
Abstract
Vasopressin is emerging as a rational therapy for vasodilatory shock states. In part 1 of the review we discussed the structure and function of the various vasopressin receptors. In part 2 we discuss vascular smooth muscle contraction pathways with an emphasis on the effects of vasopressin on ATP-sensitive K+ channels, nitric oxide pathways, and interaction with adrenergic agents. We explore the complex and contradictory studies of vasopressin on cardiac inotropy and coronary vascular tone. Finally, we summarize the clinical studies of vasopressin in shock states, which to date have been relatively small and have focused on physiologic outcomes. Because of potential adverse effects of vasopressin, clinical use of vasopressin in vasodilatory shock should await a randomized controlled trial of the effect of vasopressin's effect on outcomes such as organ failure and mortality.
Similar content being viewed by others


Introduction
Vasopressin is a hormone that is essential for both osmotic and cardiovascular homeostasis. A deficiency in vasopressin exists in some shock states and replacement of physiologic levels of vasopressin can restore vascular tone. Vasopressin is therefore emerging as a rational therapy for shock. Preliminary studies [1–12] show that infusion of low-dose vasopressin in patients who have vasodilatory shock decreases norepinephrine (noradrenaline) dose requirements, maintains blood pressure and cardiac output, decreases pulmonary vascular resistance, and increases urine output. Thus, low-dose vasopressin could improve renal and other organ function in septic shock. Paradoxically, vasopressin has also been demonstrated to cause vasodilation in some vascular beds, distinguishing this hormone from other vasoconstrictor agents.
The present review explores the vascular actions of vasopressin. In part 1 of the review we discussed the signaling pathways, distribution of vasopressin receptors, and the structural elements responsible for the functional diversity found within the vasopressin receptor family. We now explore the mechanisms of vasoconstriction and vasodilation of the vascular smooth muscle, with an emphasis on vasopressin interaction in these pathways. We discuss the seemingly contradictory studies and some new information regarding the actions of vasopressin on the heart. Finally, we summarize the clinical trials of vasopressin in vasodilatory shock states and comment on areas for future research.
Vascular smooth muscle contraction pathways and vasopressin interaction
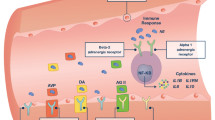
Vasopressin restores vascular tone in vasoplegic (catecholamine-resistant) shock states by at least four known mechanisms [13]: through activation of V1 vascular receptors (V1Rs); modulation of ATP-sensitive K+ channels (KATP); modulation of nitric oxide (NO); and potentiation of adrenergic and other vasoconstrictor agents. A short discussion of vascular smooth muscle contraction pathways is necessary to understand the interaction of vasopressin.
All muscle cells use calcium as a signal for contraction. Vascular smooth muscle cells are regulated by a variety of neurotransmitters and hormones; these interact with a network of signal transduction pathways that ultimately affect contractility either by affecting calcium levels in the cell or the response of the contractile apparatus to calcium. Calcium levels are increased by extracellular entry via voltage-gated calcium channels and by release from intracellular stores. At high cytosolic concentrations, calcium forms a complex with calmodulin that activates a kinase, which phosphorylates the regulatory light chain of myosin. Phosphorylated myosin activates myosin ATPase by actin and the cycling of myosin cross-bridges along actin filaments, which contracts the muscles. Vasodilation occurs when a kinase interacts with myosin phosphatase, which dephosphorylates myosin and prevents muscle contraction [14].
Vasopressin, norepinephrine, and angiotensin II act on cell surface receptors that couple with G-proteins to effect vasoconstriction. Vasopressin interacts with V1Rs, which are found in high density on vascular smooth muscle, through the Gq/11 pathway to stimulate phospholipase C and produce the intracellular messengers inositol trisphosphate (IP3) and diacylglycerol. These second messengers then activate protein kinase C and elevate intracellular free calcium to initiate contraction of vascular smooth muscle. In contrast, vasodilators such as atrial natriuretic peptide (ANP) and NO activate a cGMP-dependent kinase that, by interacting with myosin phosphatase, dephosphorylates myosin and thus prevents muscle contraction [14]. The opposing influences of these pathways are important in determining the functional state of vascular smooth muscle, and integration of this signaling is a key component in vascular homeostasis [15].
A key mechanism by which vascular smooth muscle tone is controlled is through K+ channels [16]. The resting membrane potential of vascular smooth muscle ranges from -30 mV to -60 mV. A more positive potential (depolarization) opens voltage-gated calcium channels, increasing cytosolic Ca2+ concentration, and induces vasoconstriction. Conversely, hyperpolarization closes these channels, decreases cytosolic Ca2+ concentration, and induces vasodilation [13]. The membrane potential of vascular smooth muscle is controlled by a number of ion transporters and channels, particularly K+ channels. The opening of K+ channels allows an efflux of potassium, thus hyperpolarizing the plasma membrane and preventing entry of calcium into the cell [16], even in the presence of vasoconstrictor agents [17].
Four types of K+ channels have been described (Table 1) [16]. Of these, the KATP channel is the best understood and plays a critical role in disease states such as vasodilatory shock. KATP channels are physiologically activated by decreases in cellular ATP and by increases in the cellular concentrations of hydrogen ion and lactate [18, 19]. This activation prevents opening of voltage-gated Ca2+ channels and contributes to the vasoplegia (resistance to catecholamines) that is seen in shock states.
Activation of KATP channels is a critical mechanism in the hypotension and vasodilation that are characteristic of vasodilatory shock. Agents that close KATP channels (such as sulfonylureas) have been shown to increase arterial pressure and vascular resistance in vasodilatory shock due to hypoxia [20], in septic shock [20–22], and in the late, vasodilatory phase of hemorrhagic shock [23]. An important mechanism by which vasopressin restores vascular tone in vasoplegic (catecholamine-resistant) shock states may be its ability to close KATP channels [24].
