
Abstract
A highly efficient microprojectile transformation system for sorghum (Sorghum bicolor L.) has been developed by using immature embryos (IEs) of inbred line Tx430. Co-bombardment was performed with the neomycin phosphotransferase II (nptII) gene and the green fluorescent protein (gfp) gene, both under the control of the maize ubiquitin1 (ubi1) promoter. After optimization of both tissue culture media and parameters of microprojectile transformation, 25 independent transgenic events were obtained from 121 bombarded IEs. The average transformation frequency (the total number of independent transgenic events divided by the total number of bombarded IEs) was 20.7% in three independent experiments. Transgenic events were confirmed by both PCR screening and Southern hybridization of genomic DNA from primary transgenics (T0). More than 90% of transformants were fertile and displayed normal morphology in a containment glasshouse. Co-transformation rate of the nptII and gfp genes was 72% in these experiments. The segregation of nptII and gfp in T1 progenies was observed utilizing fluorescence microscopy and geneticin selection of seedlings indicating both were inherited in the T1 generation. The transformation procedure, from initiating IEs to planting putative transgenic plantlets in the glasshouse, was completed within 11–16 weeks, and was approximately threefold more efficient than the previously reported best sorghum transformation system.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sorghum [Sorghum bicolor (L.) Moench], the fifth most important cereal after wheat, rice, maize and barley, plays a unique role in food security and renewable energy (Belton and Taylor 2004). Sorghum is a drought-tolerant crop, which can grow in marginal areas not suitable for the other cereals. It is widely used for food especially in Africa and India, and for animal consumption (grain and stover) and as a biofuel energy source in Americas and Australia. Genetic engineering has the potential to have a great influence on improving sorghum yield and digestibility (Gurel et al. 2009). However, sorghum has been widely considered as a recalcitrant major crop in terms of tissue culture and genetic transformation (Grootboom et al. 2010). In fact, sorghum transformation has lagged far behind published efficiencies of rice (Hiei and Komari 2008), maize (Ishida et al. 2007) and barley (Ibrahim et al. 2010), even though it has been almost two decades since the first study on transgenic sorghum was published (Casas et al. 1993).
Microprojectile transformation and Agrobacterium-mediated transformation are two main approaches that have been utilized to obtain transgenic sorghum. The first report of successful transgenic sorghum was published by using particle bombardment in 1993 and a transformation efficiency of 0.286% was achieved (Casas et al. 1993). Seven years later, the first Agrobacterium-mediated transgenic sorghum was reported (Zhao et al. 2000) at 2.12% efficiency. Since then, several more reports have been published (Zhao and Tomes 2003; Williams et al. 2004; Gao et al. 2005b; Nguyen et al. 2007; Gurel et al. 2009; Able et al. 2001). Recently, up to 8.3% of stable Agrobacterium-mediated transformation efficiency has been reported (Gurel et al. 2009), although this was the best single experiment among a number using Agrobacterium-mediated transformation. However, there have been few significant advances in microprojectile transformation efficiency (Grootboom et al. 2010; Raghuwanshi and Birch 2010). Grootboom et al. obtained 27 transgenic plants from 3,499 immature embryos (IEs) of the cultivar P898012 utilizing manA gene, and the transformation efficiency at 0.77% was more than twofold higher than the first published attempt (0.286%) (Casas et al. 1993). Raghuwanshi and Birch obtained 16 transgenic plants of sweet sorghum from 17046 IEs bombarded. Even though the tissue culture system was optimized, the transformation efficiency of sweet sorghum (0.09%) was much lower than the first published attempt (0.286%) of grain sorghum (Casas et al. 1993). A highly efficient gene transfer system largely rests on an effective tissue culture system and an optimal DNA delivery system. Important progress has been made in both areas (Kaeppler and Pedersen 1997; Elkonin et al. 1995; Elkonin and Pakhomova 2000; Nirwan and Kothari 2003; Able et al. 1998; Karami et al. 2009; Carvalho et al. 2004; Hill-Ambroz and Weeks 2001; Jeoung et al. 2002; Able et al. 2001), but sorghum transformation efficiency remained significantly below 10% and was far behind the transformation efficiencies of rice 50–90% (Hiei and Komari 2008), maize 50% (Ishida et al. 2007) and barley 14.8% (Ibrahim et al. 2010).
