ΔS = 0.13 %/K at 800 K). Additionally, no significant rise of the interfering background can be observed within the temperature range of the simulation.
3 Experimental setup
3.1 Exhaust simulation test rig
A test rig has been set up to characterize the spectrometer without the presence of a real engine. Nevertheless, the dimensions of the pipe are compatible with industrial SCR components regarding the flanges and the inner diameter of 153 mm. The test stand uses air and gaseous ammonia to simulate the environment downstream of SCR catalysts. A 4-kW inline air heater is used to provide a hot air stream of 330 standard liters per minute (SLPM) at a temperature of 820 K and up to 1400 SLPM at lower temperatures.
Bronkhorst digital mass flow meters are used for flow monitoring and control. While the air flow rate is set by a manual needle valve and measured by an IN-Flow type meter with a maximum range of 2500 SLPM, the ammonia flow rate is controlled by a Low-Δp-Flow type control valve with 0.5 SLPM maximum flow rate. The ammonia flow rate can be set to a constant flow rate or concentration, which is then controlled depending on the measured air flow. Concentrations ranging from 10 to 10,000 ppmV can be prepared with this setup. The ammonia injection is placed 213 mm upstream of the spectrometer measurement plane. Four 6-mm stainless steel tubes are positioned at half the pipe radius every 90° along the circumference injecting the ammonia against the air flow.
3.2 Spectrometer setup
The complete spectrometer setup consisting of various electronic and optical components is shown in Fig. 3 on the right. A Thorlabs PRO8000 mainframe with 200 mA current module and temperature module powers the laser diode and controls its temperature. The current can be modulated by an external analog signal with up to 200 kHz bandwidth and a rise and fall time of less than 2 µs. A 20-MHz function generator was used for this purpose.

Schematic of the test rig and spectrometer setup with peripheral components and spectrometer ring section, view in upstream direction, showing channel numbers and thermocouple positions
The light source is a fiber-coupled distributed feedback (DFB) diode laser manufactured by nanoplus with a nominal wavelength of 2201 nm and a maximum output power of 2 mW ex-fiber. Single-mode (SM) fiber was used to distribute the light to each of the four fiber ports. To supply light for all channels at the same time, a custom-made 1 × 4 fiber optic splitter of planar light wave circuit (PLC, LEONI Fiber Optics) design was employed, suitable for broadband application. The last element in the single-mode chain was a small aspheric beam collimator fused directly to the end of the fiber. The collimator lens was guided through a stainless steel tube into the exhaust system and sealed with high-temperature-resistant glue. The beam exits the collimator with a divergence of 0.22° resulting in a 1/e2 beam diameter of 1.05 ± 0.01 mm at a distance of 150 mm.
The core of our spectrometer is a stainless steel ring positioned between two flanges of the exhaust pipe. It is 25 mm thick and with an inner diameter of 153 mm fully compatible to the SCR exhaust system reproduced by the test stand. It provides four optical access ports to the flow with absorption path lengths between 138 mm and 147 mm, four thermocouples and a pressure sensor. Mineral-insulated thermocouples type K (Ø 0.5 mm) and a Keller PAA-35XHTC digital pressure transducer with a range of 0–10 bar were utilized to measure temperature and pressure.
On the detection side of the single-path line-of-sight, we used multi-mode (MM) fiber with a core diameter of 1 mm and a numerical aperture (NA) of 0.48 to gather as much of the laser light as possible. They are guided through a stainless steel tube similar to the SM side but without the possibility of adjustment. Directly at the fiber end, a Hamamatsu double-extended InGaAs photodiode with a typical cutoff frequency of 6 MHz was placed. The current output of the detectors was amplified with FEMTO variable gain transimpedance amplifiers. The amplifiers’ bandwidth at the gain used in combination with the photodiodes’ impedance is 4.5 MHz. The amplified signal is detected by a National Instruments PXI system with a four-channel 4 MSamples/s 16 bit PXIe-6124 data acquisition module. The phase-locked acquisition is performed via in-house developed LabVIEW software.