Another mechanism by which vasopressin exerts vascular control is through modulation of NO. The latter contributes to the hypotension and resistance to vasopressor drugs that occurs in vasodilatory shock. The vasodilating effect of NO is mediated mainly by the activation of myosin light-chain phosophatase. However, NO also activates K+ channels in the vascular smooth muscle [25, 26]. Agents that block NO synthesis during septic shock increase arterial pressure and decrease the doses of vasoconstrictor catecholamines needed to maintain arterial pressure [27]. Vasopressin may restore vascular tone in vasodilatory shock states by blunting the increase in cGMP that is induced by NO [28] and ANP [29], and by decreasing the synthesis of inducible nitric oxide synthase (NOS) that is stimulated by lipopolysaccharide [28]. This inhibition occurs via the V1R [30, 31].
Vasopressin potentiates the vasoconstrictor effects of many agents, including norepinephrine [32, 33] and angiotensin II [34–36]. The underlying mechanism of this is unknown but possibilities include coupling between G-protein-coupled receptors [36], interaction between G-proteins, and interference with G-protein-coupled receptor downregulation through arrestin trafficking.
Vasopressin has been demonstrated to cause vasodilation in numerous vascular beds [37–44] – a feature not shared by other vasoconstrictor agents. The mechanism of vasodilation has been demonstrated to be due to activation of endothelial oxytocin receptors (OTRs) [45], which in turn trigger activation of endothelial isoforms of NOS.
Whether vasopressin causes vasoconstriction or vasodilation depends on the vascular bed studied [46], which may, in turn, depend on the receptor density (V1R versus OTR), the model studied, the dose of vasopressin [47], and the duration of exposure to the hormone [48]. Indeed, the opposing influences of various pathways that determine the functional state of vascular smooth muscle is an area for further study. For example, prolonged exposure to cAMP inhibits both angiotensin II and vasopressin-stimulated phosphoinositide hydrolysis and intracellular calcium mobilization [49]. Adenylyl cyclases present a focal point for signal integration in vascular smooth muscle, and type III adenylyl cyclase has been proposed as a key subtype for cross-talk between constrictor and dilator pathways [50]. The important question is whether vasopressin can cause simultaneous vasoconstriction of some vascular beds and vasodilation of others.
Vasopressin and the heart
The actions of vasopressin on the heart are complex and the studies are seemingly contradictory. Depending on the species studied, the dose used, and the experimental model, vasopressin can cause coronary vasoconstriction or vasodilation and exert positive or negative inotropic effects. In addition to its vascular effects on coronary blood flow, vasopressin also has mitogenic and metabolic effects on the heart.
Coronary vascular tone
The effect of vasopressin on the coronary vascular bed is controversial. Several investigators have demonstrated a V1R-mediated coronary vasoconstrictor response to vasopressin [51–54] – an effect that appears to be dose dependent [55, 56] and intensified by removal of endothelium [46]. In contrast, coronary vasodilation in response to vasopressin has been demonstrated in isolated canine [57, 58] and primate [44] coronary arteries. More recently, vasopressin was demonstrated to cause coronary vasodilation in an intact animal model. A bolus injection of vasopressin significantly increased the vascular diameter of the left anterior descending artery in pigs [59]. This vasodilation was present during sinus rhythm, ventricular fibrillation, and after successful cardiopulmonary resuscitation. Vasopressin probably effects coronary vasodilation through control of endothelial tone [58], as has been demonstrated in the pulmonary vasculature [39].
A difference between the 'normal' and stressed heart in their responses to vasopressin has been reported, with vasoconstriction seen in normoxic state and vasodilation seen during hypoxia [60]. Using an isolated working rat heart model, high-dose vasopressin (777 ± 67 pg/ml) reduced coronary flow by 38.4 ± 2.6% in normoxic hearts. Myocardial function was also significantly decreased by vasopressin. In contrast, the same dose of vasopressin administered to hypoxic hearts resulted in a smaller decrease in coronary blood flow (-11.5 ± 2.8%) and an improvement in myocardial function. Interestingly, in hearts treated first with vasopressin and then with hypoxia, there was a greater degree of coronary vasodilation as compared with that observed in hearts treated with hypoxia alone. These results indicate that the vasoconstrictor effect of vasopressin on the coronary vessels, as well as its effect on the myocardium, may be dependent on oxygen tension and possibly on the redox state of the cell. In addition, vasopressin-constricted vessels appear to retain considerable vasodilatory reserve, despite evidence of ischemic conditions [60].
Several preclinical studies have evaluated vasopressin in animal models of cardiac arrest [61–64]. These studies suggested that vasopressin leads to superior resuscitation rates as compared with epinephrine (adrenaline). The improvement in restoration of spontaneous circulation is partially ascribed to an improvement in coronary blood flow [65]. However, in the setting of cardiac arrest, the improvement in coronary blood flow is probably mediated by an improvement in coronary perfusion pressure as opposed to vasopressin-mediated coronary vasodilation.
