
Abstract
The immune system in early life is tasked with transitioning from a relatively protected environment to one in which it encounters a wide variety of innocuous antigens and dangerous pathogens. The immaturity of the developing immune system, and particularly the distinct functionality of T lymphocytes in early life, has been implicated in increased susceptibility to infection. Previous work has demonstrated that immune responses in early life are skewed toward limited inflammation and atopy; however, there is mounting evidence that such responses are context- and tissue-dependent. The regulation, differentiation, and maintenance of infant T cell responses, particularly as it relates to tissue compartmentalization, remains poorly understood. How the tissue environment impacts early-life immune responses and whether the development of localized protective immune memory cell subsets are established is an emerging area of research. As infectious diseases affecting the respiratory and digestive tracts are a leading cause of morbidity and mortality worldwide in infants and young children, a deeper understanding of site-specific immunity is essential to addressing these challenges. Here, we review the current paradigms of T cell responses during infancy as they relate to tissue localization and discuss implications for the development of vaccines and therapeutics.
Similar content being viewed by others


Introduction
Neonates and infants are disproportionately susceptible to multiple viral and bacterial pathogens, many of which are encountered via mucosal and barrier sites, including the respiratory and digestive tracts, skin, and other mucosal surfaces. The worse outcome of infants to infection, as well as their limited and delayed responses to vaccines, has been primarily attributed to immaturity of the immune system in early life. Reduced functional responses by T lymphocytes have been specifically implicated due to their role in coordinating many aspects of adaptive immunity.
A major challenge during the neonatal period is to not only develop immune responses to the diverse pathogens encountered at multiple sites, but also to contain infections while at the same time preventing potentially lethal effects associated with systemic infection and immune responses. The immediate response to infection with pathogenic and non-pathogenic microorganisms involves components of the innate immune response, which are highly active during infancy, including the production of anti-microbial peptides, direct phagocytosis from neutrophils and monocytes/macrophages, and production of anti-microbial products and pro-inflammatory cytokines (for a review, see [1]). These innate immune responses are not sufficient to clear most pathogens or to prevent dissemination of infection, and adaptive immune responses characterized by specificity and long-term memory are subsequently triggered. The adaptive immune response is mobilized in early life and required for protecting the neonate, as infants with severe combined immunodeficiency (SCID) who lack lymphocytes suffer from repeated and disseminated infections [2] and ultimately succumb without immune reconstitution.
The adaptive immune response is initiated by dendritic cells (DCs) which have taken up antigens in the affected tissues and migrated to the tissue-draining lymph node(s). DCs present antigen to T cells in the context of major histocompatibility complex (MHC) molecules and simultaneously provide costimulatory signals (CD80 and CD86) necessary for T cell activation via the cell surface T cell antigen receptor (TCR) and the costimulatory receptor CD28. Following these activating signals, T cells begin to rapidly proliferate. CD4+ T cells, in particular, begin to produce IL-2, which acts in both an autocrine and paracrine manner as a third signal to further enhance proliferation and subsequent differentiation into effector T cells (Teff). Along with IL-2, the presence of additional cytokines at this time influences the commitment of newly activated T cells to defined functional subsets with particular cytokines promoting certain subsets. Among CD4+ T cells, Th1, Th2, Th17, regulatory T cell (Treg), and follicular helper T cell (Tfh) subsets have been well-described [3, 4]. CD8+ T cells appear to be less heterogeneous and differentiate primarily into cytotoxic effectors (CTLs) analogous to Th1-type CD4+ T cells [5]. Newly activated effector T cells then home to the affected tissues where they exert their effector functions. Th1-type T cells, typically generated in the context of viral infections, are robust producers of IFN-γ and TNF-α and are required to clear many types of viral and bacterial infections encountered in early life.
Following the resolution of infection, a fraction of responding effector T cells will be retained long-term as populations of memory T cells, which persist for up to a lifetime of an individual. Memory T cells are heterogeneous in their localization and maintenance within circulation, lymphoid tissues, and multiple peripheral tissues and barrier sites, including the skin, lungs, and intestines. It is now understood that a significant fraction of memory T cells in both mice and humans is comprised of non-circulating, tissue-resident memory (TRM) T cells [6,7,8]. The functional importance of TRM in protection from reinfection has been demonstrated in adult murine models at a number of tissue sites and for several pathogens [6, 9,10,11,12], and the predominant presence of TRM-phenotype cells in human tissues [13] suggests important roles for maintaining immune protection and homeostasis in humans.
The generation, function, and regulation of immune responses by T cells during early life are not well understood, in either mouse models or humans. Defining the functional capacities of early-life T cells continues to evolve with findings that infant T cells may have diverse or distinct functions depending on the nature of the infection or stimulus [14]. Moreover, the ability of early-life infections to promote tissue-localized T cell responses, lasting T cell memory, and TRM responses is only beginning to be characterized. Neonates and infants have substantial populations of T cells from birth, both in the blood and in peripheral tissues. These T cells, however, are predominantly naïve, express distinct patterns of homing receptors compared to adults, and in some cases may generate more regulatory than pro-inflammatory responses. In order to develop therapies to better treat infections and promote robust, protective responses to vaccines, it is essential to define the mechanisms that control T cell differentiation, function, tissue localization, and maintenance at the earliest life stages.
This review will discuss current paradigms in infant T cell immunity and tissue localization and how recent studies have begun to shift our understanding of T cell responses in early life with implications in the design of vaccines and therapeutics to protect this vulnerable population.
Burden of infection and immunological challenges in early life
Following birth, neonates transition from a largely sterile environment to one where they are rapidly exposed to novel innocuous antigens and microorganisms and an abundance of potentially pathogenic organisms. The majority of the earliest pathogen encounters during early life occur at the interface where our immune system meets the external environment, including skin, gastrointestinal, and respiratory tracts. As a consequence, the burden of infectious disease in early life occurs at these tissue sites. Annually, over 2 million neonates are affected by severe infections with lower respiratory tract infections (LRTIs), malaria, diarrheal illnesses, neonatal sepsis, and meningitis among the most common etiologies in this population [15, 16]. The systemic spread of infection can result in sepsis, which is the most common cause of death in infants and young children worldwide. Failure to effectively control pathogens within the respiratory tract has led it to be the most common cause of pediatric sepsis [17]. The high susceptibility of infants to disseminated infection indicates that tissue-immune responses are less well-developed and are not always sufficient to contain infection in the neonatal period.
Viral respiratory tract infections (VRTIs) are ubiquitous among children with nearly all children having experienced infection with respiratory syncytial virus (RSV) within the first 2 years of life [18]. Viruses account for the majority of LRTI in children less than 5 years of age [19], with RSV, influenza, and rhinoviruses most prevalent; however, significant morbidity and mortality also result from human metapneumovirus (HMPV), coronavirus, bocavirus, parainfluenza, enterovirus, and adenovirus [20]. RSV alone is the second leading cause of death in infants and young children and the most frequent cause of non-neonatal infant mortality [21]. The clinical features of viral LRTI typically begin with mild symptoms progressing in severity to respiratory distress and lung tissue damage [20]. Few options for treatment are currently available, with supportive care prevailing [22]. A variety of antiviral therapeutics, including immunoglobulins, siRNA-interference, fusion inhibitors, and small molecules, are currently in development at the clinical trial phase [23]. While children with underlying conditions including malnutrition, chronic lung disease, congenital heart disease, and those born extremely prematurely (< 29 weeks gestation) are at increased risk for severe illness, from LRTI [24], the majority of children who require hospitalization have none of these risk factors [25, 26], emphasizing the general vulnerability of infants to these infections.