For all measurements presented in this report, the system settings were kept constant. The laser wavelength was modulated in a 90 % asymmetric triangle shape with a modulation frequency of 1040 Hz covering a spectral range of 2.6 cm−1 on the up-ramp. We worked with a sampling rate of 2 MS/s. The correction for transmission and background emission contributions to the raw signal used a fourth order polynomial. This polynomial correction was mathematically linked with the model of a multi-line Voigt shape used for fitting the measured spectrum. Doppler and pressure broadening were calculated with coefficients from HITRAN2012 database [23] based on the measured temperature and pressure. Therefore, both the polynomial and the line shapes are strongly coupled, which limits the range of the fit to physically reasonable solutions.
All the above-mentioned instruments and components form the spectrometer. The spectrometer ring and every component directly connected to it can be used in exhaust applications with gas temperatures up to 825 K. The system is gas tight and chemically resistant, and aside from the thermocouples, no obstacles are interfering with the flow. The system can easily be applied at different SCR systems by changing the spectrometer ring as an interface for the instruments.
4 Results and discussion
First, it is necessary to investigate the spectrometer’s stability regarding averaging. A long-term evaluation of the spectrometer itself was done employing a 10,230 ppmV ammonia reference cell, which was placed into one of the optical channels in the pipe. The signal was recorded and analyzed to draw the Allan–Werle deviation [42] plot in Fig. 4. One can see that the sole influence of white noise ends at a number of 1500 averages where the slope differs from −1, which would be an option to maximize SNR. However, from long-term recordings at constant flow parameters (293 K, 1000 ppmV ammonia), it is evident that not more than a total of 80 averages should be applied when working with the exhaust simulation test rig. This can be attributed to flow phenomena and drifts in the mass flow systems as well as the air supply. We can state that the limiting factor is not the spectrometer but the test rig, since averaging 1500 scans would be possible in a stable measurement environment.

Allan–Werle deviation of measurements with the spectrometer setup, comparing a 10,230 ppmV ammonia reference cell with air flow at 293 K with 1000 ppmV ammonia
To characterize the spectrometer performance, a set of line scans was recorded at three different temperatures and concentrations. Figure 5 shows averages of 80 line scans each at concentration setpoints of 500, 1000 and 2000 ppmV and a temperature of 800 K and the corresponding multi-line Voigt fit as well as the fit residual on the left. On the right, data and line fits with residuals for temperatures of 293, 480 and 800 K at a concentration setpoint of 1000 ppmV are shown. HITRAN2012 [23] line strength data as well as self- and foreign broadening coefficients were used to calculate concentration and line widths with respect to the measured pressure and temperature. Due to the fact that NH3 was measured in a hot air gas matrix, the use of air broadening coefficients is valid. On the left of Fig. 5, one can see the absorbance scaling according to the measured values of 209, 306 and 623 ppmV as expected. The absorbance in the right graph should only follow the temperature dependence of line strength, which is the case for 293 and 480 K where 783 and 787 ppmV are measured. The 800 K setpoint with its 306 ppmV deviates from this principle. The severe underestimation of concentration at high temperatures is going to be discussed later in this article.

Measured TDLAS line profiles and model fit data of coinciding 2200.5 nm NH3 line triple for variant setpoints of ammonia concentration at a certain temperature and vice versa each after averaging 80 scans. Measured values for the left dataset are 209, 306 and 623 ppmV, which are significantly lower than the setpoints. The right dataset shows similar behavior only for highest temperature setpoint, measuring 783, 787 and 306 ppmV
The detection limits for these representative single measurements can be calculated with Eq. (3) and are shown in Table 2 for an optical path length of 142 mm (actual value for channel 1) and the intrinsic temporal resolution of 13 Hz given the modulation frequency of 1040 Hz and a number of 80 averages. It can be seen that with signal-to-noise ratios between 4 and 13, the detection limit is at least 70 ppmV at 293 K and improves with rising temperature to about 50 ppmV at 800 K.
