
Abstract
Background
We previously found that regardless of the animal injury model used resuscitation strategies that minimize fluid administration requirements lead to better outcomes. We hypothesized that a resuscitation regimen that limited the total volume of fluid administered would reduce morbidity and mortality rates in critically ill trauma patients.
Methods
We performed a double-blind randomized trial to assess the safety and efficacy of adding vasopressin to resuscitative fluid. Subjects were hypotensive adults who had sustained acute traumatic injury. Subjects were given fluid alone (control group) or fluid plus vasopressin (experimental group), first as a bolus (4 IU) and then as an intravenous infusion of 200 ml/h (vasopressin 2.4 IU/h) for 5 h.
Results
We randomly assigned 78 patients to the experimental group (n = 38) or the control group (n = 40). The groups were similar in age, sex, preexisting medical illnesses, and mechanism and severity of injury. Serum vasopressin concentrations were higher in the experimental group than in the control group at admission, after infusion of vasopressin (p = 0.01), and 12 h later. The experimental group required a significantly lower total volume of resuscitation fluid over 5 days than did the control group (p = 0.04). The mortality rate at 5 days was 13% in the experimental group and 25% in the control group (p = 0.19). The rates of adverse events, organ dysfunction, and 30-day mortality were similar.
Conclusions
This is the first trial to investigate the impact of vasopressin administration in trauma patients. Infusion of low-dose vasopressin maintained elevated serum vasopressin levels and decreased fluid requirements after injury.
Similar content being viewed by others


Introduction
Improving our understanding of the optimal resuscitation strategy for patients with hemorrhagic shock may help reduce morbidity and mortality rates among civilian and military trauma patients. Vasopressin is an endogenous hormone that is crucial for maintaining vascular tone [1, 2]. Although few studies have demonstrated that vasopressin has a clinical benefit for the resuscitation of patients with hemorrhagic shock [3], the results of numerous clinical investigations support the utility of adjunctive low-dose vasopressin for other critically ill patients, such as those who have experienced out-of-hospital cardiac arrest [4] and those with vasodilatory or septic shock [5]. It is noteworthy that vasopressin does not act as a pressor agent in normal healthy volunteers or in patients with cardiogenic shock (in which vasopressin levels are not diminished) but is effective at very low doses in improving the blood pressure of patients with vasodilatory shock (in which endogenous vasopressin levels are quite low) [6–8].
Investigators have found that vasopressin is an effective pressor during the “irreversible” phase of canine hemorrhagic shock, when animals are unresponsive to both volume replacement and catecholamine vasopressors [9–11]. Using porcine models of hemorrhage after injury, numerous investigators have demonstrated that the infusion of vasopressin dramatically improves outcomes [12–19], whereas others have not found the impact of vasopressin to be favorable [18, 20, 21], Clinical experience is limited to small case series, the results of which neither support nor refute the value of exogenous vasopressin in treating hemorrhage after injury [22–25].
Our primary objective in this study was to evaluate the safety and efficacy of a new low-dose vasopressin resuscitation regimen. We hypothesized that a protocol that minimized the total volume of resuscitation fluid would lead to lower morbidity and mortality rates among critically ill trauma patients.
Methods
Study design
This double-blind, randomized, parallel-group, controlled trial was designed to assess the safety and efficacy of vasopressin as a resuscitative fluid when administered for as long as 5 h for the treatment of shock, beginning with the arrival of the patient in the emergency department. We planned to enroll patients at three Level I trauma centers in San Antonio, Texas, over a 2-year period. We determined that enrolling 165 patients per group would provide 80% power to detect a reduction in 30-day mortality rates from 40% in the control group to 25% in the vasopressin group, assuming two-sided chi-squared testing with a significance level of 0.05. Block randomization was to be performed at each site. However, only one of the three centers (University Hospital, San Antonio) was successful in implementing the study and accruing patients.
Inclusion criteria, randomization, and consent
The study was designed to include men or women at least 18 years old who exhibited clinical evidence of an acute traumatic injury and had a systolic blood pressure lower than 90 mmHg. Patients were not eligible for inclusion in the study if they were (1) admitted to the emergency department more than 6 h after sustaining the traumatic injury; (2) had received more than 4 l of fluid since the injury; (3) were enrolled in another shock trial; (4) were asystolic or required cardiopulmonary resuscitation before they could be randomly assigned to one of the study groups; (5) were pregnant by report or suspicion; (6) were known to have “Do Not Resuscitate” orders or had some visible or identifiable evidence of objection to participation (e.g., an exclusion bracelet); or (7) had known or asserted religious objections to the administration of blood products.
Upon arrival in the emergency department, patients who met all eligibility requirements were randomly assigned to either the experimental group or the control group. A statistician prepared and maintained the master randomization code for the entire trial. A copy of this code was provided to the chairman of the Data Safety Monitoring Board (DSMB) at the start of the trial. The randomization code was not broken for any patient in this study.