Diarrheal illnesses are the other major clinical manifestation of early-life infections, with 70% of the 700,000 deaths worldwide occurring in the first 2 years of life. Rotavirus remains the most common cause of severe and fatal diarrhea worldwide [27], although a vaccine is available. Cholera is also a significant cause of diarrhea-related mortality, with peak incidence in children under 5 years of age, during epidemics. Importantly, multiple episodes of diarrheal illness have been associated with nutritional deficits resulting in long-term consequences, including growth stunting and decreased cognitive function [28]. The focus of therapy during acute infectious diarrheal illness focuses on hydration status, with therapies including antibiotics reserved for specific etiologies (i.e., cholera). Prevention is of particular importance in diarrheal illness with a focus on hygiene and vaccination [29]. The prevalence of mucosal infections in early life suggests inefficient mucosal immune responses and a need to enhance immunity in a site-specific fashion.
Peripheral seeding and initiation of early-life T cell responses
In both mice and humans, T cells have distinct phenotypic and distribution patterns in early life which change gradually with age (Table 1). Although mice are born relatively lymphopenic, humans are born with a full complement of peripheral lymphocytes [30,31,32]. The majority of T cells in human infants exhibit features of recent thymic emigrants, including high levels of T cell receptor excision circles (TRECs) as transient products of TCR gene rearrangement and expression of CD31 on the cell surface [32, 33] (Table 1). In both humans and infant mice (> 1 week of age), the majority of T cells present within blood and lymphoid and peripheral tissues in early life exhibit a naïve phenotype with high expression of L-selectin (CD62L) and low expression of the activation marker CD44 for murine naïve T cells, and expression of the CD45RA isoform and the chemokine receptor CCR7 for human naïve T cells [32, 34]. In mice, T cells during the first week of post-natal life display increased expression of activation markers, including enhanced levels of CD44, as a result of lymphopenia-induced homeostatic expansion which transitions to a more naïve-like phenotype in the second week of life and beyond [30, 31]. In humans, this early-life homeostatic expansion does not occur, suggesting that use of mice as models for human early-life T cell immunity should assess responses following this initial period of lymphopenia-induced proliferation.
The seeding of blood and tissues with newly generated naïve T cells is a feature of the neonatal immune response. Due to continuous antigen exposure as well as the abundance of new antigens acquired during early life, T cells become activated and convert to memory phenotypes involving the upregulation of differentiation markers and downregulation of lymph node homing receptors (CD44hi/CD62Lo for mouse memory T cells and CD45RO+/CCR7lo for human memory T cells). In humans, memory-phenotype cells can be detected during infancy mostly in mucosal sites such as lungs and jejunum, while during late childhood and in adults, the majority of T cells in mucosal sites and > 50% in lymphoid tissue and blood are memory phenotype [13, 32]. These distinct patterns of T cell compartmentalization can significantly impact the way by which infants respond to infectious challenge in tissues compared to adults.
A major function of naïve T cells is active immune surveillance, and such cells are essential in the host’s ability to respond to novel pathogens. Naïve T cells migrate primarily through the blood, lymph, and lymphoid tissues, a process mediated by expression of CD62L, which binds CD34, expressed by endothelial cells, and GlyCAM-1, found on the lymph node high endothelial venules, as well as CCR7 which binds the chemokines CCL19 and CCL21, both expressed in the lymph nodes. During infection, naïve T cells interact with mature, activated DCs bearing pathogen-derived antigens within the context of the lymph nodes. This interaction of naïve T cells with DCs may influence how infant T cells can respond to new antigens.
In early life, DC populations differ both in subset composition and functionality relative to adults. In both the lungs and spleen, neonatal mice exhibit reduced numbers of CD11b+ DCs (CD4+CD11b+), important in CD4+ T cell activation, relative to adults [35, 36]. Similarly, numbers of CD103+ (CD8+CD103+) DCs, responsible for activation of CD8+ T cells, are reduced in the neonatal spleen during the steady state and exhibit reduced maturation and migration from the lung to the lung-draining lymph node following viral infection [35]. In humans, frequencies of CD1c+ DCs, analogous to CD11b+ DCs in mice, are higher in the jejunum and appendix of infant and pediatric donors compared to adults [37]. Cord blood-derived DCs were found to exhibit decreased levels of MHC-II, costimulatory ligands, and IL-12 production upon stimulation compared to adult DCs [38,39,40]. In mice, however, DCs derived from neonatal spleen produced IL-12 at levels comparable to adults following stimulation [36], suggesting potential site-specific differences in DC function in early life between mice and humans. Importantly, differences in DC subsets, localization, and function in early life may have profound effects on T cell responses with reduced numbers and functionality of neonatal DCs potentially leading to reduced costimulation necessary for optimal T cell activation following infection.
While the majority of T cells in infant mouse and human tissues are naïve, populations of memory T cells can be found in the lungs, jejunum, and ileum of human infants [32], which represent the predominant sites of early antigen encounter to pathogens, commensal microorganisms, and food antigens. Interestingly, despite the local accumulation of T cell memory in the tissues, T cells present in lymph nodes draining these tissues remain predominantly naïve [32], suggesting possible in situ priming of immune responses. Similarly, neonatal mice can generate inducible bronchus-associated lymphoid tissue (iBALT) as a consequence of pulmonary inflammation [41] indicating localized T cell priming. Taken together, these findings suggest a potential role for in situ, tissue priming in the initiation of T cell responses in early life which may have important implications for vaccines.
T cell tissue homing
The interaction of T cells with DCs during priming also results in the upregulation of molecules facilitating tissue-specific homing in a process termed imprinting, where DCs derived from particular tissue sites elicit specific patterns of homing receptor expression which guide activated T cells back to those tissues [42]. In adult mouse models, T cells activated and matured in Peyer’s patches or mesenteric lymph nodes express CCR9 and the integrin α4β7, which mediate gut homing, whereas activation in lymph nodes draining non-gastrointestinal tissues, such as the skin or lung, results in upregulation of CCR4 which promotes homing to these tissues [42]. In early life, peripheral blood T cells predominantly express the gut-homing receptor α4β7 while adult peripheral blood-derived T cells express the tissue-homing receptor CCR4 [43] (Table 1). Furthermore, in response to pathogen-derived signals, the expression of homing receptors on infant, but not adult T cells, was altered [43], suggesting differential regulation of T cell homing during infection during infancy (Table 1).
The distinct patterns of homing receptor expression in early life may influence infant T cell localization in the context of infection. In a neonatal mouse model of influenza infection, recruitment of virus-specific CD8+ T cells to the lung was delayed relative to adults and neonatal T cells exhibited distinct localization patterns within the lung tissue [44, 45]. In other studies in humans and mouse models, there is evidence that enhanced recruitment of T cells to the lung may contribute to pathology. We found increased frequencies of CD8+ T cells in airway secretions from children requiring invasive mechanical ventilation due to severe respiratory infection that correlated with lung injury [46], suggesting infiltration of pathogenic CD8+ T cells into the lung. Neonatal mouse studies have also shown that RSV infection in early life resulted in enhanced T cell infiltration with increased airway hyperresponsiveness upon later reinfection [47, 48]. Autopsy studies of fatal cases of RSV and influenza demonstrate extensive immune cell infiltration dominated by neutrophils and macrophages [49, 50]. Taken together, these findings support the notion that T cell homing during infection may be distinct in early life compared to in adulthood, with suboptimal responses under some conditions and enhanced accumulation of T cells in the tissues under other conditions contributing to immune pathology.
