
Abstract
Transition metals are essential nutrients to virtually all forms of life, including bacterial pathogens. In Staphylococcus aureus, metal ions participate in diverse biochemical processes such as metabolism, DNA synthesis, regulation of virulence factors, and defense against oxidative stress. As an innate immune response to bacterial infection, vertebrate hosts sequester transition metals in a process that has been termed “nutritional immunity.” To successfully infect vertebrates, S. aureus must overcome host sequestration of these critical nutrients. The objective of this review is to outline the current knowledge of staphylococcal metal ion acquisition systems, as well as to define the host mechanisms of nutritional immunity during staphylococcal infection.
Similar content being viewed by others
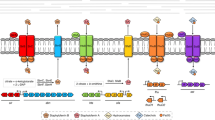
Introduction
Staphylococcus aureus is a remarkably successful human pathogen. It is the most common cause of skin and soft tissue infections [1, 2], endocarditis [3], and osteomyelitis [4] and a common cause of bloodstream infections, surgical site infections, and pneumonia. Invasive staphylococcal infections are a source of considerable morbidity and mortality [5]. Treatment of staphylococcal disease is complicated by the prevalence of antimicrobial-resistant strains. Penicillin-resistant S. aureus emerged in the 1940s [6], followed by the initial description of methicillin-resistant S. aureus (MRSA) in 1961 [7]. The development of subsequent anti-staphylococcal antibiotics has almost uniformly been followed by reports of resistance or treatment failure [8]. Additionally, attempts to design and implement effective staphylococcal vaccines have been unsuccessful thus far [9]. Also complicating the control of staphylococcal infection is the emergence of virulent, community-acquired MRSA (CA-MRSA) strains [10]. Because CA-MRSA infection can occur in those individuals with no known predisposing factors for acquisition of S. aureus, it is difficult to predict which populations are at the greatest risk.
The capability of S. aureus to cause disease is facilitated by production of a diverse array of virulence factors [11, 12]. Staphylococcal adhesins allow effective binding and colonization of host tissues. After colonization of host tissues, expression of secreted toxins and exoenzymes leads to degradation of host tissues, further tissue invasion, and metastasis to other sites. A subset of staphylococcal virulence factors counteracts the host immune response. This includes factors that destroy neutrophils or inhibit their activity [13–18], prevent activation of the complement cascade [19–22], disrupt phagocytosis [23–25], limit the efficacy of antimicrobial peptides [26–28], and interfere with or augment the function of T cells [29, 30]. The spectrum of human disease following staphylococcal infection is therefore a function of the success of the host immune system in controlling staphylococcal virulence. If the host immune system is effective in controlling staphylococcal infection, the clinical outcome is that of asymptomatic colonization or localized disease, such as a soft tissue abscess. If S. aureus succeeds in overcoming the host immune system, the result is invasive and disseminated disease. In this way, human infection by S. aureus represents a constant battle between host and pathogen.
The concept of nutritional immunity exemplifies the struggle between S. aureus and the human immune system [31]. Nutritional immunity describes the process whereby a host sequesters nutrients essential to bacterial growth, thus limiting the ability of invading pathogens to proliferate in the host environment. The most well characterized example of nutritional immunity during staphylococcal infection is iron sequestration, in which iron is maintained predominantly intracellularly or in complex with high-affinity host binding proteins [32]. The result of this sequestration is an extracellular iron concentration insufficient to support bacterial growth [33]. Since iron is necessary for bacterial proliferation, S. aureus must circumvent iron sequestration in order to successfully infect host tissues. Accordingly, staphylococci have evolved systems for the acquisition, processing, and detoxification of iron and iron-containing host molecules and proteins. Emerging evidence suggests that the principle of nutritional immunity extends to other transition metals, including zinc and manganese [34].
The objective of this review is to provide a comprehensive analysis of both the mechanisms by which S. aureus acquires transition metals, as well as the host mechanisms aimed at sequestration of these essential nutrients. We focus on the impact of metal ion acquisition on staphylococcal virulence and the host immune response to staphylococcal infection.
S. aureus overcomes nutritional immunity to obtain iron from the host
Iron is a critical nutrient for both humans and pathogenic bacteria. In humans, iron-containing compounds participate in a number of important cellular processes including energy metabolism, cellular proliferation, DNA repair, and protection against oxidative stress [35–37]. However, iron is also essential to invading bacterial pathogens; therefore vertebrates sequester iron as an innate immune response against bacterial infection. Strict regulatory control of iron homeostasis also protects the host from damage associated with either iron overload or iron deficiency. Iron excess, as manifested in hemochromatosis or in individuals requiring chronic transfusions, results in tissue damage and organ dysfunction. Conversely, iron deficiency, encountered in patients with chronic inflammation or inadequate dietary intake, results in ineffective erythropoiesis [35].
A number of factors contribute to the sequestration of iron in the vertebrate host (Fig. 1). The iron content of vertebrates is mostly intracellular, either maintained within the iron storage protein ferritin, or complexed to the tetrapyrrole ring of heme. Heme is the major iron reservoir in vertebrates, accounting for up to 80% of total body iron [38]. The majority of vertebrate iron is therefore inaccessible to predominantly extracellular pathogens such as S. aureus. Furthermore, any iron released into the extracellular environment is tightly bound to the high-affinity iron-scavenging glycoproteins transferrin and lactoferrin. Together, these mechanisms ensure that the amount of free extracellular iron that is available to invading pathogens is negligible. In response to infection, mammalian hosts can further limit iron availability through a number of mechanisms including decreased dietary iron absorption, reduced release of iron from macrophages, and release of apolactoferrin from neutrophil granules at the site of infection [32]. The importance of iron sequestration as an immune response to invading pathogens is illustrated by individuals with iron overload, who have an increased susceptibility to bacterial infection. For example, patients with hemochromatosis are more susceptible to infection by Yersinia enterocolitica, Vibrio vulnificans, and Listeria monocytogenes [39–42].

S. aureus overcomes nutritional immunity to obtain iron from the host. a During infection, staphylococcal hemolysins lyse red blood cells to release hemoglobin, which can be further degraded to heme and free iron. b In response to infection, vertebrate hosts fortify iron sequestration. Free iron is bound by transferrin and lactoferrin, free heme by hemopexin, and hemoglobin by haptoglobin to limit iron availability to invading pathogens. c S. aureus produces two siderophores, staphyloferrin A and staphyloferrin B, which bind iron with high affinity and allow competition with iron sequestration by lactoferrin and transferrin. Import of iron bound to staphyloferrin A and staphyloferrin B is mediated by HtsABC and SirABC, respectively, in processes powered by the FhuC ATPase. d The Isd system mediates acquisition of heme, the preferred iron source during S. aureus infection. IsdB and IsdH are cell-surface receptors for hemoglobin and hemoglobin-haptoglobin, respectively. IsdB and IsdH pass heme to IsdA, or alternatively to IsdC or IsdE, culminating in transport across the cell membrane by IsdDEF. In the cytoplasm, heme is degraded by IsdG or IsdI to release iron
In order to successfully infect humans, bacteria must overcome iron limitation in host tissues. Pathogenic bacteria have evolved five primary mechanisms to circumvent host iron sequestration. The first mechanism, utilized by Borrelia burgdorferi and Lactobacillus plantarum, is to substitute manganese for iron in metal-containing enzymes, eliminating the cell’s requirement for iron [43, 44]. A second mechanism is the production of secreted iron-binding compounds known as siderophores. Siderophores bind iron with high affinity and thus can effectively compete with extracellular iron sequestration by transferrin and lactoferrin. Third, bacteria utilize heme acquisition systems to obtain host iron. Bacterial heme acquisition systems involve cell surface receptors that recognize either free heme or heme bound to hemoproteins, a transport apparatus to move heme across the cell membrane into the bacterial cytoplasm, and enzymes that liberate iron from heme. A fourth mechanism used by bacteria to obtain iron is the expression of ferric or ferrous iron transporters. Finally, bacteria may express transferrin and lactoferrin receptors to obtain iron directly from high-affinity host proteins. To successfully infect humans, S. aureus utilizes siderophores and a heme acquisition system to overcome host iron sequestration (Fig. 1).
S. aureus produces siderophores to steal iron from transferrin
S. aureus produces two polycarboxylate-type siderophores, staphyloferrin A and staphyloferrin B, which were initially isolated and characterized from Staphylococcus hyicus [45]. An additional staphylococcal siderophore, aureochelin, has been proposed but not further characterized [46]. Iron acquisition by staphyloferrin A and staphyloferrin B is regulated by the ferric uptake regulator (Fur) in response to environmental iron concentrations [47–49]. Fur is an iron-dependent transcriptional regulator that is conserved among Gram-positive and Gram-negative bacteria [50]. In iron-replete conditions, Fur binds a consensus DNA sequence (Fur box) upstream of Fur-regulated genes, leading to transcriptional repression. In iron-deplete conditions, Fur no longer binds to Fur boxes, and transcription of Fur-regulated genes can proceed. In this way, S. aureus can initiate a transcriptional program based on the amount of iron available in the environment [51].
Staphyloferrin A is a 479 Da molecule encoded by the sfaABCD gene cluster [47, 52]. Following iron complexation by staphyloferrin A, transport across the staphylococcal membrane is mediated by the ABC transporter HtsABC, in a process powered by a separate ATPase, FhuC (Fig. 1) [47, 53]. The name of the htsABC operon is derived from its initial characterization as a heme transport system [54]. Subsequent crystallographic studies of HtsA demonstrate that this protein clearly binds staphyloferrin A [55]. Staphyloferrin B is a 448 Da compound composed of l-2,3-diaminopropionic acid, 1,2-diaminoethane, and α-ketoglutaric acid [56]. Staphyloferrin B is a product of the sbnABCDEFGHI operon, of which only SbnC, SbnE, SbnF, and SbnH are required for in vitro synthesis [57]. Transport of staphyloferrin B across the staphylococcal cell membrane is mediated by the staphylococcal iron-regulated transporter SirABC (Fig. 1) [58, 59]. The advantage of producing two siderophores is unclear, although there is precedence for this phenomenon in other bacteria including Pseudomonas aeruginosa (pyoverdines and pyochelin) and Bacillus anthracis (bacillibactin and petrobactin). Concomitant inactivation of the genes responsible for the production of staphyloferrin A and staphyloferrin B results in severe growth limitation in iron-deplete media [47]. Inactivation of the sbn locus alone, but not the sfa locus, also yields a growth defect in iron-restricted media [47]. Thus, under in vitro conditions, staphyloferrin B seems to have a more prominent role in staphylococcal growth during iron limitation. Yet both staphyloferrin A and staphyloferrin B can remove iron from human holotransferrin to support S. aureus growth in iron-deplete conditions [60]. Whether or not staphyloferrin A and staphyloferrin B have distinct in vivo roles remains to be tested.
In addition to siderophore-mediated iron acquisition by staphyloferrin A and B, S. aureus has the capability to liberate iron from heterologous siderophores and mammalian catecholamine hormones. Brock and Ng demonstrated that S. aureus is capable of utilizing the hydroxymate-type siderophore desferrioxamine as an iron source [61]. Uptake of desferrioxamine is dependent on the fhuCBG operon, which encodes the membrane-spanning and ATPase components of a classical traffic ATPase [62]. Subsequent experiments identified FhuD1 and FhuD2 as the lipoprotein receptors for FhuCBG-mediated siderophore uptake [63]. The ability of S. aureus to utilize desferrioxamine as an iron source is clinically relevant given its use as a chelating agent, and previous reports that this compound enhances the growth and virulence of other bacterial and fungal pathogens [64–66]. S. aureus also has the capability to utilize mammalian catecholamine hormones as an iron source. Beasley et al. demonstrated that catecholamine hormones enhance the growth of siderophore-deficient S. aureus grown in the presence of human serum or transferrin [60]. Uptake of catecholamine iron is dependent on SstABCD, a previously described iron-regulated ABC transporter [67].
Iron acquisition by siderophores contributes to staphylococcal virulence
Siderophore-mediated iron acquisition is essential to the virulence of a number of bacterial pathogens. Inactivation of siderophore production reduces virulence in B. anthracis [68], P. aeruginosa [69, 70], Y. enterocolitica [71], Legionella pneumophila [72], and Escherichia coli [73], among others. A limited number of experiments have investigated the role of siderophore-mediated iron acquisition in S. aureus. Beasley et al. examined the virulence of S. aureus possessing mutations in either sirA, hts, sbn, sfa, sst, or combinations thereof [60]. Mutation of the loci encoding staphyloferrin A or staphyloferrin B alone does not lead to a statistically significant reduction in colony recovery from the organs of intravenously infected mice. This result is in contrast to previously published data in which mutation of sbnE significantly decreased colony recovery in a murine kidney abscess model [74]. Inactivation of either the catecholamine transporter (Sst) alone, both siderophore operons (sfa and sbn), or both siderophore transporters (Hts and Sir) yields a reduction in colony recovery from the hearts of systemically infected mice. Additionally, combined inactivation of sfa, sbn, and sst results in reduced colony recovery from the heart and liver, whereas combined inactivation of sst, sirA, and hts decreases colony recovery in heart, liver, and kidney. Mutation of the gene encoding FhuC, the ABC-type ATPase required for both staphyloferrin A and B uptake, results in decreased murine morbidity, but not colony recovery, in a kidney abscess model [53]. Collectively, these results suggest that siderophore-mediated iron acquisition is important for S. aureus virulence.