An efficient genetic transformation system plays a key role in molecular breeding and understanding the genetic control of plant physiology and development. Sequencing of the sorghum genome was completed in 2009 (Paterson et al. 2009). Genome sequencing and gene discovery have gained momentum in the last decade. Yet, the absence of an efficient sorghum transformation protocol has hampered progress in sorghum genetic engineering and understanding of sorghum gene function.
The aim of the present study was to develop a highly efficient transformation system for sorghum. Focusing on an extensively used inbred sorghum line Tx430 (Gurel et al. 2009), we optimized callus induction medium (CIM), regeneration medium and rooting medium to reduce the production of phenolics from tissue and increase the callus induction rate and regeneration frequency. Other transformation variables were also optimized.
Materials and methods
Plasmids
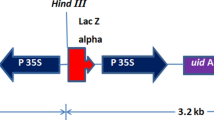
Two plasmids, pUKN and pGEM-ubi-gfp (Dugdale et al. 1998), which contain nptII encoding NPTII and gfp encoding GFP, respectively, were kindly provided by Scott Hermann, Bureau of Sugar Experimental Stations (BSES) Limited. Both genes were driven by the maize ubiquitin1 (ubi1) promoter and terminated with A. tumefaciens nopaline synthase 3′ termination signal (nos) (Fig. 1a, b).

Constructs, a the pUKN construct and b the pGEM-ubi-gfp construct
Plant materials and embryo isolation
Seeds of the inbred line Tx430 (Miller 1984) were planted in the temperature control glasshouse (18–28°C) and some were planted outside in the field during the summer. Plants were watered daily and fertilized with Osmocote® monthly. Panicles were covered with paper bags before flowering and immature seeds were harvested within 12–15 days after flowering. To sterilize immature seeds, they were soaked in 70% ethanol (v/v) while shaking at 200 rpm for 5 min. Seeds were rinsed once with sterilized water, before being soaked again in the solution containing commercial bleach (White King) 4% (v/v) sodium hypochlorite and 0.2% (v/v) surfactant Tween 20 while shaking at 200 rpm for 10 min. Finally, seeds were rinsed five times with sterilized water. IEs ranging from 1.0 to 2.0 mm in length were isolated onto 90 × 15 mm petri dishes containing CIM with their scutellums facing upward. Cultures were incubated in the dark in a tissue culture room, which was set up at 27 ± 1°C under fluorescent light of approximately 100 μmol s−1 m−2 (16 h/day).
Media for tissue culture
All media were based on MS medium (Murashige and Skoog 1962) containing 4.44 g/L MS powder with Gamborg vitamins (PhytoTechnology Laboratories), 30 g/L sucrose and 8 g/L agar. The pH of all media was adjusted to 5.7 before being autoclaved at 121°C for 15 min; 2,4-d was added to media before autoclaving. The other plant growth regulators and copper sulfate (CuSO4) were sterilized with a 0.2-μm syringe filter and added after autoclaving.
The following media were used for microprojectile transformation experiments:
Callus induction medium (CIM): MS medium supplemented with 1 g/L l-proline, 1 g/L l-asparagine, 1 g/L potassium dihydrogenphosphate (KH2PO4), 0.16 mg/L CuSO4 and 1 mg/L 2,4-d.
Regeneration medium: MS medium supplemented with 1 mg/L BAP, 1 mg/L IAA and 0.16 mg/L CuSO4.
Rooting medium: MS medium supplemented with 1 mg/L NAA, 1 mg/L IAA, 1 mg/L IBA and 0.16 mg/L CuSO4.
Selective regeneration medium: regeneration medium supplemented with 30 mg/L geneticin (G418) (disulfate salt solution, SIGMA).