T cell effector functions
T cell activation leads to the differentiation of T cells into distinct functional subsets, including Th1, Th2, Th17, Treg, and Tfh subsets. This process is governed by the inflammatory context of infection and driven by distinct transcriptional regulators promoting defined transcriptional programs [3]. In the context of early-life responses, differentiation into Th1 and Th2 subsets has been the most well-studied process. Th1-type responses are characterized by T-bet-driven expression of IFN-γ and TNF-α and the generation of CTLs, which lyse infected cells [3, 51]. Th2 effector differentiation is driven by GATA3 expression characterized by secretion of IL-4, IL-5, and IL-13, which promote eosinophil and mast cell recruitment and the generation of antibody responses important in the control of fungal and parasitic infections [3]. The generation of functional subsets of T cells has been extensively characterized in infant T cells, identifying quantitatively and qualitatively distinct responses as described below and summarized in Table 1.
Early work investigating the generation of Th1 and Th2 responses in neonates and infants revealed that the IFN-γ locus in neonatal T cells is hypermethylated [52], resulting in transcriptional repression, while the IL-4 locus is hypomethylated allowing for enhanced expression [53]. Based on these results, T cell responses in early life were designated as Th2-biased (Table 1) and provided a basis for diminished T cell responses to infection, as Th1-type responses provide protection from viral and intracellular bacterial infections. In humans, studies of cord blood-derived T cells likewise demonstrated reduced production of IFN-γ compared to adult-derived cells following activation in vitro and simultaneous production of IFN-γ and IL-4, not typically observed in adults [54, 55]. Such Th2-biased responses primed during early life could also persist into adulthood, in part through a mechanism involving apoptosis of Th1-type effectors upon antigen recall [56]. In an RSV infection and vaccination model, neonatal mice generated Th2-biased responses which led to Th2-biased memory responses in adulthood [47, 57]. Severe RSV infection in children has similarly been linked to enhanced Th2-type immune responses and subsequent wheezing episodes in later childhood [47, 58]. These studies suggested that the infection history during infancy could impact the quality of responses later in life.
Despite this evidence for Th2-skewing, a number of studies have demonstrated that neonatal/infant T cells from both humans and mice can generate Th1 responses characterized by robust IFN-γ production under appropriate conditions. While stimulation of cord blood-derived CD4+ T cells does not result in significant IFN-γ production, addition of exogenous IL-12 to the culture promotes IFN-γ secretion at levels similar to adult peripheral blood T cells [55]. Similarly, T cells derived from infant intestinal tissues produce ample IFN-γ following ex vivo stimulation [32], suggesting that human T cells in early life are fully competent to produce pro-inflammatory cytokines in the proper environment. In the context of RSV infection, robust IFN-γ-driven responses have been observed in children and nearly 80% of infants infected with RSV in the first year of life develop virus-specific CTL activity correlating with enhanced IFN-γ production and reduced IL-4 responses [59], which can also contribute to immune pathology [60]. In neonatal mice, infection with, low doses of murine leukemia virus generated protective cytotoxic T cell responses while high dose infection preferentially generated non-protective Th2 responses [61] suggesting that the nature of the initial infection may also influence the nature of protective T cell responses.
There is increasing evidence that T cells in early life may also have unique functionality not typically observed in adults. Neonatal human T cells derived from cord blood and fetal tissues undergo increased proliferation relative to adult cells [62, 63], suggesting differences in cell cycle control between infant and adult T cells. Moreover, T cells from newborn cord blood and infant peripheral blood produce high levels of the chemokine IL-8 (CXCL8) following stimulation that greatly exceeds levels secreted by adult T cells [14] (Table 1). The precise role of IL-8 in early-life T cell responses is not known, although IL-8 is a potent chemoattractant and activator of neutrophils and γδ T cells [14] and could serve to promote recruitment of these cells into infection sites. Furthermore, cord blood contains significant frequencies of γδ T cells with enhanced functionality compared to conventional αβ T cells, acting as robust producers of IFN-γ and IL-2 [64]. Taken together, these studies indicate that early-life T cell responses may exhibit context-dependent differences in function relative to adults.
Regulation of early-life T cell responses
A key challenge of early life is to balance the conflicting demands of generating appropriate, robust immune responses to pathogens and develop tolerance to innocuous and self-antigens. A number of pathways limiting T cell responsiveness in early life have been identified that are specific to either infancy and/or enhanced during early life. Erythroid-lineage CD71+ cell populations in neonatal mice and human cord blood were found to mediate immune suppression through depletion of L-arginine, an essential factor for T cell responses [65]. In addition, there are increased frequencies of Tregs present in the blood and peripheral tissues during fetal and early life which are functionally enhanced relative to adult Tregs [32, 66]. Depletion of infant Tregs promotes robust IFN-γ responses in cultures of infant CD4+ and CD8+ T cells [32], suggesting that the ability of infant T cells is not intrinsically compromised, but subject to increased inhibition. Relative to adults, infant T cells are more likely to differentiate into Tregs following stimulation in a process mediated by the RNA-binding protein Lin28b and TGF-β signaling [67, 68], revealing additional mechanisms by which early-life inflammatory responses can be dampened. Importantly, multiple suppressive mechanisms may be necessary to limit potentially detrimental inflammatory responses in the context of extensive new antigen exposure immediately after birth. The increased presence and functional capacity of infant Tregs may also serve to contain tissue-immune responses and prevent lymphocytic infiltration into multiple tissues during this critical early-life window.
Memory T cell establishment and subsets
Following the resolution of primary infection, CD4+ and CD8+ effector T cell populations undergo a rapid contraction during which approximately 90–95% of effector cells die by apoptosis. Subsets of pathogen-specific T cells, however, survive this contraction and are retained as long-lived memory T cells which can persist for life, conveying protective immunity upon secondary pathogen encounter. Factors determining which responding cells persist as memory, however, are not completely clear, and even less is known about how this process occurs in early life. There is evidence that neonatal effector T cells have increased propensity for apoptosis [62, 63], and their increased proliferation may also drive them to a reduced lifespan.
A key factor required for memory T cell generation is signaling via homeostatic and survival cytokines such as IL-7 and IL-15. In adults, naïve CD4+ and CD8+ T cells express IL-7R (a dimer of CD127 and the common gamma chain receptor CD132) [69, 70] and IL-7 signaling is important, although not sufficient, for the transition from effector to memory as memory CD4+ cells fail to develop in IL-7-deficient hosts [71, 72]. IL-15 signaling is also essential for CD8+ memory formation [73], although it is dispensable in the generation of memory CD4+ T cells [74]. IL-7 and IL-15 signaling have both been further shown to be important in the long-term maintenance of CD4+ and CD8+ memory T cells by promoting homeostatic proliferation [71, 72, 75]. In both humans and mice, T cells derived from fetal tissues or cord blood have an increased tendency to proliferate in response IL-7 and IL-15 signaling relative to adult controls [76, 77]. The increased susceptibility of neonatal T cells to apoptosis following activation can be ameliorated by IL-7 and IL-15 in vitro [62, 63], suggesting that this pathway could be targeted for enhancing memory T cell generation from neonatal T cells.
In the context of infection, cell surface markers delineating T cell memory precursors in adults have been identified. CD8+ cells expressing high levels of CD127 and low levels of the co-inhibitory receptor killer-cell lectin-like receptor G1 (KLRG1) serve as precursors to memory T cells [78, 79], while CD127(lo)KLRG1(hi) CD8+ T cells tend to be short-lived effector cells [78]. In an analogous fashion, high expression of Ly6c delineates terminally differentiated CD4+ effector cells from memory precursors [80]. At the transcriptional level, terminally differentiated CD4+ and CD8+ effector T cells express high levels of the Th1 lineage-defining transcription factor T-bet [79, 81] which has been shown in mouse infection models to drive differentiation toward terminal effector rather than memory T cells [82].