To demonstrate that every one of the four channels shows generally similar performance, Fig. 6 shows simultaneously acquired data while a constant concentration of 1000 ppmV ammonia was seeded into the hot air flow having a temperature of 480 K at the spectrometer’s position. Again, an average of 80 single scans, the corresponding fit and the residuals are displayed. The standard deviation of the residual is always lower than 2.5 × 10−4 resulting in detection limits of 53–68 for the single measurements that coincide with previous results. The differences in measured concentration indicate spatial inhomogeneities of the ammonia distribution induced by the non-steady flow field and possible differences in injected mass flow among the injection pipes.

Simultaneously acquired TDLAS line profiles and model fit data for every optical channel at the setpoint of 1000 ppmV and 480 K each after averaging 80 scans. Measured values for concentration are 801, 694, 835 and 709 ppmV, respectively. Differences indicate spatial concentration inhomogeneities of ammonia induced by the interacting air flow
To eliminate statistical fluctuations as a reason for the underestimations shown in Table 2, a set of measurements has been investigated. Figure 7 merges the mean values and their standard deviations for measuring 5 min at constant temperature and concentration setpoints. The picture drawn above can be sustained for longtime measurements at any concentration setpoint or temperature investigated and in each of the four channels. At 293 and 480 K, we underestimate the injected concentrations by 20–25 %, whereas at 800 K, we measure 50–70 % discrepancy.

Measured mole fraction of all four channels at temperature setpoints 293, 480 and 800 K and concentration setpoints 500, 1000 and 2000 ppmV; measurements recorded for 5 min and evaluated after averaging 80 single measurements; plot shows marker at mean value of mole fraction and averaged ±1σ of fit residual as error bars
Possible sources of these discrepancies can be divided into two groups: the systematic error of the spectrometer and an ammonia concentration being in fact lower than expected. Systematic errors of the concentration measurement result from uncertainties of the measured temperature, pressure and absorption length as well as erroneous spectral data. Wrong ammonia concentrations are basically possible because of ammonia dosing errors by the flow control system, ammonia adhesion in the entire system and ammonia decomposition in hot environment.
The maximum relative uncertainties of temperature, pressure and absorption length measurements are 1.2, 0.2 and 0.4 %, respectively. These errors influence the calculated concentration directly and add up to a total uncertainty of 1.8 %. One of the main reasons for the discrepancies might be the high uncertainty in line strength data of 10–20 % for the two outer lines and more than 20 % for the central line (see Table 1) whose influence on total line strength increases with rising temperature from 35 % at 300 K to 56 % at 800 K. HITRAN’s [23] air- and self-half-widths are another source of error. Unfortunately, their uncertainty is given to be “average or estimate.” To estimate the error, we compared evaluation using calculated collisional widths with having the collisional width fitted to spectral data taken from the ammonia reference cell. The collisional width was usually lower when calculated resulting in an underestimation of concentration by 15 %, so it can be assumed that a systematic underestimation results from erroneous line widths. The uncertainty of the temperature-dependence coefficients of the lines should also be taken into account as we departed far from HITRAN’s [23] reference point of 296 K. This uncertainty is again not quantified, so no conclusion can be drawn here. It must also be noted that TDLAS as a line-of-sight technique might suffer from temperature inhomogeneities along the optical path. We measured temperatures at the positions shown in Fig. 3. Typical temperature development for the setpoint of 800 K showed the highest fluctuations. The mean temperatures of all four channels vary less than 5 K, and the actual values are within a 20-K range corresponding to less than 3 % of the mean value. The temperature’s influence on line strength can be calculated to be 0.13 %/K at 800 K at its worst. This would result in an uncertainty of 2.6 % in mole fraction due to temperature inhomogeneities of 20 K.