As bacterial pathogens have evolved to use siderophores to obtain host iron, so have vertebrate hosts evolved to counteract these processes. Siderocalin, also known as lipocalin-2 or neutrophil gelatinase-associated lipocalin, is a mammalian protein capable of binding bacterial siderophores. Siderocalin binds enterobactin, a catecholate siderophore produced by E. coli, resulting in bacteriostasis in iron-limited conditions [75, 76]. In keeping with this, siderocalin-deficient mice are more susceptible to E. coli infection [76, 77]. Subsequent studies revealed that siderocalin is also important for prevention of infection by Klebsiella pneumoniae and Mycobacterium tuberculosis [78–80]. Siderocalin production is induced by a number of stimuli, including TLR4 binding [76], bacterial colonization of the nasal mucosa [81], and ischemia [82]. Thus, production of siderocalin is a host strategy to sequester iron that has been earmarked for bacterial utilization. Not to be outdone, some bacterial pathogens have evolved mechanisms to circumvent siderocalin. B. anthracis produces two siderophores, bacillibactin and petrobactin [83]. Bacillibactin is readily bound by siderocalin, but petrobactin has a unique chelating subunit that precludes siderocalin binding [84]. Salmonella Typhimurium utilizes a similar strategy to evade siderocalin by producing a glycosylated derivative of enterobactin known as salmochelin [85]. These “stealth siderophores” are yet another example of bacterial subversion of innate immunity. Stealth siderophores have not been described in S. aureus.
S. aureus preferentially acquires iron from heme and hemoproteins
Although siderophore-mediated iron acquisition promotes staphylococcal growth in iron-limited conditions in vitro and in vivo, the preferred iron source for S. aureus is heme. When S. aureus is grown in the presence of isotopically labeled iron complexed to either transferrin or heme, a fourfold to fivefold enrichment in the ratio of heme-iron to transferrin-iron is noted in bacteria as compared with the media [54]. Thus, S. aureus preferentially imports heme-iron over transferrin-iron.
In vertebrates, heme is the primary reservoir of iron, representing up to 80% of total body iron. This is in contrast to transferrin-bound iron, which represents less than 1% of total body iron [38]. Heme consists of a tetrapyrrole ring encircling an iron atom, and is produced in the bone marrow and liver. Heme is used as a prosthetic group for many enzymes, and these hemoproteins have diverse roles, including the transfer of electrons and redox reactions. The most abundant hemoprotein in vertebrates is hemoglobin, which consists of four heme moieties bound to polypeptide chains. One human erythrocyte contains approximately 280 million molecules of the hemoglobin tetramer [86]. Other hemoproteins include myoglobin, peroxidases, and cytochromes. Heme biosynthesis and catabolism must be carefully regulated, given the potential toxicity of heme and hemoprotein derivatives [87]. Hemoglobin liberated from erythrocytes is bound by the high-affinity binding protein haptoglobin and subsequently processed by the reticuloendothelial system [88]. Additionally, free heme released into plasma can be sequestered by albumin, hemopexin, and alpha-1-microglobulin [86, 88, 89]. These mechanisms protect the host from heme toxicity, while also limiting the availability of heme and hemoproteins to invading pathogens.
Given that heme is the major reservoir of iron in vertebrate hosts, it is not surprising that both Gram-positive and Gram-negative bacterial pathogens have evolved mechanisms to liberate iron from host heme [90]. Several classes of bacterial heme acquisition systems have been described [90, 91]. In S. aureus, heme acquisition is mediated by the iron-regulated surface determinant (Isd) system. The isd locus was initially identified during an examination of the S. aureus genome for homologues of the transpeptidase Sortase A, which functions to anchor proteins to the cell wall [92–94]. The only Sortase A homologue identified in S. aureus, Sortase B (srtB), is contained within an iron-regulated operon that also contains genes whose products were homologous to heme-binding proteins. Upstream of this operon are two additional genes divergently transcribed from independent promoters. Consensus Fur boxes are located upstream of each operon. Since the initial description of the isd locus, much has been learned regarding heme uptake by the Isd system. The Isd system is encoded by ten genes in five operons: isdA, isdB, isdCDEFsrtBisdG, isdH, and orfXisdI [95]. IsdB and IsdH are cell surface receptors for hemoglobin and hemoglobin-haptoglobin, respectively [96, 97]. Heme is passed from IsdB or IsdH to IsdA, also located at the cell surface [98, 99]. IsdA transfers heme to IsdC, which mediates transfer across the cell wall [99, 100]. Alternatively, IsdH and IsdB may pass heme directly to IsdC or IsdE [98]. IsdDEF is an ABC transporter in which IsdE is a heme-binding lipoprotein, IsdD is an ATPase, and IsdF is a permease. Heme is passed from IsdC to IsdE and subsequently transported into the cytoplasm (Fig. 1) [98]. Interestingly, mutation of isdDEF decreases, but does not abolish, the ability of S. aureus to grow on heme, suggesting that there are IsdDEF-independent mechanisms of heme import [101]. Mutations in the htsABC locus also reduce heme acquisition by S. aureus, but the crystal structure of HtsA strongly suggests staphyloferrin A is the ligand [54]. It is therefore possible that HtsB and HtsC function with an as yet-unidentified lipoprotein receptor to transport heme across the cell membrane.
Once heme enters the staphylococcal cytoplasm, the tetrapyrrole ring is degraded to liberate free iron by two heme-oxygenases, IsdG and IsdI (Fig. 1) [102, 103]. Unlike human heme-oxygenases which degrade heme to iron and the chromophore biliverdin, IsdG and IsdI degrade heme to iron and a novel chromophore, staphylobilin [104]. Deletion of either isdG or isdI results in a decreased ability to utilize heme, indicating that these enzymes are not functionally redundant [105]. Overexpression of either IsdG or IsdI can complement an isdGI mutant, suggesting that the relative amount of each heme-oxygenase may affect heme utilization [105]. Both isdG and isdI are transcriptionally regulated by iron, such that maximal expression occurs in iron-limited conditions. IsdG is also post-transcriptionally regulated by heme, as protein stability is enhanced in the presence of heme [105]. These findings suggested a model whereby S. aureus can fine-tune its capacity for heme utilization based on the environmental conditions encountered.
Like eukaryotes, bacteria must carefully regulate intracellular heme concentrations to ensure that nutrient iron needs are met while preventing toxicity. In S. aureus, this is accomplished by mechanisms to both sense and alleviate heme toxicity. Staphylococci exposed to subinhibitory concentrations of heme can subsequently resist heme toxicity at lethal concentrations, suggesting an adaptive response to exogenous heme [106]. Proteomic analyses revealed that 21 S. aureus proteins are regulated by heme independently of iron [48]. Included among these proteins is the ATP-binding component of a putative ABC transporter that increases 45-fold in the presence of heme. The gene encoding this protein is adjacent to a predicted permease component of the transport system. Inactivation of these genes, designated heme-regulated transporter genes hrtA and hrtB, severely impairs the ability of S. aureus to grow on high concentrations of heme. These and subsequent experiments suggest that HrtAB is a transport system dedicated to the excretion of excess heme or a toxic heme metabolite [106, 107]. The strong heme-dependent regulation of hrtAB suggests that the expression of these genes is under the control of a dedicated transcriptional regulator. Consistent with this, two genes predicted to encode a two-component system are located immediately adjacent to the hrtAB locus on the staphylococcal chromosome. Bacterial two-component systems consist of a membrane-bound kinase that can transduce environmental stimuli to the second component, a response regulator which affects transcriptional changes. The two-component system upstream of hrtAB, subsequently named the heme sensor system (HssR and HssS), is required for the adaptive response to heme toxicity and activates transcription of hrtAB [106]. Thus, S. aureus senses heme through HssRS, increasing transcription of hrtAB, which leads to alleviation of heme toxicity. However, the exact ligand of HssS and the identity of the substrate exported by HrtAB have yet to be identified.
Heme acquisition is essential to the virulence of S. aureus
The ability of staphylococci to acquire heme and hemoproteins in vivo is facilitated by lysis of host erythrocytes by pore-forming toxins such as alpha-hemolysin (encoded by hla) (Fig. 1). Inactivation of alpha-hemolysin limits the virulence of S. aureus [108–114]. Moreover, passive immunization with anti-Hla antibodies or active immunization with nontoxigenic Hla is protective in animal models of staphylococcal infection [108, 115]. After liberation of hemoproteins from host cells, the ability of S. aureus to utilize heme via the Isd system is critical for virulence. Several lines of evidence indicate that disruption of the Isd heme acquisition system at each step (surface receptor binding, transport across the cell wall, transport across the cell membrane, or release of iron from heme) leads to decreased virulence in animal models of staphylococcal infection. Mutation of isdA, isdB, and isdC, but not isdH, decreases bacterial recovery and organ abscesses in murine models of systemic infection [96, 116, 117]. Additional support for the role of IsdB in staphylococcal pathogenesis comes from experiments using mice expressing human hemoglobin. Compared to wild-type mice, mice expressing human hemoglobin are more susceptible to S. aureus infection, as evidenced by increased colony recovery from infected organs. IsdB binds human hemoglobin more readily than murine hemoglobin in vitro, and deletion of isdB abolishes the enhanced virulence of staphylococci infecting mice with human hemoglobin [118]. Collectively, these results suggest an important role for surface proteins of the Isd system in staphylococcal virulence, and have led to additional experiments investigating the vaccine potential of these proteins. IsdA and IsdB were identified as vaccine candidates using “reverse vaccinology” [119], and subsequent studies confirmed that immunization with these proteins is protective in murine models of infection [117, 120–123]. Additional human studies are needed to investigate the vaccine potential of Isd proteins. A phase one human clinical trial of the IsdB vaccine V710 is complete [124], and further trials are ongoing.
In addition to its role as a heme-binding protein, there is evidence that IsdA also contributes to staphylococcal pathogenesis through roles in both adhesion to host tissues and resistance to innate immune responses. IsdA binds both fibrinogen and fibronectin in vitro, indicating a potential role as an iron-regulated adhesin [125]. Consistent with this hypothesis, IsdA facilitates binding to human desquamated epithelial cells, and promotes nasal colonization in rats [122, 126, 127]. IsdA also plays a role in resistance to the host immune response. By decreasing staphylococcal cell hydrophobicity, IsdA increases the resistance of S. aureus to antimicrobial compounds produced by human sebum, as well as other antimicrobial peptides [128]. This is reflected in decreased survival of S. aureus isdA mutants on live human skin. Mutation of isdA also leads to increased susceptibility to hydrogen peroxide and to killing by human neutrophils [129]. Finally, IsdA protects against the bactericidal activity of lactoferrin [130]. Thus, the contribution of IsdA to staphylococcal virulence may extend beyond its role in heme acquisition.
Heme transport across the cell membrane is also required for staphylococcal virulence. Inactivation of either htsB or htsC significantly reduces heme acquisition and decreases bacterial recovery and abscess formation in mice infected systemically with S. aureus [54]. Similarly, concurrent inactivation of htsA and isdE leads to a statistically significant decrease in bacterial burden in the lung, heart and kidneys of systemically infected mice [131]. Upon entering the cytoplasm, heme is degraded by IsdG or IsdI to liberate iron, a process that also contributes to staphylococcal virulence. Mutation of either isdI or isdG results in decreased colony recovery from the hearts of systemically infected mice. In contrast, mutation of isdG, but not isdI, results in decreased recovery from infected kidneys [105]. These findings raise the possibility that IsdG and IsdI have distinct roles depending on the host environments encountered in vivo. The presence of two differentially regulated heme degrading enzymes could allow S. aureus to cope with changing iron and heme concentrations as infection progresses. During the early stages of infection, staphylococci may encounter iron-limiting conditions while not yet importing substantial amounts of heme. Under these conditions, IsdG is destabilized by the lack of intracellular heme, leaving IsdI to degrade the low levels of imported heme. After seeding and degradation of host tissues, increased heme acquisition and the concomitant increase in intracellular heme levels would stabilize IsdG, increasing the overall capacity for heme degradation.
A surprising result was obtained when assessing the virulence of S. aureus strains inactivated for the heme sensing system and heme-regulated transporter. Rather than compromising virulence, S. aureus strains possessing mutations in hrtA or hssR grow to an average of two to three logs higher bacterial density in the livers of infected mice as compared with wild-type staphylococci [106]. This hepatic hypervirulence is the result of inhibition of a liver-specific immune response. Further experiments revealed that inactivation of HrtA leads to membrane damage which triggers increased expression and secretion of immunomodulatory factors [132]. The mechanism by which mutation of hssR leads to immunomodulation remains to be characterized, but disruption of heme sensing and detoxification systems clearly impact staphylococcal pathogenesis.
The role of manganese in staphylococcal infection
The most well characterized model of nutritional immunity during bacterial infection is sequestration of iron. However, sequestration of manganese is also an important facet of the innate immune response to staphylococcal infection [34]. Manganese is an essential nutrient for both eukaryotic and prokaryotic organisms. In humans, manganese participates in diverse cellular functions including detoxification of free radicals, metabolism, bone growth, and support of hemostasis [133]. Like iron, manganese must be carefully regulated to prevent cellular damage. Manganese intoxication occurs predominantly as a result of occupational exposure, particularly in the welding, mining, and smelting industries. Excessive manganese is neurotoxic, leading to clinical manifestations ranging from mild dystonia to a constellation of extra-pyramidal symptoms resembling Parkinson’s disease [134]. The clinical manifestations of manganese deficiency are less clear. In a study of seven men fed manganese-deficient diets, a fleeting dermatitis developed in five subjects [135]. However, naturally occurring manganese deficiency has not been described in humans [133].