During the neonatal period, considerably less is known regarding transcriptional regulation of effector and memory differentiation. In a systemic infection model in neonatal mice, increased percentages of CD8+ T cells displayed a terminally differentiated CD127(lo)KLRG1(hi) phenotype with higher levels of T-bet relative to adults [83] (Table 1). These neonatal effector CD8+ T cells had shorter lifespans in vivo compared to adult counterparts [83]. For CD4+ T cells, increased T-bet induction was observed in human infant compared to adult CD4+ T cells following activation in vitro, and in mouse, CD4+ T cells recruited to the lung during acute influenza infection in infant compared to adult mice [84].Taken together, these findings suggest that T cells may be intrinsically programmed for terminal effector differentiation in early life for promoting rapid pathogen clearance that is critical during the neonatal period and takes precedence over memory generation.
Memory T cell localization and function
Memory CD4+ and CD8+ T cells are heterogeneous in terms of phenotype, localization, and function. Like effector T cells, memory T cells retain high-level expression of CD44 and human cells express the CD45RO isoform. Memory T cells are, however, heterogeneous in their expression of the lymphoid homing molecules CD62L and CCR7 which led to delineation of two subsets: CD62L+/CCR7+, central memory T cells (TCM) which localize to secondary lymphoid tissues, and CD62L-/CCR7-effector memory T cells (TEM) present in peripheral tissues [85]. Both subsets were presumed to circulate with TCM migrating through the secondary lymphoid tissues and TEM acting to survey the peripheral tissues.
In addition to TEM and TCM subsets, a distinct population of non-circulating memory T cells termed tissue-resident memory (TRM) has been recently identified to persist long-term in peripheral tissues, including brain, skin, vaginal mucosa, and lung, following infection [6, 9,10,11,12]. Similar to TEM, TRM are CD44hi and CCR7lo/CD62Llo. Both CD4+ and CD8+TRM are distinguished by expression of CD69, a cell-surface marker that is upregulated early after T cell activation and also serves tissue retention function in lymph nodes [86]. CD4+TRM also express high levels of CD11a, the alpha chain of the integrin LFA-1 [6], while CD8+ TRM express CD103, or αE integrin that pairs with β7 integrin [87], which is not significantly upregulated by CD4+ TRM in mice. Relative to circulating T cell subsets, TRM exhibit enhanced protective capacities mediated by robust in situ responses. For example, pro-inflammatory cytokines produced by TRM can promote DC maturation and recruitment of circulating memory T cells and B cells to the site of infection [88,88,90]. While precise mechanisms underlying the generation of distinct T memory subsets remain unclear, these populations are transcriptionally distinct [11]. The phenotype and migration properties of naïve, effector, TCM, TEM, and TRM subsets are outlined in Table 2.
In humans, the majority of T cells in tissues of older children and adults exhibit memory phenotypes and the majority of these memory T cells are CD69+, suggesting that they are TRM. During infancy, early mucosal memory T cells in lungs and intestines upregulate CD69 as a TRM marker to similar extents as adult mucosal memory T cells; however, CD103 expression is reduced on infant mucosal memory CD8+ T cells compared to adult mucosal memory T cells [32]. This result suggests that differentiation to a fully mature TRM phenotype may require additional exposures and/or specific factors within the adult tissue environment. In mouse models of influenza infection, there was reduced generation of CD4+ and CD8+TRM in the lung following infection during infancy compared to mice infected as adults [84], consistent with reduced TRM found in human infant lungs. This reduced TRM formation was intrinsic to infant T cells and could be partially restored by reducing T-bet expression [84]. Due to the importance of this subset in protection from repeated infections, an enhanced understanding of TRM generation in early life will be important in the generation of vaccines and therapeutics for this vulnerable population.
Vaccines and T cell responses in early life
Vaccination is arguably the most important intervention for preventing infectious disease in early life although vaccine responses in young children are often reduced in magnitude and duration compared to older children and adults [91, 92]. Traditional vaccine approaches rely on generating protective serum neutralizing antibodies, which are a correlate of protection following vaccination against common childhood diseases including diphtheria, tetanus, measles, mumps, and rubella, among others [93]. Such protection has been extremely successful in reducing morbidity and mortality. In some cases, however, circulating antibody responses are unable to provide efficient cross protection between distinct serotypes or strains of the same pathogen, such as for Haemophilus influenzae, Streptococcus pneumoniae or influenza, and in other cases, circulating antibody responses do not appear to provide consistent, lasting protective immunity leading to limited protection by antibody-based vaccines as in the case of pertussis vaccines [93].
While the ability of vaccines to elicit tissue-localized immunity is not well-understood, there is evidence that mucosal targeting of vaccines can generate robust tissue-localized immune responses. Both oral poliovaccine (OPV) and inactivated poliovaccine (IPV) induce virus-specific antibody responses; however, OPV-induced antibody responses are mostly localized to the gastrointestinal tract while IPV elicits circulating serum neutralizing antibody responses [93, 94]. Furthermore, individuals vaccinated with IPV demonstrated enhanced stool shedding upon subsequent receipt of a single OPV vaccine strain compared to those vaccinated first with OPV, suggesting differences in site-specific protection elicited by these two vaccines [94]. Similarly, administration of OPV to infants significantly enhanced neutralizing antibody titers and reduced stool shedding compared to IPV-vaccination alone [95].
Given their enhanced functionality and specific tissue localization, TRM are an important new target for vaccine development. Factors promoting protective T cell responses by vaccines, however, are not well understood and even less is known about requirements for TRM establishment and the capacities of infants to generate TRM. Recent vaccine studies in mice have demonstrated that mucosal administration of antigen or vaccination combined with local chemokines or other molecules necessary for T cell homing is important for the establishment of tissue-localized T cell responses [12, 96,96,98]. Furthermore, administration of live-attenuated vaccine formulations can establish protective TRM in several distinct tissue-localized animal disease models [97, 98]. Moreover, children vaccinated at birth with BCG, a live-attenuated vaccine, generated circulating T cells producing adult-like, Th1-mediated IFN-γ responses [99]. Significantly, this work demonstrated both the capacity of young children to generate T cell responses to vaccination as well as robust Th1-type functionality. Interestingly, neonatal mice immunized with incomplete Freund’s adjuvant generate Th2-biased responses while complete Freund’s adjuvant, containing mycobacterial-derived components, promotes Th1-polarized responses [57] illustrating that the inflammatory nature of an immunization significantly influences the quality of the subsequent T cell response, even very early in life.
Studies of influenza vaccination further highlight differing immune and specifically T cell responses to inactivated (IIV) versus live-attenuated (LAIV) vaccines early in life. Compared to older children and adults, children under four receiving IIV demonstrated reduced induction of serum-neutralizing antibody responses and antibody-secreting cells compared to older children and adults [100]. Following immunization with IIV, neonatal mice showed impaired generation of Tfh important for antibody and germinal center responses [101] that could be restored with additional stimulation by the adjuvant MF59 [102], suggesting impaired T cell help during infancy. Live-attenuated influenza virus vaccine (LAIV) elicits measurable circulating, virus-specific T cell responses in infants and young children which are not observed in adults [103]. Furthermore, in a previous study, LAIV provided enhanced protection against the incidence of laboratory-confirmed influenza and influenza-like illness in children compared to inactivated influenza vaccine (IIV) [104] and this protection was superior to that observed in adults [104]. Whether this protection was mediated by T cells in humans is not known, recent mouse studies demonstrate that LAIV generates protective lung TRM, while vaccination with IIV does not [88]. Vaccination of infant mice with LAIV resulted in reduced TRM generation compared to adults, consistent with their intrinsic impairments in TRM differentiation [84] The in vivo efficacy of LAIV in young children can vary between seasons [105], and more studies are needed to evaluate the contribution of tissue localized to circulating responses.