The maximum ammonia dosing error resulting from given uncertainties of the flow control system and the ammonia test gas is between 25 and 40 ppmV (at 500 and 2000 ppmV, respectively). Ammonia adhesion occurs but cannot be a main reason for underestimation since we performed measurements for several minutes at each setpoint without any noticeable drifts toward higher concentrations, which would otherwise be observed as adhesion is subject to saturation effects. At last, some chemical reactions exist which could decrease the amount of ammonia from the time it is injected to the time it passes the spectrometer: Firstly, there is the equilibrium reaction 2NH3 ⇌ N2 + 3H2. According to [43], the chemical equilibrium constants K E of the reaction were calculated for temperatures of 293, 480 and 800 K to be 1.36 × 10−6, 2.63 × 10−4 and 7.11 × 10−3, respectively. The increasing equilibrium constant shows that the equilibrium changes in favor of the generation of N2 and H2 with rising temperature. At 800 K, this would cause a decrease in ammonia of 60 % in equilibrium state on its own. Secondly, because of the presence of steel tubes, there could be a form of catalytic oxidation of ammonia with oxygen into water, nitrogen and nitric oxides beginning at elevated temperatures of 500–600 K [44]. Due to the short residence time of ammonia in the flow of ~1 s at the used setpoint of 230 SLPM until passing through the spectrometer, equilibrium of those reactions is not likely to exist. Therefore, the influence of ammonia decomposition cannot be conclusively evaluated, but as chemical reactions speed up at higher temperatures, it would increase likewise.
In summary, we highlighted possible systematic errors of our spectrometer mainly affected by line strengths and widths which are in a range of 20 % explaining the measurement error for setpoints 293 and 480 K in Fig. 7. Furthermore, we identified thermal decomposition of ammonia as a possible mechanism to lower the concentration causing the results of setpoint 800 K. Yet the chemical processes have an unknown impact on the actual concentration, and therefore to estimate the spectrometer’s accuracy, we disregard measurements at 800 K.
5 Conclusion
We built a fast and in situ TDLAS-based spectrometer to measure absolute ammonia mole fraction in exhaust systems. To account for harsh boundary conditions such as temperatures of up to 800 K and strong H2O and CO2 backgrounds, we selected an ammonia transition in the ν2 + ν3 band at 2200.5 nm (4544.5 cm−1). The spectrometer employs a fiber-coupled 2.2-µm DFB laser diode split into four absorption paths spanning a measurement plane perpendicular to the flow with absorption lengths of 138–147 mm. First measurements at elevated temperatures up to 800 K have been shown, and the spectrometer has been characterized. Experiments at high temperatures unveiled high underestimation of the expected ammonia concentration. Future investigations on ammonia decomposition in hot air in a stable and controlled environment such as high-temperature absorption cells combined with numerical simulations would help to understand the kinetics of this reaction. We achieved a temporal resolution of 13 Hz and detection limits down to 50 ppmV. On a path length and time-normalized scale, we are only by factor 1.75 worse than the commercially available in situ TDLAS system SIEMENS LDS6 which cannot be used to measure in 2D planes across the flow field or at catalyst’s entry and exit. Due to HITRAN2012′s [23] uncertainties of the used line data, we estimate the accuracy of the spectrometer to be approximately 20 % at this moment. Future improved line data for exhaust conditions will significantly improve the accuracy.