Bacteria, like humans, must acquire manganese and regulate its intracellular concentration. Manganese-dependent bacterial enzymes are necessary for myriad processes, including carbohydrate and amino acid metabolism, signal transduction, stringent response, and defense against oxidative stress [136]. Thus, the success of bacterial pathogens in human infection depends on the ability to obtain this critical nutrient from host tissues. Likewise, prevention or resolution of infection is aided by host mechanisms that sequester manganese from invading pathogens.
Bacterial acquisition of manganese is facilitated by high-affinity transporters
In 1995, a manganese transporter was characterized in the cyanobacterium Synechocystis 6803 [137]. The genes necessary for production of this transporter, named mntABC for manganese transporter, encode products with sequence similarity to the ABC superfamily of bacterial permeases. Further experiments confirmed MntABC as an ABC-type transporter consisting of a cytoplasmic ATP-binding protein (MntA), a transmembrane protein (MntB), and solute-binding protein (MntC). Homologues of MntABC have since been found to facilitate manganese uptake in a number of Gram-positive and Gram-negative pathogens. A second family of bacterial manganese transporters was identified during characterization of the Bacillus subtilis protein MntR, a manganese-modulated transcriptional regulator [138]. Que and Helmann discovered transposon-insertion mutants that alleviated the increased manganese susceptibility of an mntR mutant. The most commonly recovered insertions were in a gene (mntH) predicted to encode a proton-coupled metal ion transporter of the natural resistance-associated macrophage protein (Nramp) family. In a separate report, additional MntH transporters were characterized in E. coli, Salmonella Typhimurium, Burkholderia cepacia, and P. aeruginosa [139]. Unlike mammalian Nramp proteins which mediate transport of a number of cations, bacterial Nramp transporters are highly selective for manganese [140].
Compared to the knowledge of bacterial manganese import systems, considerably less is known about how bacteria respond to toxic intracellular levels of manganese. In 1973, Fisher et al. utilized radiolabeled manganese to demonstrate both uptake and efflux of manganese by B. subtilis [141]. Almost four decades later, the first bacterial manganese efflux system was characterized. The Streptococcus pneumoniae gene mntE encodes an efflux system of the cation diffusion facilitator (CDF) family [142]. Nearly all bacterial genomes encode CDF family members, most of which are involved in efflux of zinc or other heavy metal cations [143]. In contrast, the MntE exporter in S. pneumoniae is specific for manganese. Whether other bacterial pathogens express dedicated manganese efflux systems is unknown.
The S. aureus genome encodes MntABC-type (mntABC) and Nramp-type (mntH) manganese transporters, both of which have been functionally characterized. Mutation of mntA in S. aureus results in a reduced growth rate in metal-depleted minimal media, a phenotype that is reversible upon addition of manganese. In contrast, mutation of mntH does not lead to an appreciable growth defect in metal-depleted media, suggesting that MntABC may be the dominant manganese transporter in S. aureus under in vitro conditions [144]. The transcriptional regulator MntR affects expression of both mntABC and mntH. In manganese-replete conditions, transcription of mntABC is repressed by MntR. Deletion of mntR leads to constitutive transcription of mntABC irrespective of manganese levels. In contrast, mntH transcription is reduced in the absence of MntR, suggesting that MntR may be a bifunctional regulator in S. aureus, as previously described for B. subtilis [138]. In addition to regulation by MntR, mntABC is negatively regulated by PerR, a manganese and iron-dependent Fur homologue that controls responses to oxidative stress [144, 145]. It is unknown if the regulation of manganese homeostasis in S. aureus involves an efflux system. The S. aureus genome encodes CDF family proteins, but those that have been characterized are predicted to have roles in zinc efflux [146, 147].
Aerobically growing microorganisms are at risk for toxicity from reactive oxygen species, and staphylococcal manganese acquisition is crucial for defense against oxidative stress. S. aureus produces two superoxide dismutases, SodA and SodM, which detoxify superoxide anion to hydrogen peroxide [148, 149]. Hydrogen peroxide can be further reduced to water and oxygen by the enzyme catalase. The activities of SodA and SodM are dependent on manganese [148, 149]. Manganese limitation therefore renders S. aureus more susceptible to oxidative stress (Fig. 2) [150]. Additionally, S. aureus employs superoxide dismutase-independent mechanisms to detoxify superoxide, a process that is also dependent on manganese [144].

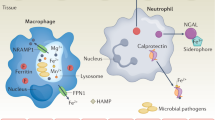
Calprotectin-mediated nutritional immunity of manganese and zinc. a In staphylococcal tissue abscesses, neutrophils release calprotectin, reactive oxygen species, and other antibacterial substances. b S. aureus acquires manganese via the manganese transporters MntABC and MntH. Zinc acquisition occurs via an undetermined mechanism. Manganese acquisition is critical for detoxification of superoxide anions via SodA and SodM, and thus contributes to resistance to killing by neutrophils and increases staphylococcal survival within abscesses. c Calprotectin binds manganese and zinc, limiting the availability of these critical nutrients in abscesses. The concomitant decrease in staphylococcal manganese and zinc acquisition cripples oxidative stress defenses and zinc-dependent processes, leading to decreased bacterial survival in the abscess
Manganese acquisition contributes to the virulence of bacterial pathogens
Disruption of manganese acquisition reduces the virulence of a number of bacterial pathogens. The Nramp-type manganese transporter MntH is a critical virulence determinant for Brucella abortus [151]. Similarly, acquisition of manganese is required for intracellular survival and virulence of Salmonella enterica [152]. Disruption of MntABC in Neisseria gonorrhoeae reduces intracellular survival and biofilm formation [153]. Several other pathogens also require manganese uptake for infection, including Enterococcus faecalis, S. pneumoniae, and Yersinia pestis [154–156]. Inactivation of the manganese efflux protein MntE in S. pneumoniae reduces nasal colonization and bloodstream invasion in mice, despite an increased resistance to reactive oxygen species [142]. This suggests that the ability to regulate intracellular concentrations of manganese may also be important for bacterial pathogenesis.
In S. aureus, mutation of mntA, mntH, or mntR in the laboratory strain 8325-4 does not lead to a significant reduction in bacterial recovery in a murine skin abscess model. In contrast, concurrent inactivation of mntA and mntH significantly decreases bacterial recovery in this model. Disruption of mntA, mntH, mntR, or both mntA and mntH also significantly reduces intracellular survival in human endothelial cells [144]. Additional evidence for the role of manganese acquisition in staphylococcal virulence came from studies of host factors that sequester manganese during infection.
Vertebrate hosts sequester manganese as an innate immune response to bacterial infection
Sequestration of manganese is an important facet of the innate immune response to bacterial infection. Two strategies utilized by vertebrate hosts to limit manganese availability to invading pathogens have been described. First, the availability of manganese to intracellular pathogens is limited by the cation transporter Nramp1. Nramp1 is an integral membrane protein expressed by professional phagocytes. Upon phagocytosis, Nramp1 is recruited to the phagosome, where it remains during the maturation of phagosomes into phagolysosomes [157]. Experiments using a fluorescent probe that is readily incorporated into the phagosome demonstrated that Nramp1 functions as a pH-dependent transporter capable of manganese efflux from the phagolysosome [158]. In this way, the availability of manganese is limiting for intracellular pathogens. Additional studies suggest that Nramp1 may also transport other cations such as iron and zinc [159, 160], and therefore may participate in limitation of multiple metals to intracellular pathogens. Consistent with this, disruption of Nramp1 in mice leads to increased susceptibility to the intracellular pathogens Salmonella Typhimurium, Mycobacterium bovis, and Leishmania donovani [161]. The importance of Nramp1 during human infection is suggested by studies linking certain polymorphisms in Nramp1 to an enhanced susceptibility to both M. tuberculosis and nontuberculous mycobacteria [162–164]. The precise mechanism by which Nramp1 polymorphisms enhance susceptibility to mycobacteria is unclear.
A second mechanism utilized by vertebrates to limit the availability of manganese to invading pathogens was discovered during a search for host factors that limit bacterial growth in abscesses [34]. One of the most abundant host proteins identified in tissue abscesses of mice infected with S. aureus was S100A8, a component of the heterodimeric protein calprotectin (also known as SA1008/SA1009, calgranulin A/B, or myeloid-related proteins 8/14). Calprotectin, which accounts for ~40% of the cytosolic protein pool of neutrophils [165–167], was originally identified based on its ability to inhibit the growth of a variety of fungal and bacterial pathogens in vitro [168, 169]. Calprotectin is also a component of neutrophil extracellular traps, fiber-like structures consisting of chromatin and neutrophil granules that bind to and facilitate killing of microorganisms [170, 171]. A number of immunomodulatory functions have been ascribed to calprotectin, including chemotactic activity for neutrophils and macrophages [166, 167], regulation of inflammation in response to vascular injury [172], amplification of the immune response via TLR4 binding [173, 174], induction of cell death [175, 176], and regulation of myeloid-derived suppressor cells [177, 178]. Previous in vitro studies suggested that the antimicrobial activity of calprotectin was related to its ability to chelate zinc, limiting the availability to pathogens [179–182]. Subsequent in vivo analyses revealed that calprotectin also limits bacterial growth in abscesses via chelation of manganese. Visceral abscesses in mice infected with S. aureus are essentially devoid of manganese and zinc. In contrast, abscesses in mice lacking calprotectin contain manganese levels equivalent to those of surrounding tissue, as well as significantly increased bacterial burdens compared with wild-type mice. The concentration of zinc does not differ significantly between S. aureus abscesses in wild-type and calprotectin-deficient mice. Collectively, these results demonstrated that manganese limitation is an important innate immune response to staphylococci in tissue abscesses. S. aureus strains possessing mutations in mntA or mntB are more sensitive to the antibacterial effects of calprotectin, further supporting the importance of manganese acquisition to staphylococcal virulence [34].
A partial mechanistic explanation of the in vivo antibacterial effects of calprotectin has been described [150]. Treatment of S. aureus with calprotectin results in enhanced susceptibility to oxidative stress through inhibition of SodA and SodM. This renders calprotectin-treated S. aureus more susceptible to killing by neutrophils, which exert antimicrobial activity partly through the generation of reactive oxygen species (Fig. 2). Mutation of staphylococcal sodA and sodM results in decreased bacterial recovery from infected organs in a murine model of systemic infection. However, in calprotectin-deficient mice there is no difference in bacterial recovery between wild-type and sodAsodM-inactivated S. aureus. Therefore, calprotectin-mediated inhibition of bacterial superoxide defenses is an important immune response to staphylococcal abscesses.
At least one bacterial species is capable of binding and inactivating calprotectin. Finegoldia magna is a Gram-positive anaerobic bacterium that is typically a human commensal, but is also capable of causing opportunistic infections [183]. A subset of clinical isolates of F. magna produce Protein L, which has domains that bind both immunoglobulins and calprotectin. F. magna strains that express Protein L are protected from calprotectin-mediated killing [184]. This is yet another example of bacterial circumvention of host immune responses aimed at limitation of nutrients. Whether or not S. aureus is capable of disrupting host-mediated manganese limitation is unknown. S. aureus produces an arsenal of factors that subvert neutrophil function, including pore-forming toxins, phenol soluble modulins, proteases, and molecules that inhibit chemotaxis [185]. It is possible that destruction of neutrophils and their antimicrobial contents allows S. aureus to avoid calprotectin-mediated manganese limitation. However, staphylococcal toxins such as the Panton-Valentine leukocidin induce the formation of neutrophil extracellular traps, which are rich in calprotectin and thus could potentiate local manganese limitation [171, 186]. Further studies are necessary to determine if staphylococcal destruction of neutrophils is an effective subversion mechanism against calprotectin-mediated metal chelation. Nevertheless, the fact that S. aureus can proliferate readily in abscesses suggests the presence of systems dedicated to overcoming calprotectin-mediated manganese limitation.
The contribution of zinc to the host-pathogen interaction
Zinc is an essential nutrient for humans. Zinc deficiency occurs in several settings, including poor nutrition, malabsoprtion, alcohol abuse, liver disease, and genetic disorders. The manifestations of zinc deficiency are numerous, and include a profound alteration of immune cell function. In an experimental human model, mild zinc deficiency resulted in a decreased CD4+/CD8+ T cell ratio, decreased IL-2 activity, and imbalance of TH1 and TH2 responses [187, 188]. Additional studies have demonstrated deleterious effects of zinc deficiency on all components of the human immune system, including neutrophils, natural killer cells, T and B lymphocytes, and monocytes [189]. Both human and animal studies have shown a correlation between zinc deficiency and enhanced susceptibility to infection. Zinc-deficient animals are more susceptible to a variety of viral, bacterial, fungal, and parasitic pathogens [190]. Conversely, human trials of zinc supplementation demonstrate protection against infectious diseases. In children, zinc supplementation reduces the duration and severity of acute diarrhea, and reduces the incidence of lower respiratory tract infection [191–193]. Zinc supplementation in patients with sickle cell disease reduces the incidence of bacterial infections, including S. aureus pneumonia. In elderly adults, zinc supplementation significantly reduces the incidence of infection [194]. Taken together, these findings indicate a critical role for zinc in protection against infectious diseases.