Inotropy
Studies of the inotropic effects of vasopressin are also controversial, and the effects appear to depend on the dose used and the model studied. In a study of an isolated working rat heart model, investigators found that high-dose vasopressin (878 pg/ml) produced significant decreases in coronary flow, myocardial oxygen consumption and left ventricular peak systolic pressure, and a small decrease in cardiac output [55]. Similarly, intracoronary infusion of vasopressin-dextran (a method employed to keep the vasopressin in the vascular compartment) in isolated perfused guinea pig hearts caused coronary vasoconstriction and negative inotropy – effects that were blocked with vasopressin antagonists and P2 purinergic receptor antagonist [66]. These results were duplicated in conscious dogs, in which an infusion of low-dose vasopressin (15 pg/ml) caused significant increases in left ventricular end-systolic pressure, end-systolic volume, total systemic resistance, and arterial elastance, whereas the heart rate and stroke volume were decreased. There was no significant change in coronary sinus blood flow. Vasopressin decreased the slope of the left ventricular end-systolic pressure–volume relation, the maximal first derivative of left ventricular pressure/end-diastolic volume relation, and the stroke work–ventricular end-diastolic relation, and shifted the relations to the right, indicating a depression of left ventricular performance [67]. The relevance of these observations in the setting of vasodilatory shock in humans, however, is not known.
It is often difficult to isolate the effects of vasopressin on inotropy from its effects on coronary blood flow. Indeed, when attempts were made to study the effects of vasopressin on the heart independently of coronary blood flow, the effects of vasopressin on inotropy were strikingly different. By maintaining constant coronary flow, the direct cardiac effects of vasopressin on an isolated rat heart preparation were determined, independent of changes in myocardial oxygen delivery elicited by coronary vasoconstriction [56]. Myocardial function was assessed at vasopressin concentrations of 0, 10, 25, 50, 100, 200, 400, and 500 pg/ml. Progressive coronary vasoconstriction was observed with increasing vasopressin concentration. In contrast, peak ventricular pressure and the first derivative of left ventricular pressure (dP/dtmax) increased at 50 and 100 pg/ml vasopressin but fell at 400 and 500 pg/ml. The maximal peak ventricular pressure and dP/dtmax responses were at 50 pg/ml, whereas at 500 pg/ml both peak ventricular pressure and dP/dtmax were reduced below control. Pretreatment with a specific V1R antagonist totally blocked both the coronary vasoconstrictor and contractility responses to vasopressin. These data suggest that, although vasopressin causes dose-related coronary vasoconstriction and negative inotropy at high vasopressin concentrations, the hormone may exert a net positive inotropic effect at low doses. It appears that the net effect of vasopressin on cardiac function in an intact preparation will depend on the concentration of vasopressin as well as on the relative balance of its effects on coronary perfusion pressure (diastolic blood pressure), coronary vascular tone, and any direct effects on the inotropic state of the myocardium.
The clinical observation that vasopressin greatly increases afterload in vasodilatory shock (systemic vascular resistance [SVR] nearly doubles) but depresses cardiac output relatively little (14%) led to speculation that vasopressin at low doses might have positive inotropic effects [3]. Furthermore, in a small trial of vasopressin in patients with heart failure and vasodilatory hypotension due to the phosphodiesterase inhibitor milrinone, vasopressin increased SVR but did not depress cardiac output [68], again suggesting a positive inotropic action. However, these conclusions are speculative because it is difficult to isolate the effects of vasopressin on contractility from its effects on coronary perfusion, heart rate, and ventricular preload. Of more importance is the net clinical benefit of these often contradictory actions. An observational study conducted in critically ill humans specifically examined the effects of low-dose vasopressin infusion on hemodynamics and cardiac performance [69]. In 41 patients with catecholamine-resistant postcardiotomy shock, continuous infusion of vasopressin was associated with a significant increase in left ventricular stroke work index and a significant decrease in heart rate, as well as vasopressor and inotropic requirements. Cardiac index and stroke volume remained unchanged despite a significant reduction in the requirement for inotropic agents. Interestingly, myocardial enzymes significantly fell in all patients and many patients with atrial arrhythmias converted on infusion. The authors concluded that low-dose vasopressin improved myocardial performance in this group of patients.
Classically, the effects of vasopressin on the heart were thought to be mediated through the V1R (vascular smooth muscle/calcium-dependent effect) or OTR (endothelial/NO effect). Neonatal rat cardiomyocytes possess V1Rs [70], and vasopressin causes a dose-dependent increase in intracellular calcium, which is dependent on extracellular magnesium and calcium concentrations, secondary to V1R activation and phospholipase-mediated IP3 generation [71]. The V1R also mediates prostacyclin and ANP release from cultured rat cardiomyocytes exposed to vasopressin [72]. OTRs were also identified in isolated rat heart, and oxytocin causes increased ANP release in perfused rat heart preparations [73]. The negative inotropic and chonotropic effects of oxytocin may be mediated by these cardiac OTRs. Blockade of cholinergic receptors and NO production attenuated the negative effects of oxytocin on cardiac function [74]. More recently it was suggested that the cardiac effects of vasopressin are due to selective activation of intravascular purinoceptors and that an intermediary of these effects is ATP [66]. Indeed, adenoviral gene transfer of the V2 renal receptor (V2R) into cardiomyocytes was shown to modulate the endogenous cAMP signal cascade and increase contractility of rat cardiomyocytes [75].
In the setting of primary cardiac dysfunction, however, it is the effect of vasopressin on SVR that may counter any potential beneficial effects on cardiac inotropy. Indeed, antagonism of vasopressin receptors has been advocated as therapy for congestive heart failure; both animal models of congestive heart failure and early clinical studies support the notion that antagonism of V1Rs and V2Rs leads to an improvement in cardiac function, probably mediated through reductions in cardiac afterload [76–78].
Cardiac hypertrophy
Vasopressin promotes cardiac hypertrophy in neonatal rat hearts via direct effects on cardiomyocyte protein synthesis secondary to IP3-mediated intracellular calcium release [79]. In the adult rat heart, vasopressin directly increased the rate of protein synthesis via the V1R, which was sensitive to amiloride – a mechanism that differs from the cAMP-dependent mechanism that is responsible for the cardiac hypertrophy induced by pressure overload [80].