Selective rooting medium: rooting medium supplemented with 30 mg/L G418.
Osmotic medium: MS medium supplemented with 0.2 M d-sorbitol and 0.2 M d-mannitol.
Kill curve experimentation
A kill curve experiment was carried out utilizing IEs at 2 weeks old after initiation on CIM. IEs were subcultured onto selective regeneration media containing G418 at 0, 10, 20, 30 and 50 mg/L (5 IEs per plate, three plates for each G418 treatment) and placed under lights. IEs were subcultured fortnightly. Four weeks later, the numbers of IEs generating shoots and surviving (the total number of IEs minus the number of necrotic IEs) were recorded.
Microprojectile transformation
IEs at 9–11 days old after initiation were used for microprojectile transformation. Six IEs were placed at the center of a shallow petri dish 15 × 90 mm containing osmotic medium and stored for 2–3 h in the dark prior to bombardment (Fig. 6a). Operation of the particle inflow gun (PIG) was conducted as described by Vain et al. (1993) (Grootboom et al. 2010). For plasmids, DNA delivery occurred via 0.6 μm gold particles (0.42 mg per shot). The distance from the filter holder to the target cells was adjusted to 18.5 cm and the helium pressure was modified to 1,000 kPa. As much as 5 μg of each pUKN plasmid and pGEM-ubi-gfp plasmid were mixed and then equally loaded into the receptacle for six shots. A 500-μm nylon mesh screen was placed 8 cm above the target tissue and a vacuum of approximately −90 kPa was generated prior to shooting with a time duration of 0.05 s. Post-bombardment, IEs were kept on osmotic medium for 3–4 h before being transferred onto CIM.
Selection of putative transgenics
After IEs recovered on CIM for 3–4 days, they were transferred to selective regeneration medium and placed under lights in a tissue culture room. IEs were then subcultured fortnightly until putative shoots grew to 4–6 cm. Subsequently, shoots were then moved to selective rooting medium for 4 weeks without subculture. Once the plantlets had a well-developed root system (>10 roots of >1 cm length), the lids of petri dishes were opened for at least 3 days to acclimatize plantlets in tissue culture room. The plantlets were transferred into the containment glasshouse and planted in plastic pots (20 L) containing 15 L of potting mix (Green Fingers). Plants were watered once per day.
Reporter gene observation
The gfp reporter gene produces a protein which fluoresces in living cells when exposed to blue light at the wavelength of 395 nm (Ormo et al. 1996). The gfp gene expression in callus was monitored under the OLYMPUS BX60 fluorescence microscope 3 and 10 days after bombardment. Each callus piece (from one IE) was then categorized (based on the number of GFP foci) into the following groups: 0–20, 21–50, 51–100 and >100. The gfp gene expression in root tips of putative transgenic plantlets and T1 seedlings was also monitored.
PCR analysis
Genomic DNA was extracted from young leaves of putative transgenic and non-transgenic line Tx430. To confirm the presence of nptII and gfp, fragments (722 bp of nptII and 732 bp of gfp) were amplified from genomic DNA using NPTII-specific primers: NPTII-1: 5′-GCTATGACTGGGCACAAC-3′; NPTII-2: 5′-GTCAAGAAG GCGATAGAAGG-3′, and GFP-specific primers: GFP-1: 5′-CCGCGGTTACTTGTACAGCTCGTCC-3′; GFP-2: 5′-CCCGGGATGGTGAGCAAGGGCGAGG-3′ (INTEGRATED DNA TECHNOLOGIES, www.idtdna.com). PCR analyses were performed in 25-μL reaction mixtures, each containing 50 ng of template DNA, 1× PCR buffer, 200 μM of each dNTP, 2.5 mM MgCl2, 0.5 μM primer and 2 U Taq DNA polymerase (NEW ENGLAND BioLabs). The PCR program utilized for the nptII gene consisted of an initial denaturation at 95°C for 5 min, followed by 35 cycles, consisting of 94°C for 30 s, 60°C for 30 s, 72°C for 50 s and a final 7-min elongation step at 72°C. Similarly for the gfp gene PCR reactions, the annealing temperature was changed to 70°C for 30 s. PCR products were separated by gel electrophoresis in 1.0% agarose gels.