Bacterial mechanisms of zinc homeostasis
Zinc is also an essential nutrient for bacterial pathogens. Zinc-binding proteins, which constitute an average of 5% of bacterial proteomes [195], participate in diverse processes such as regulation of virulence factors, metabolism, and inactivation of antibiotics. Zinc concentrations must be carefully regulated, as excess zinc can inappropriately bind to thiol groups of bacterial proteins and interfere with their function. In bacteria, zinc homeostasis is achieved through coordinated regulation of import and export. Bacteria express both high- and low-affinity zinc importers. ZnuABC is a high-affinity ABC-type zinc transporter best characterized in E. coli, consisting of a periplasmic binding protein (ZnuA), membrane permease (ZnuB), and ATPase (ZnuC) [196]. Zinc import systems homologous to Znu have been identified in several other bacterial pathogens [197]. The ZnuABC system is regulated by Zur, a metallo-regulatory protein of the Fur family, such that zinc import is repressed in zinc-replete conditions [196]. Zur has two regulatory metal binding sites, allowing for graded expression of zinc-responsive genes in accordance with zinc availability [198]. A low-affinity zinc transporter, ZupT, was identified in E. coli based on similarity to eukaryotic zinc transporters of the ZIP family [199]. However, the precise mechanism of zinc import by ZupT is unknown, as is its distribution among other bacterial genomes. Zinc export is essential for maintenance of homeostasis, and is achieved primarily through P-type ATPases which couple ATP hydrolysis to transport of zinc and other metals across the cell membrane. ZntA in E. coli mediates efflux of both zinc and cadmium [200]. Another zinc exporter (CzcCBA) was identified in Ralstonia metallidurans, and also mediates efflux of cobalt and cadmium [201]. Czc-type exporters have subsequently been postulated to mediate efflux of zinc in other bacteria, including P. aeruginosa and S. aureus [146, 202].
Efforts to identify the mechanisms of zinc import, export, and regulation in S. aureus have had mixed success. Both plasmid and chromosomally encoded zinc efflux proteins have been functionally characterized in S. aureus. Resistance to both zinc and cobalt is conferred by the chromosomally encoded czrAB operon, which encodes a metal-regulated transcriptional repressor (CzrA, also known as ZntR) and a CDF antiporter (CzrB, also known as ZntA) [146, 147, 203]. CadA is a P-type ATPase encoded by the S. aureus plasmid pI258 that mediates resistant to cadmium, lead, and zinc [204]. In contrast to these functionally characterized exporters, the proteins mediating zinc uptake in S. aureus have yet to be identified. Two genes upstream of a Zur homologue, named mreA and mreB for metal-responsive elements, were predicted to encode the ATP-binding and membrane permease components of an ABC transporter. It was postulated that mreA and mreB might encode elements of a zinc transporter based on sequence homology. However, there was no appreciable change in susceptibility to zinc limitation relative to wild type when mreA and mreB were inactivated [205]. Additional studies are needed to clarify staphylococcal mechanisms of zinc homeostasis.
Dynamic changes in zinc distribution occur in response to infection and inflammation
Imaging mass spectrometry revealed that staphylococcal murine abscesses are nearly devoid of zinc. Previous reports suggested that the antimicrobial function of calprotectin is mediated through zinc chelation, implying that calprotectin may be a mediator of zinc sequestration in staphylococcal abscesses. However, zinc levels within the staphylococcal abscess are unaffected by genetic defects in calprotectin production, raising the possibility that additional host factors contribute to zinc limitation in abscesses [34]. The acute-phase response to infection is known to alter zinc distribution in vertebrates. Administration of human IL-1 to rats leads to decreased serum zinc concentrations with concomitant redistribution to various tissues such as the liver, bone marrow, and thymus [206]. Similarly, IL-6 administration to mice leads to hypozincemia and increased hepatic zinc concentration through upregulation of the zinc transporter Zip14 in hepatocytes [207, 208]. Limitation of zinc availability may be beneficial to controlling S. aureus infection, as several processes that contribute to staphylococcal virulence are zinc dependent, including biofilm formation and superantigen activity [209, 210]. However, the role of zinc acquisition in staphylococcal pathogenesis has yet to be determined in vivo.
Exploiting transition metals to battle bacterial pathogens: the example of copper
Copper is an important cofactor for a number of bacterial enzymes, but like other transition metals, it is toxic if concentrations are not carefully regulated. The precise mechanism by which copper homeostasis is achieved in S. aureus has not been fully elucidated. Exposure to copper induces expression of copA and copZ, which encode a P-type ATPase (CopA) and copper metalochaperone (CopZ), respectively. In S. aureus, CopA is involved in copper efflux, as evidenced by increased intracellular copper concentrations in a copA mutant [211]. Expression of copA is regulated by the copper-sensitive operon repressor (CsoR) [212, 213]. Copper binding by CsoR results in a conformational change, causing release of operator DNA and de-repression of the copper-sensitive operon. CopZ has not been functionally characterized in S. aureus. In B. subtilis and L. monocytogenes, CopZ interacts and exchanges copper with CopA, and is therefore predicted to protect bacteria from the inappropriate binding of copper to cytoplasmic proteins [214, 215]. Additional studies are needed to characterize copper homeostasis in S. aureus, and to assess the role of copper acquisition in virulence.
Disruption of copper homeostasis reduces the virulence of select bacterial pathogens. Deletion of copA in S. pneumoniae causes reduced virulence in a mouse pneumonia model [216]. Similarly, inactivation of a putative copper exporter (MctB) in M. tuberculosis significantly reduces bacterial burden in the lungs of infected guinea pigs [217]. Finally, mutation of the P. aeruginosa gene cueA, an ATPase involved in protection from copper toxicity, results in a 50-fold decrease in virulence in a lethal dose murine model [218]. Collectively, these results reveal that diverse bacterial pathogens encounter potentially toxic levels of copper in vivo, and invite the question of whether modulation of copper levels in host tissues might be an innate immune response to infection. The importance of copper to the vertebrate immune response is demonstrated by copper-deficient mice, which exhibit an impaired humoral immune response and enhanced susceptibility to a variety of bacterial pathogens [219, 220]. In contrast to iron, manganese, and zinc, the levels of copper increase in response to infection and inflammation [221, 222]. This effect is partially mediated by proinflammatory cytokines, which stimulate hepatic synthesis of ceruloplasmin, the major plasma copper-binding protein [223]. Inflammatory cytokines also mediate a copper-dependent antibacterial response in macrophages. In response to interferon-gamma, macrophages increase the expression of two copper transporters, CTR1 and ATP7A. ATP7A is trafficked to the phagosome where it imports bactericidal levels of copper. Conversely, RNAi-mediated depletion of ATP7A leads to increased bacterial survival in the phagosome [220]. Therefore, in addition to nutritional immunity mediated by metal ion sequestration, vertebrate hosts can increase metal ion concentration in certain environments to kill bacterial pathogens.
The toxicity of transition metals has been utilized in the development of new antimicrobials for multi drug-resistant S. aureus. For example, copper-coated surfaces exhibit rapid killing of MRSA as compared with brass and stainless steel [224]. Similarly, orthopedic implants made of a titanium-copper alloy are antibacterial in a rabbit model of staphylococcal implant-associated infection [225]. Copper-containing biocides have shown promise as hand sanitization gels and environmental disinfectants [226, 227]. These preliminary studies reveal that copper-containing compounds are effectively antibacterial at concentrations that are not cytotoxic to human or animal cells. How exposure to toxic levels of copper kills S. aureus is unknown. In response to copper exposure, S. aureus initiates a transcriptional program characterized by induction of oxidative stress and misfolded protein responses, suggesting that copper toxicity results in increased oxidative stress and protein damage [228]. Like iron, copper can interact with hydrogen peroxide to generate damaging reactive oxygen species. S. aureus strains inactivated for the hydrogen peroxide-reducing enzymes KatA and AhpC are more sensitive to copper toxicity, further supporting oxidative stress as a mechanism for copper-mediated staphylococcal killing.
Imaging mass spectrometry, a powerful tool for the study of metal ion distribution during bacterial infection
The discovery of calprotectin’s role in manganese sequestration during staphylococcal infection was facilitated by imaging mass spectrometry (IMS). IMS uses matrix-assisted laser desorption/ionization time of flight to directly analyze a variety of molecules in tissue sections. IMS has been used to analyze proteins, peptides, lipids, drugs, and metabolites [229]. Whereas sample preparation for conventional proteomic analyses results in disruption of tissue architecture, IMS allows for demonstration of sample distribution in intact tissues. Furthermore, IMS can be combined with additional imaging modalities such as magnetic resonance imaging to provide a three-dimensional co-registration of proteomic data with anatomy [230].
IMS was instrumental in the identification of calprotectin as an innate immune effector responding to staphylococcal abscesses [34]. Examination of mice infected with S. aureus by IMS revealed that calprotectin co-localized to abscessed tissue in a neutrophil-dependent manner. Given previous reports that calprotectin functions via metal chelation, laser-ablation inductively coupled plasma mass spectrometry (LA-ICPMS) was used to demonstrate metal ion distribution and concentrations in abscessed tissue. LA-ICPMS is a highly sensitive form of mass spectroscopy capable of quantifying elemental abundance within two dimensions. As discussed above, LA-ICPMS analysis of infected murine tissues allowed determination of manganese and zinc distribution in staphylococcal abscesses, and confirmed that manganese is limited in abscessed tissue. LA-ICPMS therefore allows for determination of both spatial distribution of metal ions as well as their absolute concentrations in a range of infected tissues. These technologies will help to determine whether calprotectin-mediated metal chelation is an immune response specific to staphylococci in abscesses, or is a more general immune strategy to limit bacterial growth in a diverse range of host tissues.
In addition to the study of the host responses to bacterial infection, IMS offers a number of exciting microbiologic applications. IMS can be used to image single microbial colonies, bacteria growing in biofilms, interactions between two different microbes, and polymicrobial communities [231]. The ability to characterize microbial physiology in vivo, together with the capability to assess host immune responses, provides an opportunity for comprehensive study of bacterial infections. For example, IMS analysis of experimental staphylococcal infection could be utilized to reveal microbial colonization factors facilitating adhesion, host proteins responding to colonization and tissue damage, changes in elemental distribution in response to infection, in vivo proteome analysis of biofilms, distribution of antibiotics in infected tissues, and bacterial proteomic changes in response to antimicrobial therapy. This paradigm could be applied to a number of different host tissues and pathogens. These broad applications make IMS an exciting and powerful tool for study of bacterial infection.
Concluding remarks
The success of S. aureus as a human pathogen is facilitated by its ability to infect and survive within diverse host tissues. The ability to acquire nutrients from the host, including metal ions, is critical for staphylococcal infection. Despite innate immune mechanisms that limit the availability of transition metals, S. aureus can proliferate in vertebrate hosts through the expression of metal acquisition systems that successfully overcome nutritional immunity. Our understanding of the pathways that mediate iron acquisition in S. aureus has increased considerably. During infection, staphylococci sense low levels of iron in host tissues and respond by initiating a transcriptional program that includes expression of iron-acquisition systems. Iron acquisition is critical to S. aureus virulence, and therefore is an attractive target for the development of new antimicrobials and vaccines.
Emerging data suggest that host limitation of manganese is also an essential component of the immune response to staphylococcal infection. Calprotectin-mediated manganese sequestration limits S. aureus growth in abscesses, in part by disrupting manganese-dependent oxidative stress defenses. The expression of high-affinity manganese transporters allows S. aureus to overcome manganese sequestration and survive within the hostile environment of an abscess. Additional studies suggest that infection-induced redistribution of other metal ions such as copper and zinc also contribute to resistance to microbial infection. Unlike iron, manganese, and zinc, host copper concentrations increase in response to infection, suggesting that the human immune system may exploit the toxicity of copper as an innate immune mechanism against bacterial pathogens.
There are several questions yet to be answered regarding staphylococcal metal ion acquisition and the host mechanisms that limit metal availability. Thus far, the contribution of metal acquisition systems to staphylococcal virulence has been assessed primarily in defined organ abscesses. Although skin and soft tissue abscesses are common clinical manifestations of staphylococcal disease, infection of other tissues such as bone, lung, or heart may not be accompanied by formation of defined abscesses. Furthermore, the availability of essential metal nutrients may differ among host tissues, raising the possibility that the contribution of metal acquisition to staphylococcal pathogenesis may vary according to the route and primary focus of infection. For example, concomitant inactivation of both isdB and isdH leads to a significant reduction in bacterial recovery from the lungs, heart, and kidneys of mice infected intravenously with S. aureus. In contrast, when mice were infected intranasally with S. aureus isdBH mutants, there is no significant difference in bacterial recovery from the lungs in comparison to wild type [131]. Additional experiments are therefore needed to establish the importance of staphylococcal metal acquisition, and conversely host-mediated metal sequestration, for infection of diverse host tissues.
The kinetics of host-mediated metal sequestration during staphylococcal infection are unknown. LA-ICPMS studies reveal that mature staphylococcal abscesses are essentially devoid of manganese and zinc. However, the metal concentrations encountered by invading staphylococci prior to abscess formation have not been determined. An understanding of the role of staphylococcal metal acquisition during earlier phases of infection is valuable, as there may be opportunities for therapeutic intervention prior to dissemination and formation of destructive tissue lesions. Further experiments are also necessary to clarify the mechanisms of metal ion homeostasis in S. aureus. In contrast to our knowledge of S. aureus iron acquisition and homeostasis, little is known about how staphylococci procure and regulate the concentration of other transition metals such as copper and zinc. The availability of sensitive imaging mass spectrometry methods will facilitate further study of the struggle for metals between host and bacterium. Determination of additional mechanisms by which staphylococci overcome nutritional immunity may generate new targets for antimicrobial design and vaccine development.