Summary
V1R-mediated coronary vasoconstriction is a dose-dependent phenomenon that may be attenuated by the endothelial vasodilating properties of vasopressin action via the OTR or P2 purinergic receptor. When cardiac contractility is studied independently of coronary perfusion, vasopressin may have a positive inotropic effect at low doses. Further work is necessary to determine the significance of these observations in human hearts in both health and disease states.
Clinical application of vasopressin in shock
In health, vasopressin's role in the maintenance of resting arteriolar tone and systemic blood pressure is minor. Indeed, high concentrations of vasopressin are required before vasoconstrictor effects are seen. It is only during shock states that vasopressin's role in the maintenance of systemic blood pressure is seen. Indeed, vasopressin deficiency and hypersensitivity to the hormone's pressor effects appear to be a hallmark of vasodilatory shock states [13]. These states include vasodilatory septic shock [1–5], vasodilatory shock post-cardiopulmonary bypass [6–9, 81], vasodilatory shock due to phosphodiesterase inhibition in the treatment of heart failure [12, 68], hemodynamically unstable organ donors [11], and the late, so-called 'irreversible' phase of volume treated hemorrhagic shock [82]. The reason for the reduction in circulating concentration of vasopressin has not been fully determined. However, depletion of of neurohypophyseal stores has been observed in profound shock states [83].
The use of vasopressin clinically has followed observations that exogenous administration of vasopressin during shock is capable of restoring systemic blood pressure. Landry and coworkers [4] first demonstrated this property in five patients with advanced septic shock. Since their initial observations, several uncontrolled trials have demonstrated that vasopressin can restore blood pressure during septic shock, following cardiopulmonary bypass and following epinephrine-resistant cardiac arrest (Table 2). However, few controlled studies have been performed to evaluate properly the effectiveness of vasopressin in shock. This is a critical point because it cannot be inferred that if an agent restores blood pressure then it will also lead to an improvement in outcome. An increase in blood pressure may be being obtained at the expense of perfusion to critical organs, or it may worsen cardiac performance by impairment of ventricular output through an increase in ventricular afterload. Consequently, organ injury could worsen in the face of a restoration of blood pressure. A case in point is the manner in which NOS inhibition was embraced to treat shock in septic patients [84]. Indeed, NOS inhibitors have clinical effects that are similar to those of vasopressin. Several reports have documented an increase in blood pressure, reduction in pressor requirement, and attendant reduction in cardiac output [84–86] (a profile that resembles that of vasopressin) in patients with septic shock. However, a recent randomized controlled trial of a NOS inhibitor in septic shock was halted because of higher mortality rates in the group that received treatment [87].
At present the only blinded, systematic evaluation of vasopressin in sepsis is that recently reported by Patel and coworkers [2]. In a controlled manner, they compared the effects of vasopressin with those of norepinephrine in 24 patients with septic shock who required vasopressor infusions. Patients who received vasopressin had a significant (80%) reduction in vasopressor requirement. Interestingly, patients in the vasopressin arm experienced a doubling in urine output and a 75% increase in creatinine clearance. Based on current information, it appears that replacement of vasopressin at a fixed dose can eliminate the need for catecholamine pressors in many patients.
Vasopressin was also evaluated in the setting of hypotension following induction of anesthesia in patients chronically treated with angiotensin-converting enzyme inhibitors [88, 89]. One study compared terlipressin (a vasopressin agonist) plus ephedrine (n = 21) versus ephedrine alone (n = 19) in patients following induction of anesthesia [88]. The second study evaluated vasopressin (n = 13) compared with placebo (n = 14) in patients following cardiac bypass [89]. Both studies demonstrated that the vasopressin agonist led to better hemodynamic stability and less catecholamine use. Consequently, in patients who are refractory to conventional vasopressors (owing to chronic blockade of their renin–angiotensin system), vasopressin may offer some clinical benefit in improving hemodynamics. Indeed, the study conducted by Morales and coworkers [89] demonstrated that, among those patients chronically treated with angiotensin-converting enzyme inhibitors, the group that received vasopressin had a shorter duration of stay in the intensive care unit following induction of anesthesia. These studies must be repeated in order to evaluate these highly relevant end-points and to confirm the safety of vasopressin before widespread clinical use of this agent can be recommended.
Vasopressin has also been demonstrated to increase arterial and coronary perfusion pressure as compared with clinical doses of epinephrine in animal models of cardiac arrest. Interestingly, like epinephrine, vasopressin may also be administered via the endotracheal tube. In fact vasopressin had better hemodyamic effects than did intratracheal epinephrine in one study of a canine model of cardiac arrest [90]. Based on these favorable reports, vasopressin has been advocated for use in cardiac arrest. In 1997, Lindner and coworkers [91] reported the effects of 40 units of vasopressin versus 1 mg epinephrine in patients who had not responded to three counter-shocks in the field. Fourteen (70%) patients in the vasopressin group versus seven (35%) patients in the epinephrine group survived to hospitalization. However, in a more recent study of vasopressin in cardiac arrest, no benefit over epinephrine was found [92]. That study evaluated vasopressin versus epinephrine as the first agent given in 200 patients who suffered in-hospital cardiac arrest. The investigators found that there was no advantage with either agent with respect to 1-hour survival or survival to hospital discharge. Importantly, there was no difference between groups in Mini Mental Status Examination or cerebral performance category scores. The reason for the discrepancy between the two studies is unclear. One explanation is differences between the two populations evaluated. Lindner and coworkers [91] evaluated patients who suffered a cardiac arrest out of hospital, whereas Steill and coworkers [92] evaluated hospitalized patients. Hospitalized patients may have a different prognosis after cardiac arrest than that of their counterparts in the community. Similarly, the etiology of the cardiac arrest may also have differed between the two groups, with more patients having a primary cardiac event in the community.