Because these patients were trauma victims in shock, they could not grant informed consent for participation in the study; therefore, we enrolled them under the provisions for “exception from informed consent” (in accordance with regulation 21 CFR 50.24, Exception from Informed Consent Requirements for Emergency Research). We had previously developed a system for obtaining community consent for another study of the efficacy of shock treatment, and we adapted these procedures for this protocol. The University of Texas Health Science Center San Antonio institutional review board approved this protocol.
Study procedures
Subjects were evaluated for participation, screened, and enrolled by the principal investigator or designee within 6 h of their arrival at the emergency department. All patients received routine aggressive critical care for their traumatic injuries, including nonoperative care beginning in the prehospital setting, radiologic procedures, and the insertion of at least one additional intravenous line for crystalloid infusion. The standard Advanced Trauma Life Support (ATLS) protocol was utilized for fluid resuscitation and was initiated and managed by the trauma team, which was of course blinded to the type of study drug infused. The typical clinical parameters, such as systolic blood pressure, heart rate, and base deficit, were utilized to guide care.
Infusion of the study drug was initiated within 1 h after the patient’s systolic blood pressure fell to ≤90 mmHg. The patients received a bolus (3 cc) intravenous infusion of either saline alone (placebo) or saline plus vasopressin 4 IU, followed by intravenous infusion of saline alone or saline plus vasopressin (2.4 IU/h) at a rate of 200 ml/h via a dedicated catheter. Study medications for all patients who met inclusion criteria were prepared and labeled by an unblinded pharmacist. The use of vasopressin and desmopressin (DDAVP) was prohibited for 12 h following the study infusion. A record was kept of all potential subjects (in shock upon arrival) who were assessed by emergency department personnel.
The experimental infusion (saline plus vasopressin or saline alone as placebo) was terminated 5 h after administration of the bolus infusion. If at any time during the infusion the patient’s systolic blood pressure was > 160 mmHg continuously for 5 min, the infusion was held up for 20 min. After 20 min, if the systolic blood pressure was not > 160 mmHg, the infusion was resumed at a rate of 100 ml/h (50%). If the systolic blood pressure remained at ≤160 mmHg after an additional 20 min at this lower infusion rate, the infusion was increased to 200 ml/h (100%). Patients were monitored for adequacy of resuscitation, as determined by systolic blood pressure >100 mmHg, heart rate <120 beats per minute, changes in the Glasgow Coma Score (GCS), presence of a palpable peripheral pulse, or changes in the respiratory rate, skin color, and/or temperature. If, on the basis of these clinical signs and symptoms, it was determined that a patient in either group was not adequately responding to the randomly assigned resuscitative fluid, a second intravenous line was placed for infusion of additional fluids. At least one patient evaluation was performed at the end of the study infusion.
Study endpoints
Data regarding the endpoints of resuscitation, mortality, and morbidity were collected from all study patients. The primary safety endpoints were 24-h mortality rates, 30-day mortality rates, and the incidence of durable serious adverse events (SAEs). Secondary safety endpoints were the incidence of other SAEs and AEs, including abdominal compartment syndrome, extremity compartment syndrome, poor neurologic outcome (Glasgow Outcome Score ≤ 8), number of ventilator-free days, intravenous fluid requirements, transfusion requirements (packed red blood cells, fresh frozen plasma, platelets, cryoprecipitate), and vasopressin concentrations (at baseline, after infusion, and at 12 h). Our primary efficacy endpoint for determining whether the standard of care (saline) plus vasopressin was superior to saline alone was the 30-day mortality rate. The secondary efficacy endpoints were 24-h and 5-day mortality rates and the incidence of multiple organ dysfunction syndrome (MODS) score [26] through day 30.
Additional evaluations
For a convenience subset (eight patients from the experimental group and six from the control group), we obtained vasopressin concentrations before administering the bolus of the study drug, immediately after infusing the study drug, and 12 h after the infusion. Vasopressin concentrations were also compared with those of 10 normal referents (laboratory staff members were sampled). Vital signs and laboratory results were recorded through day 7, as required by the institution’s standard of care. MODS scores were collected on study days 3 through 30, while the patient was in the intensive care unit. Ongoing attempts were made to contact a legally authorized representative or family members to notify them of the ongoing study and obtain consent for continued participation.
Data and Safety Monitoring Board
An independent Data and Safety Monitoring Board (DSMB) served as a monitoring body for the study. Other than the study biostatistician, only voting members of the DSMB saw interim analyses of outcome data. The DSMB terminated the study before completion because of lower-than-expected accrual at the only center that was recruiting patients.