References
Edelsberg J, Taneja C, Zervos M, Haque N, Moore C, Reyes K, Spalding J, Jiang J, Oster G (2009) Trends in US hospital admissions for skin and soft tissue infections. Emerg Infect Dis 15(9):1516–1518
Moran GJ, Krishnadasan A, Gorwitz RJ, Fosheim GE, McDougal LK, Carey RB, Talan DA (2006) Methicillin-resistant S. aureus infections among patients in the emergency department. N Engl J Med 355(7):666–674. doi:10.1056/NEJMoa055356
Baddour LM, Wilson WR, Bayer AS, Fowler VG Jr, Bolger AF, Levison ME, Ferrieri P, Gerber MA, Tani LY, Gewitz MH, Tong DC, Steckelberg JM, Baltimore RS, Shulman ST, Burns JC, Falace DA, Newburger JW, Pallasch TJ, Takahashi M, Taubert KA (2005) Infective endocarditis: diagnosis, antimicrobial therapy, and management of complications: a statement for healthcare professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, and the Councils on Clinical Cardiology, Stroke, and Cardiovascular Surgery and Anesthesia, American Heart Association: endorsed by the Infectious Diseases Society of America. Circulation 111(23):e394–e434. doi:10.1161/CIRCULATIONAHA.105.165564
Lew DP, Waldvogel FA (2004) Osteomyelitis. Lancet 364(9431):369–379. doi:10.1016/S0140-6736(04)16727-5
Kallen AJ, Mu Y, Bulens S, Reingold A, Petit S, Gershman K, Ray SM, Harrison LH, Lynfield R, Dumyati G, Townes JM, Schaffner W, Patel PR, Fridkin SK (2010) Health care-associated invasive MRSA infections, 2005–2008. Jama 304(6):641–648. doi:10.1001/jama.2010.1115
Kirby WM (1944) Extraction of a highly potent penicillin inactivator from penicillin resistant staphylococci. Science 99(2579):452–453. doi:10.1126/science.99.2579.452
Barber M (1961) Methicillin-resistant staphylococci. J Clin Pathol 14:385–393
Nannini E, Murray BE, Arias CA (2010) Resistance or decreased susceptibility to glycopeptides, daptomycin, and linezolid in methicillin-resistant Staphylococcus aureus. Curr Opin Pharmacol 10(5):516–521. doi:10.1016/j.coph.2010.06.006
Schaffer AC, Lee JC (2009) Staphylococcal vaccines and immunotherapies. Infect Dis Clin North Am 23(1):153–171. doi:10.1016/j.idc.2008.10.005
David MZ, Daum RS (2010) Community-associated methicillin-resistant Staphylococcus aureus: epidemiology and clinical consequences of an emerging epidemic. Clin Microbiol Rev 23(3):616–687. doi:10.1128/CMR.00081-09
Bartlett AH, Hulten KG (2010) Staphylococcus aureus pathogenesis: secretion systems, adhesins, and invasins. Pediatr Infect Dis J 29(9):860–861. doi:10.1097/INF.0b013e3181ef2477
Gordon RJ, Lowy FD (2008) Pathogenesis of methicillin-resistant Staphylococcus aureus infection. Clin Infect Dis 46(Suppl 5):S350–S359. doi:10.1086/533591
de Haas CJ, Veldkamp KE, Peschel A, Weerkamp F, Van Wamel WJ, Heezius EC, Poppelier MJ, Van Kessel KP, van Strijp JA (2004) Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J Exp Med 199(5):687–695. doi:10.1084/jem.20031636
Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, Torres VJ (2011) Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79(3):814–825. doi:10.1111/j.1365-2958.2010.07490.x
Menestrina G, Serra MD, Prevost G (2001) Mode of action of beta-barrel pore-forming toxins of the staphylococcal alpha-hemolysin family. Toxicon 39(11):1661–1672
Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT (2002) Staphylococcus aureus extracellular adherence protein serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med 8(7):687–693. doi:10.1038/nm728
Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, Kennedy AD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M (2007) Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat Med 13(12):1510–1514. doi:10.1038/nm1656
Bestebroer J, Poppelier MJ, Ulfman LH, Lenting PJ, Denis CV, van Kessel KP, van Strijp JA, de Haas CJ (2007) Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood 109(7):2936–2943. doi:10.1182/blood-2006-06-015461
Rooijakkers SH, Ruyken M, Roos A, Daha MR, Presanis JS, Sim RB, van Wamel WJ, van Kessel KP, van Strijp JA (2005) Immune evasion by a staphylococcal complement inhibitor that acts on C3 convertases. Nat Immunol 6(9):920–927. doi:10.1038/ni1235
Lee LY, Hook M, Haviland D, Wetsel RA, Yonter EO, Syribeys P, Vernachio J, Brown EL (2004) Inhibition of complement activation by a secreted Staphylococcus aureus protein. J Infect Dis 190(3):571–579. doi:10.1086/422259
Haupt K, Reuter M, van den Elsen J, Burman J, Halbich S, Richter J, Skerka C, Zipfel PF (2008) The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement Factor H and C3b. PLoS Pathog 4(12):e1000250. doi:10.1371/journal.ppat.1000250
Langley R, Wines B, Willoughby N, Basu I, Proft T, Fraser JD (2005) The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J Immunol 174(5):2926–2933
Forsgren A, Nordstrom K (1974) Protein A from Staphylococcus aureus: the biological significance of its reaction with IgG. Ann N Y Acad Sci 236:252–266
Thakker M, Park JS, Carey V, Lee JC (1998) Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect Immun 66(11):5183–5189
Nilsson IM, Lee JC, Bremell T, Ryden C, Tarkowski A (1997) The role of staphylococcal polysaccharide microcapsule expression in septicemia and septic arthritis. Infect Immun 65(10):4216–4221
Peschel A, Jack RW, Otto M, Collins LV, Staubitz P, Nicholson G, Kalbacher H, Nieuwenhuizen WF, Jung G, Tarkowski A, van Kessel KP, van Strijp JA (2001) Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with l-lysine. J Exp Med 193(9):1067–1076
Peschel A, Otto M, Jack RW, Kalbacher H, Jung G, Gotz F (1999) Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J Biol Chem 274(13):8405–8410
Li M, Cha DJ, Lai Y, Villaruz AE, Sturdevant DE, Otto M (2007) The antimicrobial peptide-sensing system aps of Staphylococcus aureus. Mol Microbiol 66(5):1136–1147. doi:10.1111/j.1365-2958.2007.05986.x
Jonsson K, McDevitt D, McGavin MH, Patti JM, Hook M (1995) Staphylococcus aureus expresses a major histocompatibility complex class II analog. J Biol Chem 270(37):21457–21460
Lee LY, Miyamoto YJ, McIntyre BW, Hook M, McCrea KW, McDevitt D, Brown EL (2002) The Staphylococcus aureus Map protein is an immunomodulator that interferes with T cell-mediated responses. J Clin Invest 110(10):1461–1471. doi:10.1172/JCI16318
Kochan I (1973) The role of iron in bacterial infections, with special consideration of host-tubercle bacillus interaction. Curr Top Microbiol Immunol 60:1–30
Weinberg ED (2009) Iron availability and infection. Biochim Biophys Acta 1790(7):600–605. doi:10.1016/j.bbagen.2008.07.002
Braun V, Hantke K, Koster W (1998) Bacterial iron transport: mechanisms, genetics, and regulation. Met Ions Biol Syst 35:67–145
Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ, Anderson KL, Dattilo BM, Dunman PM, Gerads R, Caprioli RM, Nacken W, Chazin WJ, Skaar EP (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319(5865):962–965. doi:10.1126/science.1152449
Ganz T, Nemeth E (2006) Regulation of iron acquisition and iron distribution in mammals. Biochim Biophys Acta 1763(7):690–699. doi:10.1016/j.bbamcr.2006.03.014
Le NT, Richardson DR (2002) The role of iron in cell cycle progression and the proliferation of neoplastic cells. Biochim Biophys Acta 1603(1):31–46
Lukianova OA, David SS (2005) A role for iron-sulfur clusters in DNA repair. Curr Opin Chem Biol 9(2):145–151. doi:10.1016/j.cbpa.2005.02.006
Crichton R (2001) Inorganic biochemistry of iron metabolism: from molecular mechanisms to clinical consequences, vol 2. Wiley, West Sussex
Vadillo M, Corbella X, Pac V, Fernandez-Viladrich P, Pujol R (1994) Multiple liver abscesses due to Yersinia enterocolitica discloses primary hemochromatosis: three cases reports and review. Clin Infect Dis 18(6):938–941
Barton JC, Acton RT (2009) Hemochromatosis and Vibrio vulnificus wound infections. J Clin Gastroenterol 43(9):890–893. doi:10.1097/MCG.0b013e31819069c1
Weinberg ED (2000) Microbial pathogens with impaired ability to acquire host iron. Biometals 13(1):85–89
Manso C, Rivas I, Peraire J, Vidal F, Richart C (1997) Fatal Listeria meningitis, endocarditis and pericarditis in a patient with haemochromatosis. Scand J Infect Dis 29(3):308–309
Posey JE, Gherardini FC (2000) Lack of a role for iron in the Lyme disease pathogen. Science 288(5471):1651–1653
Archibald F (1986) Manganese: its acquisition by and function in the lactic acid bacteria. Crit Rev Microbiol 13(1):63–109. doi:10.3109/10408418609108735
Meiwes J, Fiedler HP, Haag H, Zahner H, Konetschny-Rapp S, Jung G (1990) Isolation and characterization of staphyloferrin A, a compound with siderophore activity from Staphylococcus hyicus DSM 20459. FEMS Microbiol Lett 55(1–2):201–205
Courcol RJ, Trivier D, Bissinger MC, Martin GR, Brown MR (1997) Siderophore production by Staphylococcus aureus and identification of iron-regulated proteins. Infect Immun 65(5):1944–1948
Beasley FC, Vines ED, Grigg JC, Zheng Q, Liu S, Lajoie GA, Murphy ME, Heinrichs DE (2009) Characterization of staphyloferrin A biosynthetic and transport mutants in Staphylococcus aureus. Mol Microbiol 72(4):947–963. doi:10.1111/j.1365-2958.2009.06698.x
Friedman DB, Stauff DL, Pishchany G, Whitwell CW, Torres VJ, Skaar EP (2006) Staphylococcus aureus redirects central metabolism to increase iron availability. PLoS Pathog 2(8):e87. doi:10.1371/journal.ppat.0020087
Horsburgh MJ, Ingham E, Foster SJ (2001) In Staphylococcus aureus, Fur is an interactive regulator with PerR, contributes to virulence, and is necessary for oxidative stress resistance through positive regulation of catalase and iron homeostasis. J Bacteriol 183(2):468–475. doi:10.1128/JB.183.2.468-475.2001
Hantke K (1981) Regulation of ferric iron transport in Escherichia coli K12: isolation of a constitutive mutant. Mol Gen Genet 182(2):288–292
Xiong A, Singh VK, Cabrera G, Jayaswal RK (2000) Molecular characterization of the ferric-uptake regulator, Fur, from Staphylococcus aureus. Microbiology 146(Pt 3):659–668
Cotton JL, Tao J, Balibar CJ (2009) Identification and characterization of the Staphylococcus aureus gene cluster coding for staphyloferrin A. Biochemistry 48(5):1025–1035. doi:10.1021/bi801844c
Speziali CD, Dale SE, Henderson JA, Vines ED, Heinrichs DE (2006) Requirement of Staphylococcus aureus ATP-binding cassette-ATPase FhuC for iron-restricted growth and evidence that it functions with more than one iron transporter. J Bacteriol 188(6):2048–2055. doi:10.1128/JB.188.6.2048-2055.2006
Skaar EP, Humayun M, Bae T, DeBord KL, Schneewind O (2004) Iron-source preference of Staphylococcus aureus infections. Science 305(5690):1626–1628. doi:10.1126/science.1099930
Grigg JC, Cooper JD, Cheung J, Heinrichs DE, Murphy ME (2010) The Staphylococcus aureus siderophore receptor HtsA undergoes localized conformational changes to enclose staphyloferrin A in an arginine-rich binding pocket. J Biol Chem 285(15):11162–11171. doi:10.1074/jbc.M109.097865
Drechsel H, Freund S, Nicholson G, Haag H, Jung O, Zahner H, Jung G (1993) Purification and chemical characterization of staphyloferrin B, a hydrophilic siderophore from staphylococci. Biometals 6(3):185–192
Cheung J, Beasley FC, Liu S, Lajoie GA, Heinrichs DE (2009) Molecular characterization of staphyloferrin B biosynthesis in Staphylococcus aureus. Mol Microbiol 74(3):594–608. doi:10.1111/j.1365-2958.2009.06880.x
Dale SE, Sebulsky MT, Heinrichs DE (2004) Involvement of SirABC in iron-siderophore import in Staphylococcus aureus. J Bacteriol 186(24):8356–8362. doi:10.1128/JB.186.24.8356-8362.2004
Grigg JC, Cheung J, Heinrichs DE, Murphy ME (2010) Specificity of Staphyloferrin B recognition by the SirA receptor from Staphylococcus aureus. J Biol Chem 285(45):34579–34588. doi:10.1074/jbc.M110.172924
Beasley FC, Marolda CL, Cheung J, Buac S, Heinrichs DE (2011) Staphylococcus aureus transporters Hts, Sir, and Sst capture iron liberated from human transferrin by staphyloferrin a, staphyloferrin B, and catecholamine stress hormones, respectively, and contribute to virulence. Infect Immun 79(6):2345–2355. doi:10.1128/IAI.00117-11