Taken together, these results suggest that neonates and infants are capable of responding effectively following vaccination and provide evidence that T cell responses in early life are not inherently less functional than those of adults. Identifying the immune mechanisms underlying effective host T cell responses to vaccines and how these factors differ between infants and adults is a priority in the rational design of future vaccines and therapeutics for infectious disease. Finally, determining whether vaccines elicit lasting TRM populations in early life and establishing whether circulating T cell responses can predict TRM generation following vaccination could substantially improve both vaccine development and response monitoring in childhood and throughout life.
Conclusions
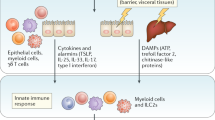
nfants and neonates are highly susceptible to pathogens encountered via the respiratory and gastrointestinal tracts, yet the regulation, differentiation, and maintenance of infant T cell responses during homeostasis and infection or vaccination remains poorly understood. Infant T cells exhibit distinct intrinsic responses at the earliest phases of activation, subsequent tissue homing, and functional differentiation. The generation of memory T cells in circulation and those localized to tissue sites as TRM has been shown to be critical for protection against virus infections, particularly at mucosal sites; however, evidence from human studies and mouse models indicates that TRM formation is significantly reduced during infancy. Given the distinct properties of infant T cells, we propose a model for how infant T cells may be intrinsically programmed for a differentiation pathway that promotes effector responses at the site of infection to promote pathogen clearance, rather than long-term memory responses, using lung and respiratory infection as an example (Fig. 1). The high proliferative capacity and apoptosis by infant T cells, together with their ability to produce IFN-γ at mucosal sites, may promote terminal effector differentiation compared to T cell responses later in life, which encompass both pathogen clearance mechanisms and in situ memory formation (Fig. 1). As a result, the generation of TRM and pathogen-specific memory T cells at the site of infection during infancy is compromised, resulting in reduced protective immunity to recurrent pathogen exposures. Further studies in mouse models and humans to elucidate the molecular mechanisms and transcriptional and epigenetic differences in early- compared to later-life T cells at distinct sites will be essential for developing specific strategies to optimize localized immunity at this critical period of development.

Model for T cell responses to infection in adulthood and early life. Diagram shows schematic of effector and memory differentiation from adult (upper) and infant (lower) T cells. Proliferation of naïve CD4+ and CD8+ T cells is enhanced relative to adults following stimulation, driving differentiation to effector T cells that may likewise be increased in infant compared to adult T cells. Contraction of infant effector T cells by apoptosis is further augmented compared to adults, resulting in decreased establishment of memory T cells, both in circulation and resident in tissues, resulting in decreased protection to repeat pathogen exposures during early life
References
Kollmann TR, Kampmann B, Mazmanian SK, Marchant A, Levy O (2017) Protecting the newborn and young infant from infectious diseases: lessons from immune ontogeny. Immunity 46(3):350–363. doi:10.1016/j.immuni.2017.03.009
Relan M, Lehman HK (2014) Common dermatologic manifestations of primary immune deficiencies. Curr Allergy Asthma Rep 14(12):480. doi:10.1007/s11882-014-0480-2
Zhu J, Yamane H, Paul WE (2010) Differentiation of effector CD4 T cell populations (*). Annu Rev Immunol 28:445–489. doi:10.1146/annurev-immunol-030409-101212
Crotty S (2011) Follicular helper CD4 T cells (TFH). Annu Rev Immunol 29:621–663. doi:10.1146/annurev-immunol-031210-101400
Harty JT, Tvinnereim AR, White DW (2000) CD8+ T cell effector mechanisms in resistance to infection. Annu Rev Immunol 18:275–308. doi:10.1146/annurev.immunol.18.1.275
Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrancois L, Farber DL (2011) Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol 187(11):5510–5514. doi:10.4049/jimmunol.1102243
Turner DL, Bickham KL, Thome JJ, Kim CY, D'Ovidio F, Wherry EJ, Farber DL (2014) Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol 7(3):501–510. doi:10.1038/mi.2013.67
Jozwik A, Habibi MS, Paras A, Zhu J, Guvenel A, Dhariwal J, Almond M, Wong EH, Sykes A, Maybeno M, Del Rosario J, Trujillo-Torralbo MB, Mallia P, Sidney J, Peters B, Kon OM, Sette A, Johnston SL, Openshaw PJ, Chiu C (2015) RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat Commun 6:10224. doi:10.1038/ncomms10224
Gebhardt T, Wakim LM, Eidsmo L, Reading PC, Heath WR, Carbone FR (2009) Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat Immunol 10(5):524–530. doi:10.1038/ni.1718
Masopust D, Choo D, Vezys V, Wherry EJ, Duraiswamy J, Akondy R, Wang J, Casey KA, Barber DL, Kawamura KS, Fraser KA, Webby RJ, Brinkmann V, Butcher EC, Newell KA, Ahmed R (2010) Dynamic T cell migration program provides resident memory within intestinal epithelium. J Exp Med 207(3):553–564. doi:10.1084/jem.20090858
Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villadangos J, Smyth G, Bevan MJ (2012) The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol 189(7):3462–3471. doi:10.4049/jimmunol.1201305
Shin H, Iwasaki A (2012) A vaccine strategy that protects against genital herpes by establishing local memory T cells. Nature 491(7424):463–467. doi:10.1038/nature11522
Thome JJC, Yudanin NA, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, Farber DL (2014) Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159:814–828
Gibbons D, Fleming P, Virasami A, Michel ML, Sebire NJ, Costeloe K, Carr R, Klein N, Hayday A (2014) Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat Med 20(10):1206–1210. doi:10.1038/nm.3670
Lawn JE, Blencowe H, Oza S, You D, Lee AC, Waiswa P, Lalli M, Bhutta Z, Barros AJ, Christian P, Mathers C, Cousens SN, Lancet Every Newborn Study G (2014) Every newborn: progress, priorities, and potential beyond survival. Lancet 384(9938):189–205. doi:10.1016/S0140-6736(14)60496-7