Statistical analyses
A group sequential procedure was to have been applied. We planned for three interim analyses of 30-day mortality, to be performed when 33%, 67%, and 100% of the expected number of 107 deaths had been observed. Boundaries for statistical testing were based on the O’Brien and Fleming alpha spending function, with the overall significance level fixed at 0.05. The first decision point in the group sequential procedure was not reached because the DSMB terminated the study after only 22% (n = 24) of the expected number of deaths had occurred. Time-to-event data were analyzed with proportional hazards models and Kaplan–Meier curves; binary data were analyzed with Pearson’s chi-squared test or Fisher’s exact test; and continuously distributed data were analyzed in original or log units with analysis of variance (ANOVA) or repeated-measures linear models. All statistical testing was two-sided with a significance level of 0.05. SAS Version 9.1.3 for Windows (SAS Institute, Cary, NC, USA) was used for all analyses.
Results
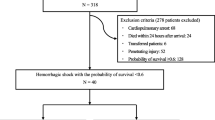
Of the 78 study patients, 38 were assigned to the experimental group and 40 to the control group (Table 1). The mean age of the patients was 47 ± 20 years; 59 (78%) were men; 64 (84%) were white; and 33 (43%) were Hispanic. There were no significant differences between groups in these demographic characteristics (Table 1).
Approximately half of the patients (n = 37, 49%) arrived by air. The primary mechanism of injury in both groups was blunt trauma (experimental group, n = 31, 84%; control group, n = 27, 69%). The history of chronic medical illness (hypertension, diabetes, liver disease, and obesity [body mass index (BMI) > 30 kg/m2] was similar in the two groups (Table 1). A similar number of patients in each group required operation or angioembolization (55% of the experimental group and 58.5% of the control group underwent a major operation; and 7.5% of the experimental group and 4.9% of the control underwent angioembolization). Of the study patients, 69 completed the 30-day follow-up period (experimental group, 32; control group, 37). Patients were enrolled from February 2007 through February 2009.
The mean Injury Severity Score (ISS) of the study patients was high (29 ± 15) (Table 2). The number of patients with Abbreviated Injury Scale (AIS) scores of ≥3, by anatomical region, are shown in Table 2. The only significant difference in AIS scores between the groups was found in the abdominal scores: a significantly larger number (p = 0.045) of patients in the control group had severe abdominal injuries. Except for this score, there were no significant differences between groups in the numbers of patients with severe injuries to the various anatomic regions or with severely depressed mentation (GCS score ≤ 8).
Serum vasopressin concentrations varied between groups across time (Fig. 1 reveals median values). Mean baseline vasopressin concentrations were 15 ± 9 pg/ml for the experimental group and 20 ± 16 pg/ml for the control group (p = 0.70). Immediately after infusion of the study drugs, the vasopressin concentrations were 23 ± 20 pg/ml for the experimental group and 7 ± 7 pg/ml for the control group (p = 0.01); and 12 h after the infusion they were 25 ± 37 pg/ml for the experimental group and 9 ± 13 pg/ml for the control group (p = 0.61). The mean systolic blood pressure was numerically higher in the experimental group following the 5-hour drug infusion than in the control group, but the mean difference between treatments was not statistically significant (p = 0.54).

Vasopressin levels over time, by treatment group. The mean difference between groups immediately after infusion was statistically significant (p = 0.01)
The experimental group required a smaller total amount of resuscitation fluids (crystalloids plus blood and blood products) than did the control group, and the differences became manifest at 6 h (at the end of infusion of the study drugs) (Fig. 2). Although there were no statistically significant differences in total fluids received early in the hospitalization (1, 6, 24, and 48 h), the patients receiving vasopressin required less total fluid than control patients (p = 0.03) and less total blood and blood products (p = 0.04) over the course of the first 120 h. Patients in the experimental group required 13.2 ± 9.8 l of fluid, whereas patients in the control group required 16.0 ± 12.8 l during the first 120 h. The total amount of blood and blood products administered over the first 120 h (5 days) to the experimental group was 3.8 ± 5 l, whereas the amount administered to the control group was 5.4 ± 6.6 l. Massive transfusions were not different in the two groups: 12 (46%) in the experimental group versus 22 (61%) in the control group (p = 0.25).

Volume of fluid administration (p = 0.03) and volume of total blood and blood products (p = 0.04) were significantly different between the control group and the experimental group over the first 120 h (5 days)
The mortality rate 24 h after admission was 13% in the experimental group and 23% in the control group (p = 0.28); and the mortality rate at 5 days was 13% in the experimental group and 25% in the control group (p = 0.19). The mortality rate at 30 days, the primary endpoint of the study, was 34% in the experimental group and 28% in the control group (p = 0.52) (Fig. 3; Tables 3, 4).

Overall survival of patients in the experimental group and control groups did not differ significantly (p = 0.64)
The incidence of MODS was similar in the two groups, affecting nine patients (24%) in the experimental group and seven patients (18%) in the control group (Table 3). The incidence of serious and moderate adverse events and the incidence of any adverse event per subject were also similar in the two groups: 13 (34%) SAEs in the experimental group and 13 (33%) in the control group; 25 (65%) AEs in the experimental group and 22 (55%) in the control group.