Administration of vasopressin to patients in low flow states (i.e. cardiogenic or hypovolemic shock) is strongly contraindicated because in these states cardiac output is severely depressed by the increase in afterload. Indeed, blockade of V1Rs and V2Rs has been advocated for treating congestive heart failure. In a rat model of congestive heart failure a single oral administration of conivaptan (a V1R and V2R blocker) increased urine volume and decreased urine osmolality in a dose-dependant manner [77]. Furthermore, conivaptan attenuated the changes in left ventricular end-diastolic pressure, and lung and right ventricular weight. The investigators stressed that vasopressin plays a significant role in elevating vascular tone through vasopressin V1Rs and plays a major role in retaining free water through V2Rs in this model of congestive heart failure.
In summary, the use of vasopressin at a low dose (0.04 units/min) is not associated with substantial decline in cardiac output. Vasopressin does not constrict the pulmonary circulation, and thus vasopressin may be preferred for patients with pulmonary hypertension. In this respect vasopressin differs from NOS inhibitors. It is hoped that, unlike early trials of NOS inhibition in sepsis, vasopressin's more favorable hemodynamic profile will translate into clinical benefit. Also, vasopressin's selective constriction of renal efferent over afferent arterioles could spare renal function in shock. Hopefully, the results of an active multicenter randomized controlled evaluation [93] will help to determine the role of vasopressin in septic shock.
Conclusion
Vasopressin is a unique vasoactive hormone that is important in control of vascular tone and has myocardial effects. Vasopressin can restore vascular tone in refractory vasodilatory shock states due to V1R activation of KATP channels, inhibitory action on NO, and potentiation of endogenous vasoconstrictors. Although animal and in vitro studies suggest that vasopressin may have negative inotropic and coronary vasoconstrictor properties, clinical studies of low-dose vasopressin to date do not demonstrate adverse cardiac effects of vasopressin. In refractory shock states, administration of vasopressin in low, physiologic doses has been associated with impressive stabilization of hemodynamics. Vasopressin is gaining popularity in diverse states such as septic shock and vasodilatory states associated with cardiac anesthesia and surgery. We stress that the clinical studies to date have been small and have focused on physiologic outcomes, and data on adverse effects are limited. Therefore, we do not recommend vasopressin as first-line therapy for vasodilatory shock. Future prospective studies are necessary to define the role of vasopressin in the therapy of vasodilatory shock.
Abbreviations
- ANP:
-
atrial natriuretic peptide
- IP3:
-
inositol trisphosphate
- KATP:
-
ATP-sensitive K+ channel
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- OTR:
-
oxytocin receptor
- SVR:
-
systemic vascular resistance
- V1R:
-
V1 vascular receptor
- V2R:
-
V2 renal receptor.
References
Holmes CL, Walley KR, Chittock DR, Lehman T, Russell JA: The effects of vasopressin on hemodynamics and renal function in severe septic shock: a case series. Intensive Care Med 2001, 27: 1416-1421. 10.1007/s001340101014
Patel BM, Chittock DR, Russell JA, Walley KR: Beneficial effects of short-term vasopressin infusion during severe septic shock. Anesthesiology 2002, 96: 576-582.
Landry DW, Levin HR, Gallant EM, Ashton RC Jr, Seo S, D'A-lessandro D, Oz MC, Oliver JA: Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation 1997, 95: 1122-1125.
Landry DW, Levin HR, Gallant EM, Seo S, D'Alessandro D, Oz MC, Oliver JA: Vasopressin pressor hypersensitivity in vasodilatory septic shock. Crit Care Med 1997, 25: 1279-1282. 10.1097/00003246-199708000-00012
Malay MB, Ashton RC Jr, Landry DW, Townsend RN: Low-dose vasopressin in the treatment of vasodilatory septic shock. J Trauma 1999, 47: 699-703.
Argenziano M, Choudhri AF, Oz MC, Rose EA, Smith CR, Landry DW: A prospective randomized trial of arginine vasopressin in the treatment of vasodilatory shock after left ventricular assist device placement. Circulation 1997, 96: II-286-II-290.
Argenziano M, Chen JM, Choudhri AF, Cullinane S, Garfein E, Weinberg AD, Smith CR Jr, Rose EA, Landry DW, Oz MC: Management of vasodilatory shock after cardiac surgery: identification of predisposing factors and use of a novel pressor agent. J Thorac Cardiovasc Surg 1998, 116: 973-980.
Argenziano M, Chen JM, Cullinane S, Choudhri AF, Rose EA, Smith CR, Edwards NM, Landry DW, Oz MC: Arginine vasopressin in the management of vasodilatory hypotension after cardiac transplantation. J Heart Lung Transplant 1999, 18: 814-817. 10.1016/S1053-2498(99)00038-8
Rosenzweig EB, Starc TJ, Chen JM, Cullinane S, Timchak DM, Gersony WM, Landry DW, Galantowicz ME: Intravenous arginine-vasopressin in children with vasodilatory shock after cardiac surgery. Circulation 1999, 100: II182-II186.
Morales DL, Gregg D, Helman DN, Williams MR, Naka Y, Landry DW, Oz MC: Arginine vasopressin in the treatment of 50 patients with postcardiotomy vasodilatory shock. Ann Thorac Surg 2000, 69: 102-106. 10.1016/S0003-4975(99)01197-2
Chen JM, Cullinane S, Spanier TB, Artrip JH, John R, Edwards NM, Oz MC, Landry DW: Vasopressin deficiency and pressor hypersensitivity in hemodynamically unstable organ donors. Circulation 1999, 100: II244-II246.