Southern hybridization analysis
Aliquots of ~10 μg total genomic DNA of each putative transgenic sample and non-transgenic Tx430 were digested with the restriction enzyme SacI-HF™ (NEW ENGLAND BioLabs) at 37°C overnight. As the positive control, 100 pg of undigested pUKN plasmid was used. The digested DNA and plasmid were then separated on a 0.8% agarose gel for 16 h at 25 V. DNA samples were then transferred to a positively charged nylon membrane (Roche) via blot apparatus overnight. The following day, the membrane was hybridized using the digoxigenin (DIG)-labeled nptII probe at 42°C overnight (probe construction was carried out using nptII primers following the PCR conditions described previously). The next day, the membrane was washed, blocked and fluorescent signal detected according to manufacturer instruction (Roche).
Transgene inheritance assay
Six PCR-confirmed T0 lines containing both nptII and gfp genes were selected for analysis of gene inheritance. Non-transgenic Tx430 seeds were used as a control. For each selected line, 25 seeds were placed onto a petri dish of 25 × 90 mm, lined with two filter papers (Whatman No. 1). Seeds were then partially covered by sterilized water and allowed to germinate in the dark at 27 ± 1°C for 3 days. The gfp gene expression in the T1 seedlings was monitored utilizing the OLYMPUS BX60 fluorescent microscope. Seedlings were then soaked in sterilized water containing 30 mg/L G418. Plates were placed under light in the tissue culture room for 1 week. Observations of the health and vigor of seedlings for each treatment were recorded, with particular attention paid to detecting any signs of necrosis among the seedlings under G418 selection.
Results
Kill curve
IEs began to display G418 stress symptoms on selective regeneration media within 1 week. IEs on regeneration medium without G148 (G0) grew healthily and showed the beginnings of shoots within 2 weeks and generated vigorous shoots within 4 weeks. On G0 medium, all IEs survived and 14 of 15 IEs produced vigorous shoots (Table 1). In contrast, none of the IEs generated shoots on G30 or G50 media, and the majority were necrotic. After 4 weeks, the results demonstrated that G418 had a significant impact on callus growth and development in selective media with different G418 concentrations (G0, G10, G20, G30 and G50) (Table 1). Even though two and six IEs produced shoots on G20 and G10 media, respectively, the shoots were shorter and weaker than those on G0 media. The pattern in the kill curve experiment was observed as follows: the higher the G418 concentration in selective regeneration media, the lower the shoot regeneration frequency was evident and the more necrotic IEs were observed (Table 1). In this experiment, 30 mg/L G418 in medium was sufficient to prevent IEs from producing shoots and was therefore selected as the optimal concentration for Tx430 transformation.
GFP observations
Strong gfp gene expression was observed in all bombarded IEs with at least 20 GFP foci. More than 100 GFP foci were spotted in about 80% of IEs (Figs. 2, 3a). When examining root tips of putative transgenic plantlets, the gfp gene expression was shown to be systemically green under the fluorescent microscope (Fig. 3c). No GFP was found in root tips of non-transgenic plantlets and the root tips of these plantlets displayed yellow color under the fluorescent microscope (Fig. 3b).

Transient gfp gene expression in bombarded Tx430 immature embryos

GFP expression under the blue light at the wavelength of 395 nm of the OLYMPUS BX60 fluorescence microscope; a Tx430 immature embryo-derived callus 3 days after bombardment; b root tip of non-transgenic Tx430 plantlet; c root tip of transgenic Tx430 plantlet; d 3-day-old T1 seedling
Molecular analysis of putative transgenic (T0) lines
PCR results confirmed the presence of the NPTII gene in all transgenic lines and no nptII escapes were observed (Fig. 4a). The result of gfp PCR analysis showed that 18 of 25 plants were positive (Fig. 4b). Overall, the co-transformation frequency of the gfp and nptII genes was 72%. Southern hybridisation confirmed the integration of the nptII gene into the genome of all T0 plants (Fig. 5). Single copy and multiple copies of nptII gene were found in various plant lines. Among the 25 transgenic lines, 24% had a single nptII gene copy, 28% had two to three copies and 48% had at least four copies.