Discussion
The principal finding of this study was that low-dose vasopressin infusion maintains elevated serum vasopressin levels and decreases the requirements for fluid administration after injury. The results of this study suggest a possible early survival advantage with the use of vasopressin after severe trauma.
Preclinical studies supporting the utility of vasopressin for treating hemorrhage
Vasopressin (antidiuretic hormone) is produced in response to hemorrhage in rats [1], dogs [2, 27–29], and cats [30]. In most species, vasopressin is generated in response to a decrease in arterial blood pressure, which is primarily related to the activation of various arterial and cardiac receptors [31–33]. The type of fluid resuscitation may affect vasopressin release because the response after saline infusion is much larger than that after hypertonic saline infusion in canine hemorrhage models [34]. In dogs, vasopressin levels decrease after an initial peak apparently because of an increase in clearance rather than a decrease in secretion [35].
Although catecholamines, angiotensin, and vasopressin contribute to the maintenance of vascular tone, and, at least in dogs, angiotensin appears to have little impact on vasopressin responses to hemorrhage [36, 37], vasopressin induces splanchnic vasoconstriction in feline hemorrhagic shock models and is complementary to other endogenous systems that maintain arterial tone [38]. Conscious dogs with hemorrhagic shock require vasopressin for maintaining blood pressure because vasopressin receptor blockade leads to hypotension [39].
Using porcine models of liver injury with uncontrolled hemorrhage, Wenzel and colleagues demonstrated that vasopressin infusions dramatically improve survival rates [12–14]. Preclinical investigations from the principal author’s (S.M.C.) laboratory have also demonstrated the benefits of low doses of vasopressin in trauma resuscitation after a variety of insults leading to shock states (brain, liver, lung, and soft tissue injuries). The infusion of vasopressin into pigs in this brain injury/hemorrhage model minimized the volume of fluid resuscitation required and was associated with markedly improved neurologic outcomes [19], improved brain oxygenation [16], and higher survival rates [17]. Other investigators have found vasopressin comparable to norepinephrine with respect to hemodynamics and brain metabolism in porcine brain injury models [18]. We also found that vasopressin significantly improves cardiopulmonary values after severe blunt chest trauma with hemorrhage [15]. Other experiences with the use of vasopressin in porcine hemorrhage models have not been as promising; some investigators have reported vasopressin-induced adverse effects on metabolic and hemodynamic function [18, 20].
Morales and colleagues found that vasopressin is an effective pressor during the irreversible phase of hemorrhagic shock in dogs unresponsive to both volume replacement and catecholamine vasopressors [9]. These findings were supported by other researchers, who found that vasopressin is superior to catecholamines in supporting hemodynamics after hemorrhage in canine models [10, 11]. The use of vasopressin alone (or any pressor alone) in the absence of resuscitative fluids is associated with poor outcomes [40].
Primate and clinical studies supporting the utility of vasopressin for treating hemorrhage
Unanesthetized monkeys exhibit a large increase in vasopressin concentrations as an immediate response to hemorrhage; the decrease in arterial pressure, rather than hypovolemia, is the prime stimulus for vasopressin liberation [41]. In contrast, other laboratories have suggested that in the conscious primate the sympathetic nervous system is the primary mechanism for controlling blood pressure; vasopressin makes only a minor contribution [42].
Vasopressin is involved in cardiovascular homeostasis and is released in response to hypotension caused by hemorrhage from the neurohypophysis, which results in dramatic increases in the plasma concentration. During the initial phase of hemorrhagic shock, vasopressin (as do other vasoconstrictors such as catecholamines and angiotensin) maintains arterial pressure. As shock worsens, vasopressin concentrations in the plasma decrease, as has been demonstrated in canine models of hemorrhagic shock of 1 h duration. In fact, immunohistochemical analysis of the neurohypophysis in dogs has demonstrated that vasopressin all but disappears after approximately 60 min of severe hemorrhagic hypotension [43]. The mechanism for this vasopressin exhaustion is unclear, but it is known that neurohypophysial stores of vasopressin may be depleted after profound osmotic stimulation and probably also after sustained baroreflex stimulation. The limited information available about vasopressin liberation in humans shows that mild hemorrhage, which does not cause a decrease in arterial blood pressure, is not associated with the liberation of vasopressin [44].
A characteristic of vasopressin that may make it an ideal choice for treating patients with hemorrhagic shock is the fact that it appears to work as a pressor agent in humans by a variety of proposed mechanisms. First, vasopressin exerts little if any pressor effect on normal healthy subjects. Second, vasopressin antagonists cause marked hypotension in hypovolemic subjects. Third, the administration of vasopressin to hypotensive or volume-depleted subjects does not result in a marked pressor response, perhaps because the V-1 receptors on vascular smooth muscle are already occupied by the endogenous hormone released by the baroreflex. Fourth, patients with vasodilatory septic shock (and animals with hemorrhagic shock) are deficient in vasopressin, probably because of a defect in the baroreflex-mediated secretion of vasopressin. Fifth, low doses of exogenous vasopressin yield a hypersensitive pressor response. Sixth, vasopressin potentiates the vasoconstrictor actions of conventional catecholamine vasopressors. Seventh, the administration of exogenous vasopressin not only increases arterial pressure but also appropriately increases serum concentrations of vasopressin. Eighth, desensitization, or down-regulation, of catecholamine α1-adrenergic receptors may occur during septic (and hemorrhagic) shock. Finally, vascular receptor binding by vasopressin may assist in restoring peripheral vascular tone if decreased synthesis, increased degradation, or modification of the catecholamine α1-adrenergic receptors leads to refractory hypotension [45].