Gold JA, Cullinane S, Chen J, Oz MC, Oliver JA, Landry DW: Vasopressin as an alternative to norepinephrine in the treatment of milrinone-induced hypotension. Crit Care Med 2000, 28: 249-252. 10.1097/00003246-200001000-00043
Landry DW, Oliver JA: The pathogenesis of vasodilatory shock. N Engl J Med 2001, 345: 588-595. 10.1056/NEJMra002709
Surks HK, Mochizuki N, Kasai Y, Georgescu SP, Tang KM, Ito M, Lincoln TM, Mendelsohn ME: Regulation of myosin phosphatase by a specific interaction with cGMP-dependent protein kinase Ialpha. Science 1999, 286: 1583-1587. 10.1126/science.286.5444.1583
Webb JG, Yates PW, Yang Q, Mukhin YV, Lanier SM: Adenylyl cyclase isoforms and signal integration in models of vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 2001, 281: H1545-H1552.
Standen NB, Quayle JM: K+channel modulation in arterial smooth muscle. Acta Physiol Scand 1998, 164: 549-557. 10.1046/j.1365-201X.1998.00433.x
Jackson WF: Ion channels and vascular tone. Hypertension 2000, 35: 173-178.
Davies NW: Modulation of ATP-sensitive K+channels in skeletal muscle by intracellular protons. Nature 1990, 343: 375-377. 10.1038/343375a0
Keung EC, Li Q: Lactate activates ATP-sensitive potassium channels in guinea pig ventricular myocytes. J Clin Invest 1991, 88: 1772-1777.
Landry DW, Oliver JA: The ATP-sensitive K+channel mediates hypotension in endotoxemia and hypoxic lactic acidosis in dog. J Clin Invest 1992, 89: 2071-2074.
Geisen K, Vegh A, Krause E, Papp JG: Cardiovascular effects of conventional sulfonylureas and glimepiride. Horm Metab Res 1996, 28: 496-507.
Gardiner SM, Kemp PA, March JE, Bennett T: Regional haemodynamic responses to infusion of lipopolysaccharide in conscious rats: effects of pre- or post-treatment with glibenclamide. Br J Pharmacol 1999, 128: 1772-1778.
Salzman AL, Vromen A, Denenberg A, Szabo C: K(ATP)-channel inhibition improves hemodynamics and cellular energetics in hemorrhagic shock. Am J Physiol 1997, 272: H688-H694.
Wakatsuki T, Nakaya Y, Inoue I: Vasopressin modulates K(+)-channel activities of cultured smooth muscle cells from porcine coronary artery. Am J Physiol 1992, 263: H491-H496.
Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA: Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature 1994, 368: 850-853. 10.1038/368850a0
Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK: Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proc Natl Acad Sci USA 1994, 91: 7583-7587.
Kilbourn R: Nitric oxide synthase inhibitors – a mechanism-based treatment of septic shock. Crit Care Med 1999, 27: 857-858. 10.1097/00003246-199905000-00003
Umino T, Kusano E, Muto S, Akimoto T, Yanagiba S, Ono S, Amemiya M, Ando Y, Homma S, Ikeda U, Shimada K, Asano Y: AVP inhibits LPS- and IL-1beta-stimulated NO and cGMP via V1 receptor in cultured rat mesangial cells. Am J Physiol 1999, 276: F433-F441.
Nambi P, Whitman M, Gessner G, Aiyar N, Crooke ST: Vasopressin-mediated inhibition of atrial natriuretic factor-stimulated cGMP accumulation in an established smooth muscle cell line. Proc Natl Acad Sci USA 1986, 83: 8492-8495.
Kusano E, Tian S, Umino T, Tetsuka T, Ando Y, Asano Y: Arginine vasopressin inhibits interleukin-1 beta-stimulated nitric oxide and cyclic guanosine monophosphate production via the V1 receptor in cultured rat vascular smooth muscle cells. J Hypertens 1997, 15: 627-632. 10.1097/00004872-199715060-00009
Yamamoto K, Ikeda U, Okada K, Saito T, Shimada K: Arginine vasopressin inhibits nitric oxide synthesis in cytokine-stimulated vascular smooth muscle cells. Hypertens Res 1997, 20: 209-216.
Karmazyn M, Manku MS, Horrobin DF: Changes of vascular reactivity induced by low vasopressin concentrations: interactions with cortisol and lithium and possible involvement of prostaglandins. Endocrinology 1978, 102: 1230-1236.
Noguera I, Medina P, Segarra G, Martinez MC, Aldasoro M, Vila JM, Lluch S: Potentiation by vasopressin of adrenergic vasoconstriction in the rat isolated mesenteric artery. Br J Pharmacol 1997, 122: 431-438.
Emori T, Hirata Y, Ohta K, Kanno K, Eguchi S, Imai T, Shichiri M, Marumo F: Cellular mechanism of endothelin-1 release by angiotensin and vasopressin. Hypertension 1991, 18: 165-170.
Caramelo C, Okada K, Tsai P, Linas SL, Schrier RW: Interaction of arginine vasopressin and angiotensin II on Ca2+in vascular smooth muscle cells. Kidney Int 1990, 38: 47-54.
Iversen BM, Arendshorst WJ: ANG II and vasopressin stimulate calcium entry in dispersed smooth muscle cells of pre-glomerular arterioles. Am J Physiol 1998, 274: F498-F508.
Bichet DG, Razi M, Lonergan M, Arthus MF, Papukna V, Kortas C, Barjon JN: Hemodynamic and coagulation responses to 1-desamino[8-D-arginine] vasopressin in patients with congenital nephrogenic diabetes insipidus. N Engl J Med 1988, 318: 881-887.