Brock JH, Ng J (1983) The effect of desferrioxamine on the growth of Staphylococcus aureus, Yersinia enterocolitica and Streptococcus faecalis in human serum: uptake of desferrioxamine-bound iron. FEMS Microbiol Lett 20(3):439–442
Sebulsky MT, Hohnstein D, Hunter MD, Heinrichs DE (2000) Identification and characterization of a membrane permease involved in iron-hydroxamate transport in Staphylococcus aureus. J Bacteriol 182(16):4394–4400
Sebulsky MT, Heinrichs DE (2001) Identification and characterization of fhuD1 and fhuD2, two genes involved in iron-hydroxamate uptake in Staphylococcus aureus. J Bacteriol 183(17):4994–5000
Neupane GP, Kim DM (2009) Comparison of the effects of deferasirox, deferiprone, and deferoxamine on the growth and virulence of Vibrio vulnificus. Transfusion 49(8):1762–1769. doi:10.1111/j.1537-2995.2009.02186.x
Lesic B, Foulon J, Carniel E (2002) Comparison of the effects of deferiprone versus deferoxamine on growth and virulence of Yersinia enterocolitica. Antimicrob Agents Chemother 46(6):1741–1745
Boelaert JR, de Locht M, Van Cutsem J, Kerrels V, Cantinieaux B, Verdonck A, Van Landuyt HW, Schneider YJ (1993) Mucormycosis during deferoxamine therapy is a siderophore-mediated infection. In vitro and in vivo animal studies. J Clin Invest 91(5):1979–1986. doi:10.1172/JCI116419
Morrissey JA, Cockayne A, Hill PJ, Williams P (2000) Molecular cloning and analysis of a putative siderophore ABC transporter from Staphylococcus aureus. Infect Immun 68(11):6281–6288
Cendrowski S, MacArthur W, Hanna P (2004) Bacillus anthracis requires siderophore biosynthesis for growth in macrophages and mouse virulence. Mol Microbiol 51(2):407–417. doi:10.1046/j.1365-2958.2003.03861.x
Cox CD (1982) Effect of pyochelin on the virulence of Pseudomonas aeruginosa. Infect Immun 36(1):17–23
Meyer JM, Neely A, Stintzi A, Georges C, Holder IA (1996) Pyoverdin is essential for virulence of Pseudomonas aeruginosa. Infect Immun 64(2):518–523
Heesemann J, Hantke K, Vocke T, Saken E, Rakin A, Stojiljkovic I, Berner R (1993) Virulence of Yersinia enterocolitica is closely associated with siderophore production, expression of an iron-repressible outer membrane polypeptide of 65,000 Da and pesticin sensitivity. Mol Microbiol 8(2):397–408
Allard KA, Dao J, Sanjeevaiah P, McCoy-Simandle K, Chatfield CH, Crumrine DS, Castignetti D, Cianciotto NP (2009) Purification of Legiobactin and importance of this siderophore in lung infection by Legionella pneumophila. Infect Immun 77(7):2887–2895. doi:10.1128/IAI.00087-09
Williams PH (1979) Novel iron uptake system specified by ColV plasmids: an important component in the virulence of invasive strains of Escherichia coli. Infect Immun 26(3):925–932
Dale SE, Doherty-Kirby A, Lajoie G, Heinrichs DE (2004) Role of siderophore biosynthesis in virulence of Staphylococcus aureus: identification and characterization of genes involved in production of a siderophore. Infect Immun 72(1):29–37
Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK (2002) The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol Cell 10(5):1033–1043
Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A (2004) Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature 432(7019):917–921. doi:10.1038/nature03104
Berger T, Togawa A, Duncan GS, Elia AJ, You-Ten A, Wakeham A, Fong HE, Cheung CC, Mak TW (2006) Lipocalin 2-deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia-reperfusion injury. Proc Natl Acad Sci U S A 103(6):1834–1839. doi:10.1073/pnas.0510847103
Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, Mak TW, Clifton MC, Strong RK, Ray P, Kolls JK (2009) Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol 182(8):4947–4956. doi:10.4049/jimmunol.0803282
Johnson EE, Srikanth CV, Sandgren A, Harrington L, Trebicka E, Wang L, Borregaard N, Murray M, Cherayil BJ (2010) Siderocalin inhibits the intracellular replication of Mycobacterium tuberculosis in macrophages. FEMS Immunol Med Microbiol 58(1):138–145. doi:10.1111/j.1574-695X.2009.00622.x
Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK (2005) Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 13(1):29–41. doi:10.1016/j.str.2004.10.009
Nelson AL, Barasch JM, Bunte RM, Weiser JN (2005) Bacterial colonization of nasal mucosa induces expression of siderocalin, an iron-sequestering component of innate immunity. Cell Microbiol 7(10):1404–1417. doi:10.1111/j.1462-5822.2005.00566.x
Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J (2005) Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest 115(3):610–621. doi:10.1172/JCI23056
Wilson MK, Abergel RJ, Raymond KN, Arceneaux JE, Byers BR (2006) Siderophores of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Biochem Biophys Res Commun 348(1):320–325. doi:10.1016/j.bbrc.2006.07.055
Abergel RJ, Wilson MK, Arceneaux JE, Hoette TM, Strong RK, Byers BR, Raymond KN (2006) Anthrax pathogen evades the mammalian immune system through stealth siderophore production. Proc Natl Acad Sci U S A 103(49):18499–18503. doi:10.1073/pnas.0607055103
Hantke K, Nicholson G, Rabsch W, Winkelmann G (2003) Salmochelins, siderophores of Salmonella enterica and uropathogenic Escherichia coli strains, are recognized by the outer membrane receptor IroN. Proc Natl Acad Sci U S A 100(7):3677–3682. doi:10.1073/pnas.0737682100
Young NS, Gerson SL, High KA (2006) Clinical hematology, vol 1. Mosby Elsevier, Philadelphia
Everse J, Hsia N (1997) The toxicities of native and modified hemoglobins. Free Radic Biol Med 22(6):1075–1099
Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, Moestrup SK (2001) Identification of the haemoglobin scavenger receptor. Nature 409(6817):198–201. doi:10.1038/35051594
Allhorn M, Berggard T, Nordberg J, Olsson ML, Akerstrom B (2002) Processing of the lipocalin alpha(1)-microglobulin by hemoglobin induces heme-binding and heme-degradation properties. Blood 99(6):1894–1901
Anzaldi LL, Skaar EP (2010) Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect Immun 78(12):4977–4989. doi:10.1128/IAI.00613-10
Tong Y, Guo M (2009) Bacterial heme-transport proteins and their heme-coordination modes. Arch Biochem Biophys 481(1):1–15. doi:10.1016/j.abb.2008.10.013
Mazmanian SK, Liu G, Ton-That H, Schneewind O (1999) Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 285(5428):760–763
Mazmanian SK, Ton-That H, Schneewind O (2001) Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol Microbiol 40(5):1049–1057
Mazmanian SK, Ton-That H, Su K, Schneewind O (2002) An iron-regulated sortase anchors a class of surface protein during Staphylococcus aureus pathogenesis. Proc Natl Acad Sci U S A 99(4):2293–2298. doi:10.1073/pnas.032523999
Hammer ND, Skaar EP (2011) Molecular mechanisms of Staphylococcus aureus iron acquisition. Annu Rev Microbiol. doi:10.1146/annurev-micro-090110-102851
Torres VJ, Pishchany G, Humayun M, Schneewind O, Skaar EP (2006) Staphylococcus aureus IsdB is a hemoglobin receptor required for heme iron utilization. J Bacteriol 188(24):8421–8429. doi:10.1128/JB.01335-06
Dryla A, Gelbmann D, von Gabain A, Nagy E (2003) Identification of a novel iron regulated staphylococcal surface protein with haptoglobin-haemoglobin binding activity. Mol Microbiol 49(1):37–53
Muryoi N, Tiedemann MT, Pluym M, Cheung J, Heinrichs DE, Stillman MJ (2008) Demonstration of the iron-regulated surface determinant (Isd) heme transfer pathway in Staphylococcus aureus. J Biol Chem 283(42):28125–28136. doi:10.1074/jbc.M802171200
Zhu H, Xie G, Liu M, Olson JS, Fabian M, Dooley DM, Lei B (2008) Pathway for heme uptake from human methemoglobin by the iron-regulated surface determinants system of Staphylococcus aureus. J Biol Chem 283(26):18450–18460. doi:10.1074/jbc.M801466200
Liu M, Tanaka WN, Zhu H, Xie G, Dooley DM, Lei B (2008) Direct hemin transfer from IsdA to IsdC in the iron-regulated surface determinant (Isd) heme acquisition system of Staphylococcus aureus. J Biol Chem 283(11):6668–6676. doi:10.1074/jbc.M708372200
Mazmanian SK, Skaar EP, Gaspar AH, Humayun M, Gornicki P, Jelenska J, Joachmiak A, Missiakas DM, Schneewind O (2003) Passage of heme-iron across the envelope of Staphylococcus aureus. Science 299(5608):906–909. doi:10.1126/science.1081147
Skaar EP, Gaspar AH, Schneewind O (2004) IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J Biol Chem 279(1):436–443. doi:10.1074/jbc.M307952200
Wu R, Skaar EP, Zhang R, Joachimiak G, Gornicki P, Schneewind O, Joachimiak A (2005) Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J Biol Chem 280(4):2840–2846. doi:10.1074/jbc.M409526200
Reniere ML, Ukpabi GN, Harry SR, Stec DF, Krull R, Wright DW, Bachmann BO, Murphy ME, Skaar EP (2010) The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol Microbiol 75(6):1529–1538. doi:10.1111/j.1365-2958.2010.07076.x
Reniere ML, Skaar EP (2008) Staphylococcus aureus haem oxygenases are differentially regulated by iron and haem. Mol Microbiol 69(5):1304–1315. doi:10.1111/j.1365-2958.2008.06363.x
Torres VJ, Stauff DL, Pishchany G, Bezbradica JS, Gordy LE, Iturregui J, Anderson KL, Dunman PM, Joyce S, Skaar EP (2007) A Staphylococcus aureus regulatory system that responds to host heme and modulates virulence. Cell Host Microbe 1(2):109–119. doi:10.1016/j.chom.2007.03.001
Stauff DL, Bagaley D, Torres VJ, Joyce R, Anderson KL, Kuechenmeister L, Dunman PM, Skaar EP (2008) Staphylococcus aureus HrtA is an ATPase required for protection against heme toxicity and prevention of a transcriptional heme stress response. J Bacteriol 190(10):3588–3596. doi:10.1128/JB.01921-07
Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, Schneewind O, DeLeo FR (2010) Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis 202(7):1050–1058. doi:10.1086/656043
Bubeck Wardenburg J, Patel RJ, Schneewind O (2007) Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect Immun 75(2):1040–1044. doi:10.1128/IAI.01313-06
Bubeck Wardenburg J, Bae T, Otto M, Deleo FR, Schneewind O (2007) Poring over pores: alpha-hemolysin and Panton-Valentine leukocidin in Staphylococcus aureus pneumonia. Nat Med 13(12):1405–1406. doi:10.1038/nm1207-1405
Ji Y, Marra A, Rosenberg M, Woodnutt G (1999) Regulated antisense RNA eliminates alpha-toxin virulence in Staphylococcus aureus infection. J Bacteriol 181(21):6585–6590
Kernodle DS, Voladri RK, Menzies BE, Hager CC, Edwards KM (1997) Expression of an antisense hla fragment in Staphylococcus aureus reduces alpha-toxin production in vitro and attenuates lethal activity in a murine model. Infect Immun 65(1):179–184
Callegan MC, Engel LS, Hill JM, O’Callaghan RJ (1994) Corneal virulence of Staphylococcus aureus: roles of alpha-toxin and protein A in pathogenesis. Infect Immun 62(6):2478–2482
Patel AH, Nowlan P, Weavers ED, Foster T (1987) Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect Immun 55(12):3103–3110
Ragle BE, Bubeck Wardenburg J (2009) Anti-alpha-hemolysin monoclonal antibodies mediate protection against Staphylococcus aureus pneumonia. Infect Immun 77(7):2712–2718. doi:10.1128/IAI.00115-09
Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM (2009) Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J 23(10):3393–3404. doi:10.1096/fj.09-135467
Kim HK, DeDent A, Cheng AG, McAdow M, Bagnoli F, Missiakas DM, Schneewind O (2010) IsdA and IsdB antibodies protect mice against Staphylococcus aureus abscess formation and lethal challenge. Vaccine 28(38):6382–6392. doi:10.1016/j.vaccine.2010.02.097
Pishchany G, McCoy AL, Torres VJ, Krause JC, Crowe JE Jr, Fabry ME, Skaar EP (2010) Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe 8(6):544–550. doi:10.1016/j.chom.2010.11.002
Maione D, Margarit I, Rinaudo CD, Masignani V, Mora M, Scarselli M, Tettelin H, Brettoni C, Iacobini ET, Rosini R, D’Agostino N, Miorin L, Buccato S, Mariani M, Galli G, Nogarotto R, Nardi Dei V, Vegni F, Fraser C, Mancuso G, Teti G, Madoff LC, Paoletti LC, Rappuoli R, Kasper DL, Telford JL, Grandi G (2005) Identification of a universal group B streptococcus vaccine by multiple genome screen. Science 309(5731):148–150. doi:10.1126/science.1109869