Landry and colleagues have further delineated the mechanism of action of vasopressin. In vascular smooth muscle, vasopressin can inhibit both adenosine triphosphate (ATP)-sensitive potassium (KATP) channels and nitric oxide (NO)-induced accumulation of cyclic guanosine monophosphate (cGMP). The activation of KATP channels contributes to various forms of shock, and the activation of NO synthesis appears to contribute to hypotension. Thus, vasopressin inhibits vasodilatory mechanisms that contribute to both hypotension and vascular hyporeactivity during the late phase of hemorrhagic shock. A second factor that is believed to contribute to the pressor effectiveness of exogenous vasopressin in hemorrhagic shock is inappropriately low levels of plasma vasopressin, which may be caused by depletion of secretory stores in the neurohypophysis [9]. An excellent two-part review of vasopressin receptors and clinical physiology was recently published [46, 47].
Although few published studies have reported that vasopressin is beneficial for the resuscitation of patients with hemorrhagic shock, the results of numerous clinical investigations support the effectiveness of adjunctive low-dose vasopressin for other critically ill patients, such as those experiencing out-of-hospital cardiac arrest [4] and those with vasodilatory or septic shock [5]. Vasopressin has not been shown to be beneficial, however, for patients who experience in-hospital cardiac arrest [48]. Wenzel and colleagues reported that vasopressin is superior to epinephrine (the previous standard of care) in resuscitating patients who have experienced out-of-hospital cardiopulmonary arrest [4]. Patel and colleagues found that patients with septic shock who were taking norepinephrine when they were randomly assigned to receive vasopressin (but not placebo) could be spared the administration of conventional vasopressors [6]. In a prospective, randomized, controlled study, Dünser and coworkers studied arginine vasopressin as a treatment for advanced vasodilatory shock [5]. For patients with catecholamine-resistant vasodilatory shock, arginine vasopressin with norepinephrine was found to be superior to norepinephrine alone. Subsequently, in a large multicenter randomized trial comparing vasopressin to norepinephrine infusion in patients with septic shock, Russell and colleagues demonstrated that low-dose vasopressin reduced mortality rates from 35.7 to 26.5% (p = 0.05) in a prospectively defined stratum of less-severe septic shock (approximately half of their patients) [49].
A few case studies (totaling 17 patients) have reported various levels of clinical success in association with the administration of low-dose vasopressin to aid in the resuscitation of critically ill patients after massive hemorrhage that is refractory to other treatments [22–25]. Wenzel and colleagues reviewed the potential benefit of vasopressin in hemorrhagic shock and described the planned “VITRIS” trial [50]. In this multicenter European VITRIS trial, adult trauma patients with presumed hemorrhagic shock who do not respond to the physician-directed standard of care (intubation, fluid resuscitation, and catecholamine vasopressors) administered for 10 min at the scene the injury are randomly assigned to receive as many as three injections of either 10 IU of vasopressin or a placebo. The primary study endpoint will be the hospital admission rate.
Who experiences vasopressin deficiency?
Some researchers have suggested that vasopressin deficiency contributes to the syndrome of irreversible shock [51]. Unfortunately, few findings have been published about vasopressin after hemorrhage in humans, probably because of the complexity of the assay and the difficulty involved in performing studies on trauma patients. Westermann and colleagues sampled the blood of trauma patients upon arrival at the emergency department and then at 4, 6, and 24 h after admission [52]. The concentration of vasopressin, which is normally quite low in healthy control subjects (0.92 ± 0.38 pg/ml) [53], was elevated upon admission (43.2 ± 84.9 pg/ml) and then decreased over the ensuing 24 h. Westermann and colleagues did not characterize which patients experienced a vasopressin deficiency (i.e., vasopressin liberation was ineffective in maintaining perfusion) and therefore did not attempt to determine which patients were likely to respond to the administration of exogenous vasopressin. In subsequent studies, we plan to characterize the endogenous production of various vasoactive mediators so we can better understand the factors that lead to vasopressin deficiency.
It is unclear whether all of our patients exhibited vasopressin insufficiency at baseline. Although initial vasopressin concentrations were 10 times normal, these concentrations were approximately one-fourth of that expected in response to acute injury [41]. Although low-dose vasopressin successfully maintained baseline vasopressin concentrations and was associated with a decrease in total fluid requirements, it is unclear whether the amount administered was in fact optimal. The lack of a sustained impact on 30-day mortality rates or the incidence of organ dysfunction was surprising, considering the fact that the mortality rate was approximately 50% lower in the vasopressin group than in the control group at 1 and 5 days after injury.