References
European Union, “Commission Regulation (EU) No 459/2012,”, Off. J. Eur. Union 55(L 142), 16–24 (2012). doi:10.3000/19770677.L_2012.142.eng
The European Parliament and the Council of the European Union, “Regulation (EC) No 595/2009,” 2009
Environmental Protection Agency, “Control of Air Pollution From Motor Vehicles: Tier 3 Motor Vehicle Emission and Fuel Standards; Final Rule,” Fed. Regist. 79(81), 2014
ISO/TC 22/SC 5, “ISO 22241-1:2006—Diesel engines—NOx reduction agent AUS 32—Part 1: quality requirements.” 2006
W.-P. Trautwein, DGMK Research Report 616-1—AdBlue as a Reducing Agent for the Decrease of NOx Emissions from Diesel Engines of Commercial Vehicles. Hamburg, 2003
C. Ciardelli, I. Nova, E. Tronconi, D. Chatterjee, B. Bandl-Konrad, M. Weibel, B. Krutzsch, Reactivity of NO/NO2–NH3 SCR system for diesel exhaust aftertreatment: identification of the reaction network as a function of temperature and NO2 feed content. Appl. Catal. B Environ. 70(1–4), 80–90 (2007). doi:10.1016/j.apcatb.2005.10.041
ACEA, Selective Catalytic Reduction (Final Report), 2003, pp. 1–13
M. Wallin, C.-J. Karlsson, M. Skoglundh, A. Palmqvist, Selective catalytic reduction of NOx with NH3 over zeolite H-ZSM-5: influence of transient ammonia supply. J. Catal. 218(2), 354–364 (2003). doi:10.1016/S0021-9517(03)00148-9
H. Sjövall, L. Olsson, E. Fridell, R.J. Blint, Selective catalytic reduction of NOx with NH3 over Cu-ZSM-5—The effect of changing the gas composition. Appl. Catal. B Environ. 64(3–4), 180–188 (2006). doi:10.1016/j.apcatb.2005.12.003
R. Claps, F.V. Englich, D.P. Leleux, D. Richter, F.K. Tittel, R.F. Curl, Ammonia detection by use of near-infrared diode-laser-based overtone spectroscopy. Appl. Opt. 40(24), 4387 (2001). doi:10.1364/AO.40.004387
M.E. Webber, R. Claps, F.V. Englich, F.K. Tittel, J.B. Jeffries, R.K. Hanson, Measurements of NH3 and CO2 with distributed-feedback diode lasers near 2.0 μm in bioreactor vent gases. Appl. Opt. 40(24), 4395 (2001). doi:10.1364/AO.40.004395
D.J. Miller, K. Sun, L. Tao, M.A. Khan, M.A. Zondlo, Open-path, quantum cascade-laser-based sensor for high-resolution atmospheric ammonia measurements. Atmos. Meas. Tech. 7(1), 81–93 (2014). doi:10.5194/amt-7-81-2014
M.E. Webber, D.S. Baer, R.K. Hanson, Ammonia monitoring near 1.5 µm with diode-laser absorption sensors. Appl. Opt. 40(12), 2031–2042 (2001)
R.M. Mihalcea, M.E. Webber, D.S. Baer, R.K. Hanson, G.S. Feller, W.B. Chapman, Diode-laser absorption measurements of CO2, H2O, N2O, and NH3 near 2.0 μm. Appl. Phys. B Lasers Opt. 67(3), 283–288 (1998). doi:10.1007/s003400050507
X. Chao, J.B. Jeffries, R.K. Hanson, Development of laser absorption techniques for real-time, in situ dual-species monitoring (NO/NH3, CO/O2) in combustion exhaust. Proc. Combust. Inst. 34(2), 3583–3592 (2013). doi:10.1016/j.proci.2012.05.024
H. Teichert, T. Fernholz, V. Ebert, Simultaneous in situ measurement of CO, H2O, and gas temperatures in a full-sized coal-fired power plant by near-infrared diode lasers. Appl. Opt. 42(12), 2043–2051 (2003)
H.E. Schlosser, J. Wolfrum, V. Ebert, B.A. Williams, R.S. Sheinson, J.W. Fleming, In situ determination of molecular oxygen concentrations in full-scale fire-suppression tests using tunable diode laser absorption spectroscopy. Proc. Combust. Inst. 29(1), 353–360 (2002). doi:10.1016/S1540-7489(02)80047-5