Stranger-Jones YK, Bae T, Schneewind O (2006) Vaccine assembly from surface proteins of Staphylococcus aureus. Proc Natl Acad Sci U S A 103(45):16942–16947. doi:10.1073/pnas.0606863103
Kuklin NA, Clark DJ, Secore S, Cook J, Cope LD, McNeely T, Noble L, Brown MJ, Zorman JK, Wang XM, Pancari G, Fan H, Isett K, Burgess B, Bryan J, Brownlow M, George H, Meinz M, Liddell ME, Kelly R, Schultz L, Montgomery D, Onishi J, Losada M, Martin M, Ebert T, Tan CY, Schofield TL, Nagy E, Meineke A, Joyce JG, Kurtz MB, Caulfield MJ, Jansen KU, McClements W, Anderson AS (2006) A novel Staphylococcus aureus vaccine: iron surface determinant B induces rapid antibody responses in rhesus macaques and specific increased survival in a murine S. aureus sepsis model. Infect Immun 74(4):2215–2223. doi:10.1128/IAI.74.4.2215-2223.2006
Clarke SR, Brummell KJ, Horsburgh MJ, McDowell PW, Mohamad SA, Stapleton MR, Acevedo J, Read RC, Day NP, Peacock SJ, Mond JJ, Kokai-Kun JF, Foster SJ (2006) Identification of in vivo-expressed antigens of Staphylococcus aureus and their use in vaccinations for protection against nasal carriage. J Infect Dis 193(8):1098–1108. doi:10.1086/501471
Ebert T, Smith S, Pancari G, Clark D, Hampton R, Secore S, Towne V, Fan H, Wang XM, Wu X, Ernst R, Harvey BR, Finnefrock AC, Wang F, Tan C, Durr E, Cope L, Anderson A, An Z, McNeely T (2010) A fully human monoclonal antibody to Staphylococcus aureus iron regulated surface determinant B (IsdB) with functional activity in vitro and in vivo. Hum Antibodies 19(4):113–128. doi:10.3233/HAB-2010-0235
Harro C, Betts R, Orenstein W, Kwak EJ, Greenberg HE, Onorato MT, Hartzel J, Lipka J, DiNubile MJ, Kartsonis N (2010) Safety and immunogenicity of a novel Staphylococcus aureus vaccine: results from the first study of the vaccine dose range in humans. Clin Vaccine Immunol 17(12):1868–1874. doi:10.1128/CVI.00356-10
Clarke SR, Wiltshire MD, Foster SJ (2004) IsdA of Staphylococcus aureus is a broad spectrum, iron-regulated adhesin. Mol Microbiol 51(5):1509–1519. doi:10.1111/j.1365-2958.2003.03938.x
Corrigan RM, Miajlovic H, Foster TJ (2009) Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol 9:22. doi:10.1186/1471-2180-9-22
Clarke SR, Andre G, Walsh EJ, Dufrene YF, Foster TJ, Foster SJ (2009) Iron-regulated surface determinant protein A mediates adhesion of Staphylococcus aureus to human corneocyte envelope proteins. Infect Immun 77(6):2408–2416. doi:10.1128/IAI.01304-08
Clarke SR, Mohamed R, Bian L, Routh AF, Kokai-Kun JF, Mond JJ, Tarkowski A, Foster SJ (2007) The Staphylococcus aureus surface protein IsdA mediates resistance to innate defenses of human skin. Cell Host Microbe 1(3):199–212. doi:10.1016/j.chom.2007.04.005
Palazzolo-Ballance AM, Reniere ML, Braughton KR, Sturdevant DE, Otto M, Kreiswirth BN, Skaar EP, DeLeo FR (2008) Neutrophil microbicides induce a pathogen survival response in community-associated methicillin-resistant Staphylococcus aureus. J Immunol 180(1):500–509
Clarke SR, Foster SJ (2008) IsdA protects Staphylococcus aureus against the bactericidal protease activity of apolactoferrin. Infect Immun 76(4):1518–1526. doi:10.1128/IAI.01530-07
Mason WJ, Skaar EP (2009) Assessing the contribution of heme-iron acquisition to Staphylococcus aureus pneumonia using computed tomography. PLoS One 4(8):e6668. doi:10.1371/journal.pone.0006668
Attia AS, Benson MA, Stauff DL, Torres VJ, Skaar EP (2010) Membrane damage elicits an immunomodulatory program in Staphylococcus aureus. PLoS Pathog 6(3):e1000802. doi:10.1371/journal.ppat.1000802
Aschner JL, Aschner M (2005) Nutritional aspects of manganese homeostasis. Mol Aspects Med 26(4–5):353–362. doi:10.1016/j.mam.2005.07.003
Aschner M, Guilarte TR, Schneider JS, Zheng W (2007) Manganese: recent advances in understanding its transport and neurotoxicity. Toxicol Appl Pharmacol 221(2):131–147. doi:10.1016/j.taap.2007.03.001
Friedman BJ, Freeland-Graves JH, Bales CW, Behmardi F, Shorey-Kutschke RL, Willis RA, Crosby JB, Trickett PC, Houston SD (1987) Manganese balance and clinical observations in young men fed a manganese-deficient diet. J Nutr 117(1):133–143
Jakubovics NS, Jenkinson HF (2001) Out of the iron age: new insights into the critical role of manganese homeostasis in bacteria. Microbiology 147(Pt 7):1709–1718
Bartsevich VV, Pakrasi HB (1995) Molecular identification of an ABC transporter complex for manganese: analysis of a cyanobacterial mutant strain impaired in the photosynthetic oxygen evolution process. EMBO J 14(9):1845–1853
Que Q, Helmann JD (2000) Manganese homeostasis in Bacillus subtilis is regulated by MntR, a bifunctional regulator related to the diphtheria toxin repressor family of proteins. Mol Microbiol 35(6):1454–1468
Kehres DG, Zaharik ML, Finlay BB, Maguire ME (2000) The NRAMP proteins of Salmonella typhimurium and Escherichia coli are selective manganese transporters involved in the response to reactive oxygen. Mol Microbiol 36(5):1085–1100
Papp-Wallace KM, Maguire ME (2006) Manganese transport and the role of manganese in virulence. Annu Rev Microbiol 60:187–209. doi:10.1146/annurev.micro.60.080805.142149
Fisher S, Buxbaum L, Toth K, Eisenstadt E, Silver S (1973) Regulation of manganese accumulation and exchange in Bacillus subtilis W23. J Bacteriol 113(3):1373–1380
Rosch JW, Gao G, Ridout G, Wang YD, Tuomanen EI (2009) Role of the manganese efflux system mntE for signalling and pathogenesis in Streptococcus pneumoniae. Mol Microbiol 72(1):12–25. doi:10.1111/j.1365-2958.2009.06638.x
Nies DH (2003) Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev 27(2–3):313–339
Horsburgh MJ, Wharton SJ, Cox AG, Ingham E, Peacock S, Foster SJ (2002) MntR modulates expression of the PerR regulon and superoxide resistance in Staphylococcus aureus through control of manganese uptake. Mol Microbiol 44(5):1269–1286
Horsburgh MJ, Clements MO, Crossley H, Ingham E, Foster SJ (2001) PerR controls oxidative stress resistance and iron storage proteins and is required for virulence in Staphylococcus aureus. Infect Immun 69(6):3744–3754. doi:10.1128/IAI.69.6.3744-3754.2001
Kuroda M, Hayashi H, Ohta T (1999) Chromosome-determined zinc-responsible operon czr in Staphylococcus aureus strain 912. Microbiol Immunol 43(2):115–125
Xiong A, Jayaswal RK (1998) Molecular characterization of a chromosomal determinant conferring resistance to zinc and cobalt ions in Staphylococcus aureus. J Bacteriol 180(16):4024–4029
Clements MO, Watson SP, Foster SJ (1999) Characterization of the major superoxide dismutase of Staphylococcus aureus and its role in starvation survival, stress resistance, and pathogenicity. J Bacteriol 181(13):3898–3903
Valderas MW, Hart ME (2001) Identification and characterization of a second superoxide dismutase gene (sodM) from Staphylococcus aureus. J Bacteriol 183(11):3399–3407. doi:10.1128/JB.183.11.3399-3407.2001
Kehl-Fie TE, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro KA, Chazin WJ, Skaar EP (2011) Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10(2):158–164. doi:10.1016/j.chom.2011.07.004
Anderson ES, Paulley JT, Gaines JM, Valderas MW, Martin DW, Menscher E, Brown TD, Burns CS, Roop RM 2nd (2009) The manganese transporter MntH is a critical virulence determinant for Brucella abortus 2308 in experimentally infected mice. Infect Immun 77(8):3466–3474. doi:10.1128/IAI.00444-09
Boyer E, Bergevin I, Malo D, Gros P, Cellier MF (2002) Acquisition of Mn(II) in addition to Fe(II) is required for full virulence of Salmonella enterica serovar Typhimurium. Infect Immun 70(11):6032–6042
Lim KH, Jones CE, vanden Hoven RN, Edwards JL, Falsetta ML, Apicella MA, Jennings MP, McEwan AG (2008) Metal binding specificity of the MntABC permease of Neisseria gonorrhoeae and its influence on bacterial growth and interaction with cervical epithelial cells. Infect Immun 76(8):3569–3576. doi:10.1128/IAI.01725-07
Singh KV, Coque TM, Weinstock GM, Murray BE (1998) In vivo testing of an Enterococcus faecalis efaA mutant and use of efaA homologs for species identification. FEMS Immunol Med Microbiol 21(4):323–331
Berry AM, Paton JC (1996) Sequence heterogeneity of PsaA, a 37-kilodalton putative adhesin essential for virulence of Streptococcus pneumoniae. Infect Immun 64(12):5255–5262
Bearden SW, Perry RD (1999) The Yfe system of Yersinia pestis transports iron and manganese and is required for full virulence of plague. Mol Microbiol 32(2):403–414
Gruenheid S, Pinner E, Desjardins M, Gros P (1997) Natural resistance to infection with intracellular pathogens: the Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med 185(4):717–730
Jabado N, Jankowski A, Dougaparsad S, Picard V, Grinstein S, Gros P (2000) Natural resistance to intracellular infections: natural resistance-associated macrophage protein 1 (Nramp1) functions as a pH-dependent manganese transporter at the phagosomal membrane. J Exp Med 192(9):1237–1248
Forbes JR, Gros P (2003) Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane. Blood 102(5):1884–1892. doi:10.1182/blood-2003-02-0425
Goswami T, Bhattacharjee A, Babal P, Searle S, Moore E, Li M, Blackwell JM (2001) Natural-resistance-associated macrophage protein 1 is an H+/bivalent cation antiporter. Biochem J 354(Pt 3):511–519
Vidal S, Tremblay ML, Govoni G, Gauthier S, Sebastiani G, Malo D, Skamene E, Olivier M, Jothy S, Gros P (1995) The Ity/Lsh/Bcg locus: natural resistance to infection with intracellular parasites is abrogated by disruption of the Nramp1 gene. J Exp Med 182(3):655–666
van Crevel R, Parwati I, Sahiratmadja E, Marzuki S, Ottenhoff TH, Netea MG, van der Ven A, Nelwan RH, van der Meer JW, Alisjahbana B, van de Vosse E (2009) Infection with Mycobacterium tuberculosis Beijing genotype strains is associated with polymorphisms in SLC11A1/NRAMP1 in Indonesian patients with tuberculosis. J Infect Dis 200(11):1671–1674. doi:10.1086/648477
Jin J, Sun L, Jiao W, Zhao S, Li H, Guan X, Jiao A, Jiang Z, Shen A (2009) SLC11A1 (formerly NRAMP1) gene polymorphisms associated with pediatric tuberculosis in China. Clin Infect Dis 48(6):733–738. doi:10.1086/597034
Koh WJ, Kwon OJ, Kim EJ, Lee KS, Ki CS, Kim JW (2005) NRAMP1 gene polymorphism and susceptibility to nontuberculous mycobacterial lung diseases. Chest 128(1):94–101. doi:10.1378/chest.128.1.94
Dale I, Fagerhol MK, Naesgaard I (1983) Purification and partial characterization of a highly immunogenic human leukocyte protein, the L1 antigen. Eur J Biochem 134(1):1–6
Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N (1991) Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem 266(12):7706–7713
Yui S, Nakatani Y, Mikami M (2003) Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull 26(6):753–760
Steinbakk M, Naess-Andresen CF, Lingaas E, Dale I, Brandtzaeg P, Fagerhol MK (1990) Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 336(8718):763–765
Sohnle PG, Collins-Lech C, Wiessner JH (1991) Antimicrobial activity of an abundant calcium-binding protein in the cytoplasm of human neutrophils. J Infect Dis 163(1):187–192
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535. doi:10.1126/science.1092385
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkmann V, Jungblut PR, Zychlinsky A (2009) Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 5(10):e1000639. doi:10.1371/journal.ppat.1000639
Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, Sukhova GK, Packard RR, Hogg N, Libby P, Simon DI (2009) Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 120(5):427–436. doi:10.1161/CIRCULATIONAHA.108.814582
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13(9):1042–1049. doi:10.1038/nm1638
Ehrchen JM, Sunderkotter C, Foell D, Vogl T, Roth J (2009) The endogenous Toll-like receptor 4 agonist S100A8/S100A9 (calprotectin) as innate amplifier of infection, autoimmunity, and cancer. J Leukoc Biol 86(3):557–566. doi:10.1189/jlb.1008647