Global Burden of Disease Pediatrics C, Kyu HH, Pinho C, Wagner JA, Brown JC, Bertozzi-Villa A, Charlson FJ, Coffeng LE, Dandona L, Erskine HE, Ferrari AJ, Fitzmaurice C, Fleming TD, Forouzanfar MH, Graetz N, Guinovart C, Haagsma J, Higashi H, Kassebaum NJ, Larson HJ, Lim SS, Mokdad AH, Moradi-Lakeh M, Odell SV, Roth GA, Serina PT, Stanaway JD, Misganaw A, Whiteford HA, Wolock TM, Wulf Hanson S, Abd-Allah F, Abera SF, Abu-Raddad LJ, AlBuhairan FS, Amare AT, Antonio CA, Artaman A, Barker-Collo SL, Barrero LH, Benjet C, Bensenor IM, Bhutta ZA, Bikbov B, Brazinova A, Campos-Nonato I, Castaneda-Orjuela CA, Catala-Lopez F, Chowdhury R, Cooper C, Crump JA, Dandona R, Degenhardt L, Dellavalle RP, Dharmaratne SD, Faraon EJ, Feigin VL, Furst T, Geleijnse JM, Gessner BD, Gibney KB, Goto A, Gunnell D, Hankey GJ, Hay RJ, Hornberger JC, Hosgood HD, Hu G, Jacobsen KH, Jayaraman SP, Jeemon P, Jonas JB, Karch A, Kim D, Kim S, Kokubo Y, Kuate Defo B, Kucuk Bicer B, Kumar GA, Larsson A, Leasher JL, Leung R, Li Y, Lipshultz SE, Lopez AD, Lotufo PA, Lunevicius R, Lyons RA, Majdan M, Malekzadeh R, Mashal T, Mason-Jones AJ, Melaku YA, Memish ZA, Mendoza W, Miller TR, Mock CN, Murray J, Nolte S, Oh IH, Olusanya BO, Ortblad KF, Park EK, Paternina Caicedo AJ, Patten SB, Patton GC, Pereira DM, Perico N, Piel FB, Polinder S, Popova S, Pourmalek F, Quistberg DA, Remuzzi G, Rodriguez A, Rojas-Rueda D, Rothenbacher D, Rothstein DH, Sanabria J, Santos IS, Schwebel DC, Sepanlou SG, Shaheen A, Shiri R, Shiue I, Skirbekk V, Sliwa K, Sreeramareddy CT, Stein DJ, Steiner TJ, Stovner LJ, Sykes BL, Tabb KM, Terkawi AS, Thomson AJ, Thorne-Lyman AL, Towbin JA, Ukwaja KN, Vasankari T, Venketasubramanian N, Vlassov VV, Vollset SE, Weiderpass E, Weintraub RG, Werdecker A, Wilkinson JD, Woldeyohannes SM, Wolfe CD, Yano Y, Yip P, Yonemoto N, Yoon SJ, Younis MZ, Yu C, El Sayed Zaki M, Naghavi M, Murray CJ, Vos T (2016) Global and national burden of diseases and injuries among children and adolescents between 1990 and 2013: findings from the global burden of disease 2013 study. JAMA Pediatr 170 (3):267–287. doi:10.1001/jamapediatrics.2015.4276
Randolph AG, McCulloh RJ (2014) Pediatric sepsis: important considerations for diagnosing and managing severe infections in infants, children, and adolescents. Virulence 5(1):179–189. doi:10.4161/viru.27045
Glezen WP, Taber LH, Frank AL, Kasel JA (1986) Risk of primary infection and reinfection with respiratory syncytial virus. Am J Dis Child 140(6):543–546
Jain S, Finelli L, Team CES (2015) Community-acquired pneumonia among U.S. children. N Engl J Med 372(22):2167–2168. doi:10.1056/NEJMc1504028
Meissner HC (2016) Viral bronchiolitis in children. N Engl J Med 374(1):62–72. doi:10.1056/NEJMra1413456
Geoghegan S, Erviti A, Caballero MT, Vallone F, Zanone SM, Losada JV, Bianchi A, Acosta PL, Talarico LB, Ferretti A, Grimaldi LA, Sancilio A, Duenas K, Sastre G, Rodriguez A, Ferrero F, Barboza E, Gago GF, Nocito C, Flamenco E, Perez AR, Rebec B, Ferolla FM, Libster R, Karron RA, Bergel E, Polack FP (2017) Mortality due to respiratory syncytial virus. Burden and risk factors. Am J Respir Crit Care Med 195(1):96–103. doi:10.1164/rccm.201603-0658OC
American Academy of Pediatrics Subcommittee on D, Management of B (2006) Diagnosis and management of bronchiolitis. Pediatrics 118(4):1774–1793. doi:10.1542/peds.2006-2223
Mazur N, Martinon-Torres F, Baraldi E, Fauroux B, Greenough A, Heikkinen T, Manzoni P, Mejias A, Nair H, Papadopoulos NG, Polack FP, Ramilo O, Sharland M, Stein R, Madhi SA, Bont L, Syncytial CR (2015) Lower respiratory tract infection caused by respiratory syncytial virus: current management and new therapeutics. Lancet Resp Med 3(11):888–900. doi:10.1016/S2213-2600(15)00255-6
American Academy of Pediatrics Committee on Infectious D, American Academy of Pediatrics Bronchiolitis Guidelines C (2014) Updated guidance for palivizumab prophylaxis among infants and young children at increased risk of hospitalization for respiratory syncytial virus infection. Pediatrics 134(2):415–420. doi:10.1542/peds.2014-1665
Hall CB, Weinberg GA, Iwane MK, Blumkin AK, Edwards KM, Staat MA, Auinger P, Griffin MR, Poehling KA, Erdman D, Grijalva CG, Zhu Y, Szilagyi P (2009) The burden of respiratory syncytial virus infection in young children. N Engl J Med 360(6):588–598. doi:10.1056/NEJMoa0804877
Stockman LJ, Curns AT, Anderson LJ, Fischer-Langley G (2012) Respiratory syncytial virus-associated hospitalizations among infants and young children in the United States, 1997-2006. Pediatr Infect Dis J 31(1):5–9. doi:10.1097/INF.0b013e31822e68e6
Walker CL, Rudan I, Liu L, Nair H, Theodoratou E, Bhutta ZA, O'Brien KL, Campbell H, Black RE (2013) Global burden of childhood pneumonia and diarrhoea. Lancet 381(9875):1405–1416. doi:10.1016/S0140-6736(13)60222-6
Guerrant RL, Oria RB, Moore SR, Oria MO, Lima AA (2008) Malnutrition as an enteric infectious disease with long-term effects on child development. Nutr Rev 66(9):487–505. doi:10.1111/j.1753-4887.2008.00082.x
Alam NH, Ashraf H (2003) Treatment of infectious diarrhea in children. Paediatr Drugs 5(3):151–165
Garcia AM, Fadel SA, Cao S, Sarzotti M (2000) T cell immunity in neonates. Immunol Res 22(2–3):177–190. doi:10.1385/IR:22:2-3:177
Le Campion A, Bourgeois C, Lambolez F, Martin B, Leaument S, Dautigny N, Tanchot C, Penit C, Lucas B (2002) Naive T cells proliferate strongly in neonatal mice in response to self-peptide/self-MHC complexes. Proc Natl Acad Sci U S A 99(7):4538–4543. doi:10.1073/pnas.062621699
Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, Gordon C, Granot T, Griesemer A, Lerner H, Kato T, Farber DL (2016) Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat Med 22(1):72–77. doi:10.1038/nm.4008
Junge S, Kloeckener-Gruissem B, Zufferey R, Keisker A, Salgo B, Fauchere JC, Scherer F, Shalaby T, Grotzer M, Siler U, Seger R, Gungor T (2007) Correlation between recent thymic emigrants and CD31+ (PECAM-1) CD4+ T cells in normal individuals during aging and in lymphopenic children. Eur J Immunol 37(11):3270–3280. doi:10.1002/eji.200636976
Torow N, Yu K, Hassani K, Freitag J, Schulz O, Basic M, Brennecke A, Sparwasser T, Wagner N, Bleich A, Lochner M, Weiss S, Forster R, Pabst O, Hornef MW (2015) Active suppression of intestinal CD4(+)TCRalphabeta(+) T-lymphocyte maturation during the postnatal period. Nat Commun 6:7725. doi:10.1038/ncomms8725
Ruckwardt TJ, Malloy AM, Morabito KM, Graham BS (2014) Quantitative and qualitative deficits in neonatal lung-migratory dendritic cells impact the generation of the CD8+ T cell response. PLoS Pathog 10(2):e1003934. doi:10.1371/journal.ppat.1003934
Sun CM, Fiette L, Tanguy M, Leclerc C, Lo-Man R (2003) Ontogeny and innate properties of neonatal dendritic cells. Blood 102(2):585–591. doi:10.1182/blood-2002-09-2966
Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, Miron M, Kumar BV, Griesemer A, Ho SH, Lerner H, Thome JJ, Connors T, Reizis B, Farber DL (2017) Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity 46(3):504–515. doi:10.1016/j.immuni.2017.02.019
Hunt DW, Huppertz HI, Jiang HJ, Petty RE (1994) Studies of human cord blood dendritic cells: evidence for functional immaturity. Blood 84(12):4333–4343
Petty RE, Hunt DW (1998) Neonatal dendritic cells. Vaccine 16(14–15):1378–1382
Langrish CL, Buddle JC, Thrasher AJ, Goldblatt D (2002) Neonatal dendritic cells are intrinsically biased against Th-1 immune responses. Clin Exp Immunol 128(1):118–123