This study had several limitations. First, our sample size was small, and we were forced to terminate the trial early because of accrual problems. The two other trauma centers were unable to obtain institutional reviews board approval during a reasonable period of time owing to the additional regulatory issues inherent in military facilities. The tremendous study cost incurred (round-the-clock, in-hospital research coordinators) and the slow accrual rate at the single trauma center enrolling patients led the DSMB to terminate the study. It was determined that completion of the study would have stretched over many years and considerably exceeded the available funding resources. However, even though our population objectives were not met, we were able to make some interesting observations.
Second, we could not demonstrate the safety of vasopressin therapy because of our limited sample size. Although we did not detect even a trend toward an increased incidence of adverse events in the vasopressin group, it is possible that even low-dose vasopressin may have harmed some patients. Some retrospective reviews have suggested that the early use of vasopressors during resuscitation is deleterious and may increase mortality rates [54] or the incidence of complications such as ischemic skin lesions [55]. Certainly, sicker patients would typically be the ones who would receive vasoactive medications when the standard fluid resuscitation measures fail to work.
Third, we did not control our analyses for the use of steroids, which have been recently associated with decreased mortality rates and a decreased incidence of organ dysfunction when used in conjunction with low-dose vasopressin in the setting of septic shock [56]. Steroid infusion is rarely used in the care of trauma patients unless adrenal insufficiency is suspected.
Fourth, the dose and duration of vasopressin infusion used in this study may have been insufficient to overcome vasopressin insufficiency because recent findings have suggested that a higher dose may be more effective for patients in vasodilatory shock [57]. However, we were able to reduce the volume of fluid infused.
Finally, we did not take into account the diurnal variation in endogenous vasopressin production by varying our resuscitation regimen according to the time of day [58]. A subsequent larger study is needed to determine the impact of time of day on vasopressin levels and subsequent outcome.
Conclusions
This is the first trial to investigate the impact of vasopressin administration in trauma patients. Our double-blind randomized clinical trial demonstrated that the infusion of low-dose vasopressin could maintain elevated serum vasopressin concentrations and decreased fluid requirements after injury. The administration of vasopressin was associated with a possible early survival advantage; this finding suggests the need for a larger, adequately powered, multicenter trial to determine the potential benefits of this hormone in treating critically ill trauma patients.
References
Brown LM, Ginsburg M (1956) Effect of anaesthetics and haemorrhage on the release of neurohypophysial antidiuretic hormone. Br J Pharmacol Chemother 11:236–244
Weinstein H, Berne RM, Sachs H (1960) Vasopressin in blood: effect of hemorrhage. Endocrinology 66:712–718
Cohn SM (2007) Potential benefit of vasopressin in resuscitation of hemorrhagic shock. J Trauma 62:S56–S57
Wenzel V, Krismer AC, Arntz HR et al (2004) A comparison of vasopressin and epinephrine for out-of-hospital cardiopulmonary resuscitation. N Engl J Med 350:105–113
Dünser MW, Mayr AJ, Ulmer H et al (2003) Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation 107:2313–2319
Patel BM, Chittock DR, Russell JA et al (2002) Beneficial effects of short-term vasopressin infusion during severe septic shock. Anesthesiology 96:576–582
Landry DW, Levin HR, Gallant EM et al (1997) Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med 25:1279–1282
Sharshar T, Blanchard A, Paillard M et al (2003) Circulating vasopressin levels in septic shock. Crit Care Med 31:1752–1758
Morales D, Madigan J, Cullinane S et al (1999) Reversal by vasopressin of intractable hypotension in the late phase of hemorrhagic shock. Circulation 100:226–229
Yoo JH, Kim MS, Park HM (2006) Hemodynamic characteristics of vasopressin in dogs with severe hemorrhagic shock. J Vet Med Sci 68:967–972
Yoo JH, Kim MS, Eom KD et al (2007) Vasopressor therapy using vasopressin prior to crystalloid resuscitation in irreversible hemorrhagic shock under isoflurane anesthesia in dogs. J Vet Med Sci 69:459–464