PCR detection of transgenes in putative transgenic sorghum lines. a PCR for the nptII gene and b PCR for the gfp gene. Lines from left to right: M 1 kb DNA Ladder (NEW ENGLAND BioLabs). E empty (no template), N non-transgenic Tx430, P plasmid, 1–12 12 samples of putative transgenic lines

Southern blot hybridization of 14 putative transgenic lines with nptII probe. Lines from left to right: M DNA ladder. N NC, P equivalent single copy control (pUKN plasmid), 1–14 individual transgenic lines
Selection of putative transgenics
IEs recovered and grew quickly on CIM in the dark 3 days after bombardment. The growth of IEs was clearly constrained on selective regeneration medium by the application of G418. Two weeks after selection, most of the IEs displayed necrotic symptoms due to G418. However, a few IEs started to generate shoots. In 4–8 weeks, the transgenic sectors proliferated and produced healthy shoot selection medium (Fig. 6b). Vigorous roots of putative transgenic plantlets were generated on selective rooting medium (Fig. 6c). As much as 100% of all plantlets transferred to pots in the containment glasshouse environment survived (Fig. 6d). The transformation efficiency varied from 18.4 to 25% (mean efficiency of 20.7%) in the three independent experiments (Table 2).

The development of putative transgenic sorghum plants. a The initiation stage—immature embryo-derived calli ready for microprojectile transformation; b putative transgenic shoots on selective (30 mg/L G418) regeneration medium at 6 weeks; c putative transgenic plantlet on selective (30 mg/L G418) rooting medium at 4 weeks; d putative transgenic plants in containment glasshouse
Transgene inheritance assay
The gfp gene expression in T1 seedlings was observed under the fluorescence microscope in all tested lines (Fig. 3d). The seedlings with the gfp gene expression were uniformly fluorescent green. However, null segregants of the gfp gene was also observed and appeared yellow. Similarly, the expression of the nptII gene in T1 seedlings was confirmed by their survival under G418 selection. T1 null segregants of the nptII gene and non-transgenic Tx430 seedlings were necrotic in G418 solution after 1 week (Fig. 7a). T1 seedlings with the nptII gene expression were healthy and grew vigorously (Fig. 7b).

G418 germination assay. a T1 susceptible seedlings under 30 mg/L G418 selection after 1 week and b T1 resistant seedlings under 30 mg/L G418 selection after 1 week
Discussion
Sorghum transformation has been widely considered as challenging since the first transgenic sorghum was reported in 1993 (Casas et al. 1993). Here, we report the highest recorded sorghum transformation efficiency to date of 20.7%. This result is more than 70 times higher than the first successful transformation rate of 0.286% reported via microprojectile transformation, and more than 25 times higher than the biolistic sorghum transformation efficiencies cited by Grootboom et al. (2010) (0.11–0.77%) and Raghuwanshi and Birch (2010) (0.09%). We have more than doubled the best published frequency of Agrobacterium-mediated transformation of up to 8.3% (Gurel et al. 2009). The enhancement of sorghum transformation efficiency can be largely attributed to three crucial factors: (1) tissue culture system; (2) DNA delivery system; and (3) selection strategy.