Walker BR, Haynes J Jr, Wang HL, Voelkel NF: Vasopressin-induced pulmonary vasodilation in rats. Am J Physiol 1989, 257: H415-H422.
Evora PR, Pearson PJ, Schaff HV: Arginine vasopressin induces endothelium-dependent vasodilatation of the pulmonary artery. V1-receptor-mediated production of nitric oxide. Chest 1993, 103: 1241-1245.
Suzuki Y, Satoh S, Oyama H, Takayasu M, Shibuya M: Regional differences in the vasodilator response to vasopressin in canine cerebral arteries in vivo. Stroke 1993, 24: 1049-1053.
Rudichenko VM, Beierwaltes WH: Arginine vasopressin-induced renal vasodilation mediated by nitric oxide. J Vasc Res 1995, 32: 100-105.
Tamaki T, Kiyomoto K, He H, Tomohiro A, Nishiyama A, Aki Y, Kimura S, Abe Y: Vasodilation induced by vasopressin V2 receptor stimulation in afferent arterioles. Kidney Int 1996, 49: 722-729.
Okamura T, Toda M, Ayajiki K, Toda N: Receptor subtypes involved in relaxation and contraction by arginine vasopressin in canine isolated short posterior ciliary arteries. J Vasc Res 1997, 34: 464-472.
Okamura T, Ayajiki K, Fujioka H, Toda N: Mechanisms underlying arginine vasopressin-induced relaxation in monkey isolated coronary arteries. J Hypertens 1999, 17: 673-678. 10.1097/00004872-199917050-00011
Thibonnier M, Conarty DM, Preston JA, Plesnicher CL, Dweik RA, Erzurum SC: Human vascular endothelial cells express oxytocin receptors. Endocrinology 1999, 140: 1301-1309. 10.1210/en.140.3.1301
Garcia-Villalon AL, Garcia JL, Fernandez N, Monge L, Gomez B, Dieguez G: Regional differences in the arterial response to vasopressin: role of endothelial nitric oxide. Br J Pharmacol 1996, 118: 1848-1854.
Holmes CL, Patel BM, Russell JA, Walley KR: Physiology of vasopressin relevant to management of septic shock. Chest 2001, 120: 989-1002. 10.1378/chest.120.3.989
Liard JF: Does vasopressin-induced vasoconstriction persist during prolonged infusion in dogs? Am J Physiol 1987, 252: R668-R673.
Dixon BS: Cyclic AMP selectively enhances bradykinin receptor synthesis and expression in cultured arterial smooth muscle. Inhibition of angiotensin II and vasopressin response. J Clin Invest 1994, 93: 2535-2544.
Zhang J, Sato M, Duzic E, Kubalak SW, Lanier SM, Webb JG: Adenylyl cyclase isoforms and vasopressin enhancement of agonist-stimulated cAMP in vascular smooth muscle cells. Am J Physiol 1997, 273: H971-H980.
Serradeil-Le Gal C, Villanova G, Boutin M, Maffrand JP, Le Fur G: Effects of SR 4 a non-peptide antagonist of vasopressin V1a receptors, on vasopressin-induced coronary vasoconstriction in conscious rabbits. Fundam Clin Pharmacol 9059, 9: 17-24.
Maturi MF, Martin SE, Markle D, Maxwell M, Burruss CR, Speir E, Greene R, Ro YM, Vitale D, Green MV, et al.: Coronary vasoconstriction induced by vasopressin. Production of myocardial ischemia in dogs by constriction of nondiseased small vessels. Circulation 1991, 83: 2111-2121.
Bax WA, Van der Graaf PH, Stam WB, Bos E, Nisato D, Saxena PR: [Arg8]vasopressin-induced responses of the human isolated coronary artery: effects of non-peptide receptor antagonists. Eur J Pharmacol 1995, 285: 199-202. 10.1016/0014-2999(95)00503-D
Fernandez N, Garcia JL, Garcia-Villalon AL, Monge L, Gomez B, Dieguez G: Coronary vasoconstriction produced by vasopressin in anesthetized goats. Role of vasopressin V1 and V2 receptors and nitric oxide. Eur J Pharmacol 1998, 342: 225-233. 10.1016/S0014-2999(97)01504-5
Boyle WA III, Segel LD: Direct cardiac effects of vasopressin and their reversal by a vascular antagonist. Am J Physiol 1986, 251: H734-H741.
Walker BR, Childs ME, Adams EM: Direct cardiac effects of vasopressin: role of V1- and V2-vasopressinergic receptors. Am J Physiol 1988, 255: H261-H265.
Vanhoutte PM, Katusic ZS, Shepherd JT: Vasopressin induces endothelium-dependent relaxations of cerebral and coronary, but not of systemic arteries. J Hypertens Suppl 1984, 2: S421-S422.
Katusic ZS, Shepherd JT, Vanhoutte PM: Vasopressin causes endothelium-dependent relaxations of the canine basilar artery. Circ Res 1984, 55: 575-579.
Wenzel V, Kern KB, Hilwig RW, et al.: The left anterior descending coronary artery dilates after arginine vasopressin during normal sinus rhythm, and ventricular fibrillation with cardiopulmonary resuscitation [abstract]. Circulation 2001, 104: 2974.
Boyle WA III, Segel LD: Attenuation of vasopressin-mediated coronary constriction and myocardial depression in the hypoxic heart. Circ Res 1990, 66: 710-721.