Voelckel WG, Raedler C, Wenzel V et al (2003) Arginine vasopressin, but not epinephrine, improves survival in uncontrolled hemorrhagic shock after liver trauma in pigs. Crit Care Med 31:1160–1165
Stadlbauer KH, Wagner-Berger HG, Raedler C et al (2003) Vasopressin, but not fluid resuscitation, enhances survival in a liver trauma model with uncontrolled and otherwise lethal hemorrhagic shock in pigs. Anesthesiology 98:699–704
Raedler C, Voelckel WG, Wenzel V et al (2004) Treatment of uncontrolled hemorrhagic shock after liver trauma: fatal effects of fluid resuscitation versus improved outcome after vasopressin. Anesth Analg 98:1759–1766
Feinstein AJ, Cohn SM, King DR et al (2005) Early vasopressin improves short-term survival after pulmonary contusion. J Trauma 59:876–882 (discussion 82–83)
Dudkiewicz M, Proctor KG (2008) Tissue oxygenation during management of cerebral perfusion pressure with phenylephrine or vasopressin. Crit Care Med 36:2641–2650
Sanui M, King DR, Feinstein AJ et al (2006) Effects of arginine vasopressin during resuscitation from hemorrhagic hypotension after traumatic brain injury. Crit Care Med 34:433–438
Meybohm P, Cavus E, Bein B et al (2007) Small volume resuscitation: a randomized controlled trial with either norepinephrine or vasopressin during severe hemorrhage. J Trauma 62:640–646
Feinstein AJ, Patel MB, Sanui M et al (2005) Resuscitation with pressors after traumatic brain injury. J Am Coll Surg 201:536–545
Johnson KB, Pearce FJ, Jeffreys N et al (2006) Impact of vasopressin on hemodynamic and metabolic function in the decompensatory phase of hemorrhagic shock. J Cardiothorac Vasc Anesth 20:167–172
Meybohm P, Cavus E, Dorges V et al (2008) Release of protein S100B in haemorrhagic shock: effects of small volume resuscitation combined with arginine vasopressin. Resuscitation 76:449–456
Shelly MP, Greatorex R, Calne RY et al (1988) The physiological effects of vasopressin when used to control intra-abdominal bleeding. Intensive Care Med 14:526–531
Sharma RM, Setlur R (2005) Vasopressin in hemorrhagic shock. Anesth Analg 101:833–834
Tsuneyoshi I, Onomoto M, Yonetani A et al (2005) Low-dose vasopressin infusion in patients with severe vasodilatory hypotension after prolonged hemorrhage during general anesthesia. J Anesth 19:170–173
Krismer AC, Wenzel V, Voelckel WG et al (2005) Employing vasopressin as an adjunct vasopressor in uncontrolled traumatic hemorrhagic shock: three cases and a brief analysis of the literature. Anaesthesist 54:220–224
Marshall JC, Cook DJ, Christou NV et al (1995) Multiple organ dysfunction score: a reliable descriptor of a complex clinical outcome. Crit Care Med 23:1638–1652
Share L (1968) Control of plasma ADH titer in hemorrhage: role of atrial and arterial receptors. Am J Physiol 215:1384–1389
Rocha ESM Jr, Rosenberg M (1969) The release of vasopressin in response to haemorrhage and its role in the mechanism of blood pressure regulation. J Physiol 202:535–557
Ota K, Kimura T, Matsui K et al (1988) Effects of hemorrhage on vasopressin and Met-Enk releases in blood and cerebrospinal fluid in dogs. Am J Physiol 255:R731–R736
Beleslin D, Bisset GW, Haldar J et al (1967) The release of vasopressin without oxytocin in response to haemorrhage. Proc R Soc B 166:443–458
Claybaugh JR, Share L (1973) Vasopressin, renin, and cardiovascular responses to continuous slow hemorrhage. Am J Physiol 224:519–523
Shen YT, Cowley AW Jr, Vatner SF (1991) Relative roles of cardiac and arterial baroreceptors in vasopressin regulation during hemorrhage in conscious dogs. Circ Res 68:1422–1436
Thrasher TN, Keil LC (1998) Arterial baroreceptors control blood pressure and vasopressin responses to hemorrhage in conscious dogs. Am J Physiol 275:R1843–R1857
Saito T, Yoshida S (1971) Levels of antidiuretic hormone in plasma after hemorrhage and infusion of hypertonic saline in dogs. Endocrinology 88:1511–1513
Errigton ML, Rocha e Silva M Jr (1971) The secretion and clearance of vasopressin during the development of irreversible haemorrhagic shock. J Physiol 217:43P–45P
Shade RE, Share L (1975) Vasopressin release during nonhypotensive hemorrhage and angiotensin II infusion. Am J Physiol 228:149–154
Cowley AW Jr, Switzer SJ, Guinn MM (1980) Evidence and quantification of the vasopressin arterial pressure control system in the dog. Circ Res 46:58–67
Hock CE, Su JY, Lefer AM (1984) Role of AVP in maintenance of circulatory homeostasis during hemorrhagic shock. Am J Physiol 246:H174–H179
Schwartz J, Reid IA (1981) Effect of vasopressin blockade on blood pressure regulation during hemorrhage in conscious dogs. Endocrinology 109:1778–1780