Rangel-Moreno J, Carragher DM, de la Luz G-HM, Hwang JY, Kusser K, Hartson L, Kolls JK, Khader SA, Randall TD (2011) The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nat Immunol 12(7):639–646. doi:10.1038/ni.2053
Mora JR, Bono MR, Manjunath N, Weninger W, Cavanagh LL, Rosemblatt M, Von Andrian UH (2003) Selective imprinting of gut-homing T cells by Peyer’s patch dendritic cells. Nature 424(6944):88–93
Crespo M, Martinez DG, Cerissi A, Rivera-Reyes B, Bernstein HB, Lederman MM, Sieg SF, Luciano AA (2012) Neonatal T-cell maturation and homing receptor responses to toll-like receptor ligands differ from those of adult naive T cells: relationship to prematurity. Pediatr Res 71(2):136–143. doi:10.1038/pr.2011.26
You D, Ripple M, Balakrishna S, Troxclair D, Sandquist D, Ding L, Ahlert TA, Cormier SA (2008) Inchoate CD8+ T cell responses in neonatal mice permit influenza-induced persistent pulmonary dysfunction. J Immunol 181(5):3486–3494
Lines JL, Hoskins S, Hollifield M, Cauley LS, Garvy BA (2010) The migration of T cells in response to influenza virus is altered in neonatal mice. J Immunol 185(5):2980–2988. doi:10.4049/jimmunol.0903075
Connors TJ, Ravindranath TM, Bickham KL, Gordon CL, Zhang F, Levin B, Baird JS, Farber DL (2016) Airway CD8(+) T cells are associated with lung injury during infant viral respiratory tract infection. Am J Respir Cell Mol Biol 54(6):822–830. doi:10.1165/rcmb.2015-0297OC
Culley FJ, Pollott J, Openshaw PJ (2002) Age at first viral infection determines the pattern of T cell-mediated disease during reinfection in adulthood. J Exp Med 196(10):1381–1386
Dakhama A, Park JW, Taube C, Joetham A, Balhorn A, Miyahara N, Takeda K, Gelfand EW (2005) The enhancement or prevention of airway hyperresponsiveness during reinfection with respiratory syncytial virus is critically dependent on the age at first infection and IL-13 production. J Immunol 175(3):1876–1883
Welliver TP, Garofalo RP, Hosakote Y, Hintz KH, Avendano L, Sanchez K, Velozo L, Jafri H, Chavez-Bueno S, Ogra PL, McKinney L, Reed JL, Welliver RC Sr (2007) Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J Infect Dis 195(8):1126–1136. doi:10.1086/512615
Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS (2007) The histopathology of fatal untreated human respiratory syncytial virus infection. Mod Pathol 20(1):108–119. doi:10.1038/modpathol.3800725
Sullivan BM, Juedes A, Szabo SJ, von Herrath M, Glimcher LH (2003) Antigen-driven effector CD8 T cell function regulated by T-bet. Proc Natl Acad Sci U S A 100(26):15818–15823. doi:10.1073/pnas.2636938100
White GP, Watt PM, Holt BJ, Holt PG (2002) Differential patterns of methylation of the IFN-gamma promoter at CpG and non-CpG sites underlie differences in IFN-gamma gene expression between human neonatal and adult CD45RO-T cells. J Immunol 168(6):2820–2827
Rose S, Lichtenheld M, Foote MR, Adkins B (2007) Murine neonatal CD4+ cells are poised for rapid Th2 effector-like function. J Immunol 178(5):2667–2678
Lewis DB, Yu CC, Meyer J, English BK, Kahn SJ, Wilson CB (1991) Cellular and molecular mechanisms for reduced interleukin 4 and interferon-gamma production by neonatal T cells. J Clin Invest 87(1):194–202. doi:10.1172/JCI114970
Wu CY, Demeure C, Kiniwa M, Gately M, Delespesse G (1993) IL-12 induces the production of IFN-gamma by neonatal human CD4 T cells. J Immunol 151(4):1938–1949
Li L, Lee HH, Bell JJ, Gregg RK, Ellis JS, Gessner A, Zaghouani H (2004) IL-4 utilizes an alternative receptor to drive apoptosis of Th1 cells and skews neonatal immunity toward Th2. Immunity 20(4):429–440
Forsthuber T, Yip HC, Lehmann PV (1996) Induction of TH1 and TH2 immunity in neonatal mice. Science 271(5256):1728–1730
Pala P, Bjarnason R, Sigurbergsson F, Metcalfe C, Sigurs N, Openshaw PJ (2002) Enhanced IL-4 responses in children with a history of respiratory syncytial virus bronchiolitis in infancy. Eur Respir J 20(2):376–382
Mbawuike IN, Wells J, Byrd R, Cron SG, Glezen WP, Piedra PA (2001) HLA-restricted CD8+ cytotoxic T lymphocyte, interferon-gamma, and interleukin-4 responses to respiratory syncytial virus infection in infants and children. J Infect Dis 183(5):687–696. doi:10.1086/318815
Cannon MJ, Openshaw PJ, Askonas BA (1988) Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med 168(3):1163–1168
Sarzotti M, Robbins DS, Hoffman PM (1996) Induction of protective CTL responses in newborn mice by a murine retrovirus. Science 271(5256):1726–1728
Fukui T, Katamura K, Abe N, Kiyomasu T, Iio J, Ueno H, Mayumi M, Furusho K (1997) IL-7 induces proliferation, variable cytokine-producing ability and IL-2 responsiveness in naive CD4+ T-cells from human cord blood. Immunol Lett 59(1):21–28
Hassan J, Reen DJ (1998) IL-7 promotes the survival and maturation but not differentiation of human post-thymic CD4+ T cells. Eur J Immunol 28(10):3057–3065. doi:10.1002/(SICI)1521-4141(199810)28:10<3057:: AID-IMMU3057>3.0.CO;2-Z
Gibbons DL, Haque SF, Silberzahn T, Hamilton K, Langford C, Ellis P, Carr R, Hayday AC (2009) Neonates harbour highly active gammadelta T cells with selective impairments in preterm infants. Eur J Immunol 39(7):1794–1806. doi:10.1002/eji.200939222
Elahi S, Ertelt JM, Kinder JM, Jiang TT, Zhang X, Xin L, Chaturvedi V, Strong BS, Qualls JE, Steinbrecher KA, Kalfa TA, Shaaban AF, Way SS (2013) Immunosuppressive CD71+ erythroid cells compromise neonatal host defence against infection. Nature 504(7478):158–162. doi:10.1038/nature12675
Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, Galkina SA, Weinberg K, Stoddart CA, McCune JM (2010) Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science 330(6011):1695–1699. doi:10.1126/science.1196509
Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, Busch MP, Lee TH, Nixon DF, McCune JM (2008) Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 322(5907):1562–1565. doi:10.1126/science.1164511
Bronevetsky Y, Burt TD, McCune JM (2016) Lin28b regulates fetal regulatory T cell differentiation through modulation of TGF-beta signaling. J Immunol 197(11):4344–4350. doi:10.4049/jimmunol.1601070
Rathmell JC, Farkash EA, Gao W, Thompson CB (2001) IL-7 enhances the survival and maintains the size of naive T cells. J Immunol 167(12):6869–6876
Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD (2001) IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A 98(15):8732–8737
Li J, Huston G, Swain SL (2003) IL-7 promotes the transition of CD4 effectors to persistent memory cells. J Exp Med 198(12):1807–1815
Schluns KS, Kieper WC, Jameson SC, Lefrancois L (2000) Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol 1:426–432
Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L (2002) Cutting edge: requirement for IL-15 in the generation of primary and memory antigen-specific CD8 T cells. J Immunol 168(10):4827–4831
Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD (2002) Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8(+) cells but are not required for memory phenotype CD4(+) cells. J Exp Med 195(12):1523–1532
Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, Mathis D, Butz EA (2002) Cytokine requirements for acute and basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med 195(12):1515–1522
Schonland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, Goronzy JJ, Weyand CM (2003) Homeostatic control of T-cell generation in neonates. Blood 102(4):1428–1434. doi:10.1182/blood-2002-11-3591
Schuler T, Hammerling GJ, Arnold B (2004) Cutting edge: IL-7-dependent homeostatic proliferation of CD8+ T cells in neonatal mice allows the generation of long-lived natural memory T cells. J Immunol 172(1):15–19
Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R (2003) Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol 4(12):1191–1198
Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM (2007) Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27(2):281–295
Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM (2011) Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35(4):633–646. doi:10.1016/j.immuni.2011.08.016
Hua L, Yao S, Pham D, Jiang L, Wright J, Sawant D, Dent AL, Braciale TJ, Kaplan MH, Sun J (2013) Cytokine-dependent induction of CD4+ T cells with cytotoxic potential during influenza virus infection. J Virol 87(21):11884–11893. doi:10.1128/JVI.01461-13
Laidlaw BJ, Zhang N, Marshall HD, Staron MM, Guan T, Hu Y, Cauley LS, Craft J, Kaech SM (2014) CD4(+) T cell help guides formation of CD103(+) lung-resident memory CD8(+) T cells during influenza viral infection. Immunity 41(4):633–645. doi:10.1016/j.immuni.2014.09.007
Smith NL, Wissink E, Wang J, Pinello JF, Davenport MP, Grimson A, Rudd BD (2014) Rapid proliferation and differentiation impairs the development of memory CD8+ T cells in early life. J Immunol 193(1):177–184. doi:10.4049/jimmunol.1400553
Zens KD, Chen JK, Guyer RS, Wu FL, Cvetkovski F, Miron M, Farber DL (2017) Reduced generation of lung tissue-resident memory T cells during infancy. J Exp Med (In Press)
Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions [see comments]. Nature 401(6754):708–712
Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, Matloubian M (2006) CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440(7083):540–544. doi:10.1038/nature04606
Mueller SN, Gebhardt T, Carbone FR, Heath WR (2013) Memory T cell subsets, migration patterns, and tissue residence. Annu Rev Immunol 31:137–161. doi:10.1146/annurev-immunol-032712-095954
Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D (2014) T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346(6205):98–101. doi:10.1126/science.1254536
Ariotti S, Hogenbirk MA, Dijkgraaf FE, Visser LL, Hoekstra ME, Song JY, Jacobs H, Haanen JB, Schumacher TN (2014) T cell memory. Skin-resident memory CD8(+) T cells trigger a state of tissue-wide pathogen alert. Science 346(6205):101–105. doi:10.1126/science.1254803
Glennie ND, Yeramilli VA, Beiting DP, Volk SW, Weaver CT, Scott P (2015) Skin-resident memory CD4+ T cells enhance protection against Leishmania major infection. J Exp Med 212(9):1405–1414. doi:10.1084/jem.20142101
Lieberman JM, Greenberg DP, Wong VK, Partridge S, Chang SJ, Chiu CY, Ward JI (1995) Effect of neonatal immunization with diphtheria and tetanus toxoids on antibody responses to Haemophilus influenzae type b conjugate vaccines. J Pediatr 126(2):198–205
Vekemans J, Ota MO, Wang EC, Kidd M, Borysiewicz LK, Whittle H, McAdam KP, Morgan G, Marchant A (2002) T cell responses to vaccines in infants: defective IFNgamma production after oral polio vaccination. Clin Exp Immunol 127(3):495–498
Plotkin SA, Orenstein WA, Offit PA (2013) Vaccines. Sixth edition. edn, Elsevier Saunders, Philadelphia, Pa
Onorato IM, Modlin JF, McBean AM, Thoms ML, Losonsky GA, Bernier RH (1991) Mucosal immunity induced by enhance-potency inactivated and oral polio vaccines. J Infect Dis 163(1):1–6
Modlin JF, Halsey NA, Thoms ML, Meschievitz CK, Patriarca PA (1997) Humoral and mucosal immunity in infants induced by three sequential inactivated poliovirus vaccine-live attenuated oral poliovirus vaccine immunization schedules. Baltimore Area Polio Vaccine Study Group J Infect Dis 175(Suppl 1):S228–S234
Hammerschmidt SI, Friedrichsen M, Boelter J, Lyszkiewicz M, Kremmer E, Pabst O, Forster R (2011) Retinoic acid induces homing of protective T and B cells to the gut after subcutaneous immunization in mice. J Clin Invest 121(8):3051–3061. doi:10.1172/JCI44262
Zens KD, Chen J-K, Farber DL (2016) Vaccine generated lung tissue resident memory T cells provide heterosubtypic protection to influenza infection. J Clin Invest Insight 1(10).
Perdomo C, Zedler U, Kuhl AA, Lozza L, Saikali P, Sander LE, Vogelzang A, Kaufmann SH, Kupz A (2016) Mucosal BCG vaccination induces protective lung-resident memory T cell populations against tuberculosis. MBio 7(6). doi:10.1128/mBio.01686-16
Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, De Groote D, Corrah T, Bennett S, Wheeler J, Huygen K, Aaby P, McAdam KP, Newport MJ (1999) Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J Immunol 163(4):2249–2255
Sasaki S, Jaimes MC, Holmes TH, Dekker CL, Mahmood K, Kemble GW, Arvin AM, Greenberg HB (2007) Comparison of the influenza virus-specific effector and memory B-cell responses to immunization of children and adults with live attenuated or inactivated influenza virus vaccines. J Virol 81(1):215–228
Debock I, Jaworski K, Chadlaoui H, Delbauve S, Passon N, Twyffels L, Leo O, Flamand V (2013) Neonatal follicular Th cell responses are impaired and modulated by IL-4. J Immunol 191(3):1231–1239. doi:10.4049/jimmunol.1203288
Mastelic Gavillet B, Eberhardt CS, Auderset F, Castellino F, Seubert A, Tregoning JS, Lambert PH, de Gregorio E, Del Giudice G, Siegrist CA (2015) MF59 mediates its B cell adjuvanticity by promoting T follicular helper cells and thus germinal center responses in adult and early life. J Immunol 194(10):4836–45. doi:10.4049/jimmunol.1402071
He XS, Holmes TH, Mahmood K, Kemble GW, Dekker CL, Arvin AM, Greenberg HB (2008) Phenotypic changes in influenza-specific CD8+ T cells after immunization of children and adults with influenza vaccines. J Infect Dis 197(6):803–811. doi:10.1086/528804
Belshe RB, Edwards KM, Vesikari T, Black SV, Walker RE, Hultquist M, Kemble G, Connor EM, Group C-TCES (2007) Live attenuated versus inactivated influenza vaccine in infants and young children. N Engl J Med 356(7):685–696. doi:10.1056/NEJMoa065368
Bernstein HHK, D. W. (2016) Intranasal FluMISSED its target. AAP News & Journals Gateway. http://www.Aappublications.Org/news/2016/07/12/LAIV071216. 2016
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is a contribution to the special issue on Immunocompetence of the Newborn – Guest Editors: Arnaud Marchant and Tobias Kollmann
Rights and permissions
About this article

Cite this article
Zens, K.D., Connors, T. & Farber, D.L. Tissue compartmentalization of T cell responses during early life. Semin Immunopathol 39, 593–604 (2017). https://doi.org/10.1007/s00281-017-0648-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-017-0648-7