The tissue culture system plays a fundamental role in the success of sorghum microprojectile transformation and the Agrobacterium-mediated transformation system. There are many factors which directly influence the effectiveness of the tissue culture system, from the method of initiating explants to the procedure of planting putative transgenic plantlets in containment glasshouses. Genotype selection is of paramount importance to obtain an efficient tissue culture system. For example, P898012, C401 and Tx430 have been widely studied (Gao et al. 2005b; Sato et al. 2004). In our laboratory, we also routinely transform genotype 296B and SA281, although these do not allow for such a high efficiency as Tx430 (data not shown). Choosing the appropriate explant is also important to constantly produce reliable embryogenic callus. IEs have been shown to be the most successful and productive explants for sorghum tissue culture (Elkonin and Pakhomova 2000; Grootboom et al. 2010; Gurel et al. 2009). Optimization of the callus induction medium, regeneration medium and rooting medium is another approach to improve tissue culture performance. M11 medium, which contains MS medium supplemented with 1 g/L KH2PO4, has been shown to be more effective than MS medium for sorghum tissue culture (Sato et al. 2004; Elkonin and Pakhomova 2000). The addition of l-proline and l-asparagine has improved the production of sorghum embryogenic callus and reduced the incidence of toxic phenolic compounds in medium (Elkonin et al. 1995). Furthermore, the presence of high levels of copper in CIM and regeneration medium has been shown to enhance the callus induction rate and regeneration frequency (Nirwan and Kothari 2003). Based on reports in literature, we optimized the usage of additional ingredients including CuSO4, KH2PO4, l-proline, l-asparagine and plant growth regulators including 2,4-d, IAA, IBA, NAA and BAP in callus induction medium, regeneration medium and rooting medium. After optimization of the sorghum tissue culture system for inbred line Tx430, an efficient tissue culture system was established. More than 95% of IEs form callus, and of these more than 70% regenerate shoots with no further loss of in vitro plantlets during the transition from the tissue culture environment to containment glasshouse conditions.
Promoters and reporter genes play an important role in optimizing the DNA delivery system. There are many promoters which have been studied for plant transformation, but the strength and suitability of promoters are variable and result in different levels of gene expression in target tissue (Kumar et al. 2011; Tadesse et al. 2003; Able et al. 2001). It was reported that the number of GUS foci was significantly higher from ubi1 than from actin1 and CaMV 35S (Able et al. 2001). Similarly, this result was further refined utilizing GUS histochemical staining and enzymatic activity assay, showing the strength of promoters in descending order: ubi1 > act1D > adh1 > CaMV 35S (Tadesse et al. 2003). Recently, CaMV 35S promoter activity was reported to be very low in small T0 sorghum transgenic plants (Kumar et al. 2011). The ubi1 promoter has proven to be the most successful promoter in sorghum transformation in recent years (Grootboom et al. 2010; Raghuwanshi and Birch 2010; Gurel et al. 2009). GFP is a highly versatile reporter gene, because the gfp gene expression can be monitored any time in living cells under a fluorescence microscope in a non-destructive manner (Chiu et al. 1996). Hence, visual examination may be used to optimize helium pressure, shooting distance and other parameters very rapidly, and non-destructively by counting the GFP foci. The same tissues may then be used for regeneration of stable transformants, which is not possible with other maker genes requiring destructive or toxic enzyme assays. Visual marker genes like gfp can even be used for sorghum transformation without using antibiotics or herbicides as the selection agents (Gao et al. 2005a).
Transgenic sorghum plants have been successfully obtained from IEs with the aid of three selective markers (Kumar et al. 2011; Grootboom et al. 2010; Gurel et al. 2009). The herbicide-resistance bar gene was used in the first transgenic sorghum report (Casas et al. 1993). The phosphomannose isomerase (pmi) gene, which converts mannose-6-phosphate to fructose-6-phosphate (Joersbo et al. 1998), was proven to be an efficient selective marker for sorghum Agrobacterium-mediated transformation (Gurel et al. 2009), and the hygromycin phosphotransferase (hpt) gene, which gives the plant resistance to the antibiotic hygromycin, was reported to be a good selective marker for sorghum transformation (Kumar et al. 2011; Carvalho et al. 2004). The neomycin phosphotransferase (nptII) gene has been shown to be successful in sugarcane microprojectile transformation (Bower and Birch 1992). It was also shown to be an efficient selective marker for sorghum microprojectile transformation during the present study.