Driessen B, Zarucco L, Gunther RA et al (2007) Effects of low-volume hemoglobin glutamer-200 versus normal saline and arginine vasopressin resuscitation on systemic and skeletal muscle blood flow and oxygenation in a canine hemorrhagic shock model. Crit Care Med 35:2101–2109
Arnauld E, Czernichow P, Fumoux F et al (1977) The effects of hypotension and hypovolaemia on the liberation of vasopressin during haemorrhage in the unanaesthetized monkey (Macaca mulatta). Pflugers Arch 371:193–200
Cornish KG, Barazanji MW, Iaffaldano R (1990) Neural and hormonal control of blood pressure in conscious monkeys. Am J Physiol 258:H107–H112
Landry DW, Oliver JA (2001) The pathogenesis of vasodilatory shock. N Engl J Med 345:588–595
Goetz KL, Bond GC, Smith WE (1974) Effect of moderate hemorrhage in humans on plasma ADH and renin. Proc Soc Exp Biol Med 145:277–280
Malay MB, Ashton RC Jr, Landry DW et al (1999) Low-dose vasopressin in the treatment of vasodilatory septic shock. J Trauma 47:699–703 (discussion 703–705)
Holmes CL, Landry DW, Granton JT (2003) Science review: vasopressin and the cardiovascular system. Part 1. Receptor physiology. Crit Care 7:427–434
Holmes CL, Landry DW, Granton JT (2004) Science review: vasopressin and the cardiovascular system. Part 2. Clinical physiology. Crit Care 8:15–23
Stiell IG, Hebert PC, Wells GA et al (2001) Vasopressin versus epinephrine for inhospital cardiac arrest: a randomised controlled trial. Lancet 358:105–109
Russell JA, Walley KR, Singer J et al (2008) Vasopressin versus norepinephrine infusion in patients with septic shock. N Engl J Med 358:877–887
Wenzel V, Raab H, Dunser MW (2008) Arginine vasopressin: a promising rescue drug in the treatment of uncontrolled haemorrhagic shock. Best Pract Res Clin Anaesthesiol 22:299–316
Robin JK, Oliver JA, Landry DW (2003) Vasopressin deficiency in the syndrome of irreversible shock. J Trauma 54:S149–S154
Westermann I, Dunser MW, Haas T et al (2007) Endogenous vasopressin and copeptin response in multiple trauma patients. Shock 28:644–649
Jochberger S, Mayr VD, Luckner G et al (2006) Serum vasopressin concentrations in critically ill patients. Crit Care Med 34:293–299
Sperry JL, Minei JP, Frankel HL et al (2008) Early use of vasopressors after injury: caution before constriction. J Trauma 64:9–14
Dunser MW, Mayr AJ, Tur A et al (2003) Ischemic skin lesions as a complication of continuous vasopressin infusion in catecholamine-resistant vasodilatory shock: incidence and risk factors. Crit Care Med 31:1394–1398
Russell JA, Walley KR, Gordon AC et al (2009) Interaction of vasopressin infusion, corticosteroid treatment, and mortality of septic shock. Crit Care Med 37:811–818
Luckner G, Mayr VD, Jochberger S et al (2007) Comparison of two dose regimens of arginine vasopressin in advanced vasodilatory shock. Crit Care Med 35:2280–2285
George CP, Messerli FH, Genest J et al (1975) Diurnal variation of plasma vasopressin in man. J Clin Endocrinol Metab 41:332–338
Acknowledgments
This trial could not have been conducted without the support of Michael Given, PhD and the Office of Naval Research (grant W81XWH-08-1-0013). The authors are also grateful for the support of J. Hillman, B. Miller, and J. Palacios, University Hospital Pharmacy; the nursing staff of the University Hospital Emergency Center, Operating Room, and Surgical Trauma Intensive Care Unit. We also thank L. Kirkman (University Hospital Corporate Communications), R. Holloway (Webmaster), S. Smith, and L. Kelly. C. Wade and L. Baer performed the vasopressin assays at the Army Institute of Surgical Research, San Antonio, Texas.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
In addition to the authors on the title page, the following individuals and institutions participated in the trial as members of the UTHSCSA Clinical Trials Group.
-
Study design: M.G. Corneille, J.G. Myers
-
Data acquisition: M.G. Corneille, J.G. Myers, D. Mueller, P.P. Lopez, S.E. Wolf, G.G. Goodwiler, B.J. Eastridge, J.D. Gourlas, S. Granger, J. Bini, J. Oh
-
Data Safety Management Board: B.A. Pruitt (Medical Monitor); D. Sessler (Chair); F. Moore, B. Pollock, A. Anzueto, K. Grathwohl
-
Research coordinators: M. DeRosa, C. Brougher, M.R. Gildea, R. Sambucini, S. Hargis, D. Oakes
-
Informatics and biostatistical support: B. Sanns, C. Louden, L. Sanchez
Rights and permissions
About this article
Cite this article
Cohn, S.M., McCarthy, J., Stewart, R.M. et al. Impact of Low-dose Vasopressin on Trauma Outcome: Prospective Randomized Study. World J Surg 35, 430–439 (2011). https://doi.org/10.1007/s00268-010-0875-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00268-010-0875-8