V. Ebert, H. Teichert, P. Strauch, T. Kolb, H. Seifert, J. Wolfrum, Sensitive in situ detection of CO and O2 in a rotary kiln-based hazardous waste incinerator using 760 nm and new 2.3 μm diode lasers. Proc. Combust. Inst. 30(1), 1611–1618 (2005). doi:10.1016/j.proci.2004.08.224
A.R. Awtry, J.W. Fleming, V. Ebert, Simultaneous diode-laser-based in situ measurement of liquid water content and oxygen mole fraction in dense water mist environments. Opt. Lett. 31(7), 900 (2006). doi:10.1364/OL.31.000900
B.T. Fisher, A.R. Awtry, R.S. Sheinson, J.W. Fleming, Flow behavior impact on the suppression effectiveness of sub-10-µm water drops in propane/air co-flow non-premixed flames. Combustion Institute 31, 2731–2739 (2007)
C. Schulz, A. Dreizler, V. Ebert, J. Wolfrum, in Combustion Diagnostics in Handbook of Experimental Fluid Mechanics, ed. by C. Tropea, A.L. Yarin, J.F. Foss (Springer, Heidelberg, 2007), pp. 1241–1316
V. Ebert, J. Wolfrum, Absorption in Optical Measurements—Techniques and Applications, 2000, pp. 273–312
L.S. Rothman, I.E. Gordon, Y. Babikov, A. Barbe, D. Chris Benner, P.F. Bernath, M. Birk, L. Bizzocchi, V. Boudon, L.R. Brown, A. Campargue, K. Chance, E.A. Cohen, L.H. Coudert, V.M. Devi, B.J. Drouin, A. Fayt, J.-M. Flaud, R.R. Gamache, J.J. Harrison, J.-M. Hartmann, C. Hill, J.T. Hodges, D. Jacquemart, A. Jolly, J. Lamouroux, R.J. Le Roy, G. Li, D.A. Long, O.M. Lyulin, C.J. Mackie, S.T. Massie, S. Mikhailenko, H.S.P. Müller, O.V. Naumenko, A.V. Nikitin, J. Orphal, V. Perevalov, A. Perrin, E.R. Polovtseva, C. Richard, M.A.H. Smith, E. Starikova, K. Sung, S. Tashkun, J. Tennyson, G.C. Toon, V.G. Tyuterev, G. Wagner, The HITRAN2012 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 130, 4–50 (2013). doi:10.1016/j.jqsrt.2013.07.002
E.E. Whiting, An empirical approximation to the Voigt profile. J. Quant. Spectrosc. Radiat. Transf. 8(6), 1379–1384 (1968). doi:10.1037/a0021968
B. Buchholz, N. Böse, V. Ebert, Absolute validation of a diode laser hygrometer via intercomparison with the German national primary water vapor standard. Appl. Phys. B 116(4), 883–899 (2014). doi:10.1007/s00340-014-5775-4
L. Ma, X. Li, S.T. Sanders, A.W. Caswell, S. Roy, D.H. Plemmons, J.R. Gord, 50-kHz-rate 2D imaging of temperature and H2O concentration at the exhaust plane of a J85 engine using hyperspectral tomography. Opt. Express 21(1), 1152–1162 (2013)
F. Wang, K.F. Cen, N. Li, J.B. Jeffries, Q.X. Huang, J.H. Yan, Y. Chi, Two-dimensional tomography for gas concentration and temperature distributions based on tunable diode laser absorption spectroscopy. Meas. Sci. Technol. 21(4), 045301 (2010). doi:10.1088/0957-0233/21/4/045301
G. Totschnig, M. Lackner, R. Shau, M. Ortsiefer, High-speed vertical-cavity surface-emitting laser (VCSEL) absorption spectroscopy of ammonia (NH 3) near 1.54 μm. Appl. Phys. B Lasers Opt. B76(5), 603–608 (2003). doi:10.1007/s00340-003-1102-1
L. Li, R.M. Lees, L.-H. Xu, External cavity tunable diode laser spectra of the ν1 + 2ν4 stretch-bend combination bands of 14NH3 and 15NH3. J. Mol. Spectrosc. 243(2), 219–226 (2007). doi:10.1016/j.jms.2007.04.003
B. Lins, F. Pflaum, R. Engelbrecht, B. Schmauss, Absorption line strengths of 15NH3 in the near infrared spectral region. Appl. Phys. B 102(2), 293–301 (2010). doi:10.1007/s00340-010-4217-1
G. Dooly, H. Manap, S. O’Keeffe, E. Lewis, Highly selective optical fibre ammonia sensor for use in agriculture. Procedia Eng. 25, 1113–1116 (2011). doi:10.1016/j.proeng.2011.12.274