Ghavami S, Kerkhoff C, Chazin WJ, Kadkhoda K, Xiao W, Zuse A, Hashemi M, Eshraghi M, Schulze-Osthoff K, Klonisch T, Los M (2008) S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochim Biophys Acta 1783(2):297–311. doi:10.1016/j.bbamcr.2007.10.015
Li C, Chen H, Ding F, Zhang Y, Luo A, Wang M, Liu Z (2009) A novel p53 target gene, S100A9, induces p53-dependent cellular apoptosis and mediates the p53 apoptosis pathway. Biochem J 422(2):363–372. doi:10.1042/BJ20090465
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G (2008) Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 181(7):4666–4675
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205(10):2235–2249. doi:10.1084/jem.20080132
Sohnle PG, Hunter MJ, Hahn B, Chazin WJ (2000) Zinc-reversible antimicrobial activity of recombinant calprotectin (migration inhibitory factor-related proteins 8 and 14). J Infect Dis 182(4):1272–1275. doi:10.1086/315810
Sohnle PG, Collins-Lech C, Wiessner JH (1991) The zinc-reversible antimicrobial activity of neutrophil lysates and abscess fluid supernatants. J Infect Dis 164(1):137–142
Clohessy PA, Golden BE (1995) Calprotectin-mediated zinc chelation as a biostatic mechanism in host defence. Scand J Immunol 42(5):551–556
Lusitani D, Malawista SE, Montgomery RR (2003) Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infect Immun 71(8):4711–4716
Murdoch DA (1998) Gram-positive anaerobic cocci. Clin Microbiol Rev 11(1):81–120
Akerstrom B, Bjorck L (2009) Bacterial surface protein L binds and inactivates neutrophil proteins S100A8/A9. J Immunol 183(7):4583–4592. doi:10.4049/jimmunol.0901487
DeLeo FR, Diep BA, Otto M (2009) Host defense and pathogenesis in Staphylococcus aureus infections. Infect Dis Clin North Am 23(1):17–34. doi:10.1016/j.idc.2008.10.003
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, Robbins SM, Green FH, Surette MG, Sugai M, Bowden MG, Hussain M, Zhang K, Kubes P (2010) A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J Immunol 185(12):7413–7425. doi:10.4049/jimmunol.1000675
Prasad AS, Meftah S, Abdallah J, Kaplan J, Brewer GJ, Bach JF, Dardenne M (1988) Serum thymulin in human zinc deficiency. J Clin Invest 82(4):1202–1210. doi:10.1172/JCI113717
Beck FW, Prasad AS, Kaplan J, Fitzgerald JT, Brewer GJ (1997) Changes in cytokine production and T cell subpopulations in experimentally induced zinc-deficient humans. Am J Physiol 272(6 Pt 1):E1002–E1007
Prasad AS (2008) Zinc in human health: effect of zinc on immune cells. Mol Med 14(5–6):353–357. doi:10.2119/2008-00033.Prasad
Shankar AH, Prasad AS (1998) Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr 68(2 Suppl):447S–463S
Sazawal S, Black RE, Bhan MK, Bhandari N, Sinha A, Jalla S (1995) Zinc supplementation in young children with acute diarrhea in India. N Engl J Med 333(13):839–844. doi:10.1056/NEJM199509283331304
Sazawal S, Black RE, Jalla S, Mazumdar S, Sinha A, Bhan MK (1998) Zinc supplementation reduces the incidence of acute lower respiratory infections in infants and preschool children: a double-blind, controlled trial. Pediatrics 102(1 Pt 1):1–5
Lassi ZS, Haider BA, Bhutta ZA (2010) Zinc supplementation for the prevention of pneumonia in children aged 2 months to 59 months. Cochrane Database Syst Rev (12):CD005978. doi:10.1002/14651858.CD005978.pub2
Prasad AS, Beck FW, Bao B, Fitzgerald JT, Snell DC, Steinberg JD, Cardozo LJ (2007) Zinc supplementation decreases incidence of infections in the elderly: effect of zinc on generation of cytokines and oxidative stress. Am J Clin Nutr 85(3):837–844
Andreini C, Banci L, Bertini I, Rosato A (2006) Zinc through the three domains of life. J Proteome Res 5(11):3173–3178. doi:10.1021/pr0603699
Patzer SI, Hantke K (1998) The ZnuABC high-affinity zinc uptake system and its regulator Zur in Escherichia coli. Mol Microbiol 28(6):1199–1210
Blencowe DK, Morby AP (2003) Zn(II) metabolism in prokaryotes. FEMS Microbiol Rev 27(2–3):291–311
Shin JH, Jung HJ, An YJ, Cho YB, Cha SS, Roe JH (2011) Graded expression of zinc-responsive genes through two regulatory zinc-binding sites in Zur. Proc Natl Acad Sci U S A 108(12):5045–5050. doi:10.1073/pnas.1017744108
Grass G, Wong MD, Rosen BP, Smith RL, Rensing C (2002) ZupT is a Zn(II) uptake system in Escherichia coli. J Bacteriol 184(3):864–866
Rensing C, Mitra B, Rosen BP (1997) The zntA gene of Escherichia coli encodes a Zn(II)-translocating P-type ATPase. Proc Natl Acad Sci U S A 94(26):14326–14331
Nies DH, Silver S (1989) Plasmid-determined inducible efflux is responsible for resistance to cadmium, zinc, and cobalt in Alcaligenes eutrophus. J Bacteriol 171(2):896–900
Hassan MT, van der Lelie D, Springael D, Romling U, Ahmed N, Mergeay M (1999) Identification of a gene cluster, czr, involved in cadmium and zinc resistance in Pseudomonas aeruginosa. Gene 238(2):417–425
Singh VK, Xiong A, Usgaard TR, Chakrabarti S, Deora R, Misra TK, Jayaswal RK (1999) ZntR is an autoregulatory protein and negatively regulates the chromosomal zinc resistance operon znt of Staphylococcus aureus. Mol Microbiol 33(1):200–207
Nucifora G, Chu L, Misra TK, Silver S (1989) Cadmium resistance from Staphylococcus aureus plasmid pI258 cadA gene results from a cadmium-efflux ATPase. Proc Natl Acad Sci U S A 86(10):3544–3548
Lindsay JA, Foster SJ (2001) Zur: a Zn(2+)-responsive regulatory element of Staphylococcus aureus. Microbiology 147(Pt 5):1259–1266
Cousins RJ, Leinart AS (1988) Tissue-specific regulation of zinc metabolism and metallothionein genes by interleukin 1. FASEB J 2(13):2884–2890
Gabay C, Kushner I (1999) Acute-phase proteins and other systemic responses to inflammation. N Engl J Med 340(6):448–454. doi:10.1056/NEJM199902113400607
Liuzzi JP, Lichten LA, Rivera S, Blanchard RK, Aydemir TB, Knutson MD, Ganz T, Cousins RJ (2005) Interleukin-6 regulates the zinc transporter Zip14 in liver and contributes to the hypozincemia of the acute-phase response. Proc Natl Acad Sci U S A 102(19):6843–6848. doi:10.1073/pnas.0502257102
Pless DD, Ruthel G, Reinke EK, Ulrich RG, Bavari S (2005) Persistence of zinc-binding bacterial superantigens at the surface of antigen-presenting cells contributes to the extreme potency of these superantigens as T-cell activators. Infect Immun 73(9):5358–5366. doi:10.1128/IAI.73.9.5358-5366.2005
Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB (2008) A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A 105(49):19456–19461. doi:10.1073/pnas.0807717105
Sitthisak S, Knutsson L, Webb JW, Jayaswal RK (2007) Molecular characterization of the copper transport system in Staphylococcus aureus. Microbiology 153(Pt 12):4274–4283. doi:10.1099/mic.0.2007/009860-0
Grossoehme N, Kehl-Fie TE, Ma Z, Adams KW, Cowart DM, Scott RA, Skaar EP, Giedroc DP (2011) Control of copper resistance and inorganic sulfur metabolism by paralogous regulators in Staphylococcus aureus. J Biol Chem 286(15):13522–13531. doi:10.1074/jbc.M111.220012
Liu T, Ramesh A, Ma Z, Ward SK, Zhang L, George GN, Talaat AM, Sacchettini JC, Giedroc DP (2007) CsoR is a novel Mycobacterium tuberculosis copper-sensing transcriptional regulator. Nat Chem Biol 3(1):60–68. doi:10.1038/nchembio844
Banci L, Bertini I, Ciofi-Baffoni S, Del Conte R, Gonnelli L (2003) Understanding copper trafficking in bacteria: interaction between the copper transport protein CopZ and the N-terminal domain of the copper ATPase CopA from Bacillus subtilis. Biochemistry 42(7):1939–1949. doi:10.1021/bi027096p
Radford DS, Kihlken MA, Borrelly GP, Harwood CR, Le Brun NE, Cavet JS (2003) CopZ from Bacillus subtilis interacts in vivo with a copper exporting CPx-type ATPase CopA. FEMS Microbiol Lett 220(1):105–112
Shafeeq S, Yesilkaya H, Kloosterman TG, Narayanan G, Andrew PW, Kuipers OP, Morrissey JA (2011) The cop operon is required for copper homeostasis and contributes to virulence in Streptococcus pneumoniae. Mol Microbiol. doi:10.1111/j.1365-2958.2011.07758.x
Wolschendorf F, Ackart D, Shrestha TB, Hascall-Dove L, Nolan S, Lamichhane G, Wang Y, Bossmann SH, Basaraba RJ, Niederweis M (2011) Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 108(4):1621–1626. doi:10.1073/pnas.1009261108
Schwan WR, Warrener P, Keunz E, Stover CK, Folger KR (2005) Mutations in the cueA gene encoding a copper homeostasis P-type ATPase reduce the pathogenicity of Pseudomonas aeruginosa in mice. Int J Med Microbiol 295(4):237–242
Prohaska JR, Lukasewycz OA (1981) Copper deficiency suppresses the immune response of mice. Science 213(4507):559–561
White C, Lee J, Kambe T, Fritsche K, Petris MJ (2009) A role for the ATP7A copper-transporting ATPase in macrophage bactericidal activity. J Biol Chem 284(49):33949–33956. doi:10.1074/jbc.M109.070201
Arredondo M, Nunez MT (2005) Iron and copper metabolism. Mol Aspects Med 26(4–5):313–327. doi:10.1016/j.mam.2005.07.010
Edvinsson M, Frisk P, Molin Y, Hjelm E, Ilback NG (2008) Trace element balance is changed in infected organs during acute Chlamydophila pneumoniae infection in mice. Biometals 21(2):229–237. doi:10.1007/s10534-007-9114-7
Ramadori G, Van Damme J, Rieder H, Buschenfelde KH Meyer zum (1988) Interleukin 6, the third mediator of acute-phase reaction, modulates hepatic protein synthesis in human and mouse. Comparison with interleukin 1 beta and tumor necrosis factor-alpha. Eur J Immunol 18(8):1259–1264. doi:10.1002/eji.1830180817
Noyce JO, Michels H, Keevil CW (2006) Potential use of copper surfaces to reduce survival of epidemic meticillin-resistant Staphylococcus aureus in the healthcare environment. J Hosp Infect 63(3):289–297. doi:10.1016/j.jhin.2005.12.008
Shirai T, Tsuchiya H, Shimizu T, Ohtani K, Zen Y, Tomita K (2009) Prevention of pin tract infection with titanium-copper alloys. J Biomed Mater Res B Appl Biomater 91(1):373–380. doi:10.1002/jbm.b.31412
Hall TJ, Wren MW, Jeanes A, Gant VA (2009) A comparison of the antibacterial efficacy and cytotoxicity to cultured human skin cells of 7 commercial hand rubs and Xgel, a new copper-based biocidal hand rub. Am J Infect Control 37(4):322–326. doi:10.1016/j.ajic.2008.09.011
Gant VA, Wren MW, Rollins MS, Jeanes A, Hickok SS, Hall TJ (2007) Three novel highly charged copper-based biocides: safety and efficacy against healthcare-associated organisms. J Antimicrob Chemother 60(2):294–299. doi:10.1093/jac/dkm201
Baker J, Sitthisak S, Sengupta M, Johnson M, Jayaswal RK, Morrissey JA (2010) Copper stress induces a global stress response in Staphylococcus aureus and represses sae and agr expression and biofilm formation. Appl Environ Microbiol 76(1):150–160. doi:10.1128/AEM.02268-09
Seeley EH, Caprioli RM (2011) MALDI imaging mass spectrometry of human tissue: method challenges and clinical perspectives. Trends Biotechnol 29(3):136–143. doi:10.1016/j.tibtech.2010.12.002
Sinha TK, Khatib-Shahidi S, Yankeelov TE, Mapara K, Ehtesham M, Cornett DS, Dawant BM, Caprioli RM, Gore JC (2008) Integrating spatially resolved three-dimensional MALDI IMS with in vivo magnetic resonance imaging. Nat Methods 5(1):57–59. doi:10.1038/nmeth1147
Watrous JD, Dorrestein PC (2011) Imaging mass spectrometry in microbiology. Nat Rev Microbiol 9(9):683–694. doi:10.1038/nrmicro2634
Acknowledgments
We thank members of the Skaar laboratory for critically evaluating this manuscript. Work in the Skaar laboratory is supported by National Institutes of Health grants AI0169233, AI073843, and AI091771. E.P.S. is a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Diseases. J.E.C. is supported by National Institutes of Health grant 5T32HD060554-03.
Author information
Authors and Affiliations
Corresponding author
Additional information
This article is published as part of the Special Issue on Immunopathology of staphylococcal infections [34:3].
Rights and permissions
About this article
Cite this article
Cassat, J.E., Skaar, E.P. Metal ion acquisition in Staphylococcus aureus: overcoming nutritional immunity. Semin Immunopathol 34, 215–235 (2012). https://doi.org/10.1007/s00281-011-0294-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-011-0294-4