Genetic modification technologies play an important role in modern agriculture. An efficient genetic transformation system greatly enhances the ability of scientists to study gene function and improve economically important traits in crop plants. A high frequency of reliable sorghum transformation makes genetic engineering technology more practical and feasible, which will be critical in an era where the demand for food and biomaterials is ever expanding, and the amount of arable land is limited.
References
Able JA, Rathus C, Gray S, Nguyen TV, Godwin ID (1998) Transformation of sorghum using the Particle Inflow Gun (PIG). Int Sorghum Millets Newsl 39:98–100
Able JA, Rathus C, Godwin ID (2001) The investigation of optimal bombardment parameters for transient and stable transgene expression in sorghum. In Vitro Cell Dev Biol Plant 37(3):341–348
Belton PS, Taylor JRN (2004) Sorghum and millets: protein sources for Africa. Trends Food Sci Technol 15(2):94–98
Bower R, Birch RG (1992) Transgenic sugarcane plants via microprojectile bombardment. Plant J 2(3):409–416
Carvalho CHS, Zehr UB, Gunaratna N, Anderson J, Kononowicz HH, Hodges TK, Axtell JD (2004) Agrobacterium-mediated transformation of sorghum: factors that affect transformation efficiency. Genet Mol Biol 27(2):259–269
Casas AM, Kononowicz AK, Zehr UB, Tomes DT, Axtell JD, Butler LG, Bressan RA, Hasegawa PM (1993) Transgenic sorghum plants via microprojectile bombardment. Proc Natl Acad Sci USA 90(23):11212–11216
Chiu WL, Niwa Y, Zeng W, Hirano T, Kobayashi H, Sheen J (1996) Engineered GFP as a vital reporter in plants. Curr Biol 6(3):325–330
Dugdale B, Beetham PR, Becker DK, Harding RM, Dale JL (1998) Promoter activity associated with the intergenic regions of banana bunchy top virus DNA-1 to -6 in transgenic tobacco and banana cells. J Gen Virol 79:2301–2311
Elkonin LA, Pakhomova NV (2000) Influence of nitrogen and phosphorus on induction embryogenic callus of sorghum. Plant Cell Tissue Organ Cult 61(2):115–123
Elkonin LA, Lopushanskaya RF, Pakhomova NV (1995) Initiation and maintenance of friable, embryogenic callus of sorghum (sorghum-biocor(L) moench) by amino-acids. Maydica 40(2):153–157
Gao Z, Jayaraj J, Muthukrishnan S, Claflin L, Liang GH (2005a) Efficient genetic transformation of Sorghum using a visual screening marker. Genome 48(2):321–333
Gao Z, Xie X, Ling Y, Muthukrishnan S, Liang GH (2005b) Agrobacterium tumefaciens-mediated sorghum transformation using a mannose selection system. Plant Biotechnol J 3(6):591–599
Grootboom AW, Mkhonza NL, O’Kennedy MM, Chakauya E, Kunert K, Chikwamba RK (2010) Biolistic mediated sorghum (Sorghum bicolor L. Moench) transformation via mannose and bialaphos based selection systems. Int J Bot 6(2):1811–9719
Gurel S, Gurel E, Kaur R, Wong J, Meng L, Tan H-Q, Lemaux PG (2009) Efficient, reproducible Agrobacterium-mediated transformation of sorghum using heat treatment of immature embryos. Plant Cell Rep 28(3):429–444
Hiei Y, Komari T (2008) Agrobacterium-mediated transformation of rice using immature embryos or calli induced from mature seed. Nat Protoc 3(5):824–834
Hill-Ambroz KL, Weeks JT (2001) Comparison of constitutive promoters for sorghum [Sorghum bicolor (L.) Moench] transformation. Cereal Res Commun 29(1-2):17–24
Ibrahim AS, El-Shihy OM, Fahmy AH (2010) Highly efficient Agrobacterium tumefaciens-mediated transformation of elite Egyptian barley cultivars. Am Eurasian J Sustain Agric 4(3):403–413
Ishida Y, Hiei Y, Komari T (2007) Agrobacterium-mediated transformation of maize. Nat Protoc 2(7):1614–1621