A.A. Kosterev, R.F. Curl, F.K. Tittel, R. Köhler, C. Gmachl, F. Capasso, D.L. Sivco, A.Y. Cho, Transportable automated ammonia sensor based on a pulsed thermoelectrically cooled quantum-cascade distributed feedback laser. Appl. Opt. 41(3), 573 (2002). doi:10.1364/AO.41.000573
K. Sun, L. Tao, D.J. Miller, M.A. Khan, M.A. Zondlo, Inline multi-harmonic calibration method for open-path atmospheric ammonia measurements. Appl. Phys. B 110(2), 213–222 (2012). doi:10.1007/s00340-012-5231-2
K. Owen, A. Farooq, A calibration-free ammonia breath sensor using a quantum cascade laser with WMS 2f/1f. Appl. Phys. B 116(2), 371–383 (2013). doi:10.1007/s00340-013-5701-1
C.S. Goldenstein, R.M. Spearrin, J.B. Jeffries, R.K. Hanson, Infrared laser absorption sensors for multiple performance parameters in a detonation combustor. Proc. Combust. Inst. 35(3), 3739–3747 (2015). doi:10.1016/j.proci.2014.05.027
R.M. Spearrin, C.S. Goldenstein, I.A. Schultz, J.B. Jeffries, R.K. Hanson, Simultaneous sensing of temperature, CO, and CO2 in a scramjet combustor using quantum cascade laser absorption spectroscopy. Appl. Phys. B 117(2), 689–698 (2014). doi:10.1007/s00340-014-5884-0
J.B. McManus, J.H. Shorter, D.D. Nelson, M.S. Zahniser, D.E. Glenn, R.M. McGovern, Pulsed quantum cascade laser instrument with compact design for rapid, high sensitivity measurements of trace gases in air. Appl. Phys. B 92(3), 387–392 (2008). doi:10.1007/s00340-008-3129-9
K. Sun, L. Tao, D.J. Miller, M.A. Khan, M.A. Zondlo, On-road ammonia emissions characterized by mobile, open-path measurements. Environ. Sci. Technol. 48(7), 3943–3950 (2014). doi:10.1021/es4047704
Š. Urban, N. Tu, K. Narahari Rao, G. Guelachvili, Analysis of high-resolution Fourier transform spectra of 14NH3 at 2.3 μm. J. Mol. Spectrosc. 133(2), 312–330 (1989). doi:10.1016/0022-2852(89)90195-1
M.J. Down, C. Hill, S.N. Yurchenko, J. Tennyson, L.R. Brown, I. Kleiner, Re-analysis of ammonia spectra: updating the HITRAN 14NH3 database. J. Quant. Spectrosc. Radiat. Transf. 130, 260–272 (2013). doi:10.1016/j.jqsrt.2013.05.027
K. Sung, L.R. Brown, X. Huang, D.W. Schwenke, T.J. Lee, S.L. Coy, K.K. Lehmann, Extended line positions, intensities, empirical lower state energies and quantum assignments of NH3 from 6300 to 7000 cm − 1. J. Quant. Spectrosc. Radiat. Transf. 113(11), 1066–1083 (2012). doi:10.1016/j.jqsrt.2012.02.037
P. Werle, R. Mücke, F. Slemr, The limits of signal averaging in atmospheric trace-gas monitoring by tunable diode-laser absorption spectroscopy (TDLAS). Appl. Phys. B Photophysics Laser Chem. 57(2), 131–139 (1993). doi:10.1007/BF00425997
P.W. Atkins, Physical Chemistry, 5th edn. (Oxford University Press, Oxford, 1994)
L. Gang, Catalytic Oxidation of Ammonia to Nitrogen (Technische Universiteit Eindhoven, Eindhoven, 2002)
Acknowledgments
The authors acknowledge financial support and collaboration in the UNICO project funded by the federal state of Hessen and the Technische Universität Darmstadt, the Center of Smart Interfaces of the Technische Universität Darmstadt, as well as the Robert Bosch GmbH. The authors appreciate the collaboration with Dr. Philippe Leick regarding trace gas diagnostics in systems with exhaust aftertreatment.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Stritzke, F., Diemel, O. & Wagner, S. TDLAS-based NH3 mole fraction measurement for exhaust diagnostics during selective catalytic reduction using a fiber-coupled 2.2-µm DFB diode laser. Appl. Phys. B 119, 143–152 (2015). https://doi.org/10.1007/s00340-015-6073-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00340-015-6073-5