Jeoung JM, Krishnaveni S, Muthukrishnan S, Trick HN, Liang GH (2002) Optimization of sorghum transformation parameters using genes for green fluorescent protein and beta-glucuronidase as visual markers. Hereditas 137(1):20–28
Joersbo M, Donaldson I, Kreiberg J, Petersen SG, Brunstedt J, Okkels FT (1998) Analysis of mannose selection used for transformation of sugar beet. Mol Breed 4(2):111–117
Kaeppler HF, Pedersen JF (1997) Evaluation of 41 elite and exotic inbred Sorghum genotypes for high quality callus production. Plant Cell Tissue Organ Cult 48(1):71–75
Karami O, Esna-Ashari M, Kurdistani GK, Aghavaisi B (2009) Agrobacterium-mediated genetic transformation of plants: the role of host. Biologia Plantarum 53(2):201–212
Kumar V, Campbell LM, Rathore KS (2011) Rapid recovery- and characterization of transformants following Agrobacterium-mediated T-DNA transfer to sorghum. Plant Cell Tissue Organ Cult 104(2):137–146
Miller FR (1984) Registration of RTx 430 sorghum parental line. Crop Sci 24(6):1224–1224
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15(3):473–497
Nguyen TV, Thu TT, Claeys M, Angenon G (2007) Agrobacterium-mediated transformation of sorghum (Sorghum bicolor (L.) Moench) using an improved in vitro regeneration system. Plant Cell Tissue Organ Cult 91(2):155–164
Nirwan RS, Kothari SL (2003) High copper levels improve callus induction and plant regeneration in Sorghum bicolor (L.) Moench. In Vitro Cell Dev Biol Plant 39(2):161–164
Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ (1996) Crystal structure of the Aequorea victoria green fluorescent protein. Science 273(5280):1392–1395
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang HB, Wang XY, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang LF, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Mehboob ur R, Ware D, Westhoff P, Mayer KFX, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457(7229):551–556
Raghuwanshi A, Birch RG (2010) Genetic transformation of sweet sorghum. Plant Cell Rep 29(9):997–1005
Sato S, Clemente T, Dweikat I (2004) Identification of an elite sorghum genotype with high in vitro performance capacity. In Vitro Cell Dev Biol Plant 40(1):57–60
Tadesse Y, Sagi L, Swennen R, Jacobs M (2003) Optimisation of transformation conditions and production of transgenic sorghum (Sorghum bicolor) via microparticle bombardment. Plant Cell Tissue Organ Cult 75(1):1–18
Vain P, Keen N, Murillo J, Rathus C, Nemes C, Finer JJ (1993) Development of the particle inflow gun. Plant Cell Tissue Organ Cult 33(3):237–246
Williams SB, Gray SJ, Laidlaw HKC, Godwin ID (2004) Particle inflow gun-mediated transformation of Sorghum bicolor. In: Transgenic crops of the world: essential protocols. Kluwer Academic Publishers, Dordrecht, pp 89–102
Zhao Z, Tomes D (2003) Sorghum transformation. Genet Transform Plants 23:91–107
Zhao ZY, Cai T, Tagliani L, Miller M, Wang N, Pang H, Rudert M, Schroeder S, Hondred D, Seltzer J, Pierce D (2000) Agrobacterium-mediated sorghum transformation. Plant Mol Biol 44(6):789–798
Acknowledgments
We wish to thank the ARC (Australian Research Council) and Pacific Seeds for their financial support on Linkage project LP0883808. We also are grateful to the Bureau of Sugar Experimental Stations (BSES) Limited for providing the plasmids utilized in this study. Our thanks also extend to Dr. Bradley Campbell and Dr. Yue Sun for their efforts during the editing process.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by P. Lakshmanan.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Liu, G., Godwin, I.D. Highly efficient sorghum transformation. Plant Cell Rep 31, 999–1007 (2012). https://doi.org/10.1007/s00299-011-1218-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-011-1218-4