Wenzel V, Lindner KH, Baubin MA, Voelckel WG: Vasopressin decreases endogenous catecholamine plasma concentrations during cardiopulmonary resuscitation in pigs. Crit Care Med 2000, 28: 1096-1100. 10.1097/00003246-200004000-00031
Raedler C, Voelckel WG, Wenzel V, Bahlmann L, Baumeier W, Schmittinger CA, Herff H, Krismer AC, Lindner KH, Lurie KG: Vasopressor response in a porcine model of hypothermic cardiac arrest is improved with active compression-decompression cardiopulmonary resuscitation using the inspiratory impedance threshold valve. Anesth Analg 2002, 95: 1496-1502.
Voelckel WG, Lurie KG, McKnite S, Zielinski T, Lindstrom P, Peterson C, Wenzel V, Lindner KH, Benditt D: Effects of epinephrine and vasopressin in a piglet model of prolonged ventricular fibrillation and cardiopulmonary resuscitation. Crit Care Med 2002, 30: 957-962. 10.1097/00003246-200205000-00001
Voelckel WG, Lurie KG, McKnite S, Zielinski T, Lindstrom P, Peterson C, Wenzel V, Lindner KH: Comparison of epinephrine with vasopressin on bone marrow blood flow in an animal model of hypovolemic shock and subsequent cardiac arrest. Crit Care Med 2001, 29: 1587-1592. 10.1097/00003246-200108000-00015
Wenzel V, Lindner KH, Krismer AC, Miller EA, Voelckel WG, Lingnau W: Repeated administration of vasopressin but not epinephrine maintains coronary perfusion pressure after early and late administration during prolonged cardiopulmonary resuscitation in pigs. Circulation 1999, 99: 1379-1384.
Zenteno-Savin T, Sada-Ovalle I, Ceballos G, Rubio R: Effects of arginine vasopressin in the heart are mediated by specific intravascular endothelial receptors. Eur J Pharmacol 2000, 410: 15-23. 10.1016/S0014-2999(00)00853-0
Cheng CP, Igarashi Y, Klopfenstein HS, Applegate RJ, Shihabi Z, Little WC: Effect of vasopressin on left ventricular performance. Am J Physiol 1993, 264: H53-H60.
Gold J, Cullinane S, Chen J, Seo S, Oz MC, Oliver JA, Landry DW: Vasopressin in the treatment of milrinone-induced hypotension in severe heart failure. Am J Cardiol 2000, 85: 506-508. 10.1016/S0002-9149(99)00783-3
Dunser MW, Mayr AJ, Stallinger A, Ulmer H, Ritsch N, Knotzer H, Pajk W, Mutz NJ, Hasibeder WR: Cardiac performance during vasopressin infusion in postcardiotomy shock. Intensive Care Med 2002, 28: 746-751. 10.1007/s00134-002-1265-y
Xu YJ, Gopalakrishnan V: Vasopressin increases cytosolic free [Ca2+] in the neonatal rat cardiomyocyte. Evidence for V1 subtype receptors. Circ Res 1991, 69: 239-245.
Liu P, Hopfner RL, Xu YJ, Gopalakrishnan V: Vasopressin-evoked [Ca2+]i responses in neonatal rat cardiomyocytes. J Cardiovasc Pharmacol 1999, 34: 540-546. 10.1097/00005344-199910000-00010
Van der Bent V, Church DJ, Vallotton MB, Meda P, Kem DC, Capponi AM, Lang U: [Ca2+]i and protein kinase C in vasopressin-induced prostacyclin and ANP release in rat cardiomyocytes. Am J Physiol 1994, 266: H597-H605.
Gutkowska J, Jankowski M, Lambert C, Mukaddam-Daher S, Zingg HH, McCann SM: Oxytocin releases atrial natriuretic peptide by combining with oxytocin receptors in the heart. Proc Natl Acad Sci USA 1997, 94: 11704-11709. 10.1073/pnas.94.21.11704
Mukaddam-Daher S, Yin YL, Roy J, Gutkowska J, Cardinal R: Negative inotropic and chronotropic effects of oxytocin. Hypertension 2001, 38: 292-296.
Laugwitz KL, Ungerer M, Schoneberg T, Weig HJ, Kronsbein K, Moretti A, Hoffmann K, Seyfarth M, Schultz G, Schomig A: Adenoviral gene transfer of the human V2 vasopressin receptor improves contractile force of rat cardiomyocytes. Circulation 1999, 99: 925-933.
Udelson JE, Smith WB, Hendrix GH, Painchaud CA, Ghazzi M, Thomas I, Ghali JK, Selaru P, Chanoine F, Pressler ML, Konstam MA: Acute hemodynamic effects of conivaptan, a dual V(1A) and V(2) vasopressin receptor antagonist, in patients with advanced heart failure. Circulation 2001, 104: 2417-2423.
Wada K, Tahara A, Arai Y, Aoki M, Tomura Y, Tsukada J, Yatsu T: Effect of the vasopressin receptor antagonist conivaptan in rats with heart failure following myocardial infarction. Eur J Pharmacol 2002, 450: 169-177. 10.1016/S0014-2999(02)02101-5
Yatsu T, Kusayama T, Tomura Y, Arai Y, Aoki M, Tahara A, Wada K, Tsukada J: Effect of conivaptan, a combined vasopressin V(1a) and V(2) receptor antagonist, on vasopressin-induced cardiac and haemodynamic changes in anaesthetised dogs. Pharmacol Res 2002, 46: 375-381. 10.1016/S1043661802002062
Xu Y, Hopfner RL, McNeill JR, Gopalakrishnan V: Vasopressin accelerates protein synthesis in neonatal rat cardiomyocytes. Mol Cell Biochem 1999, 195: 183-190. 10.1023/A:1006961330375
Fukuzawa J, Haneda T, Kikuchi K: Arginine vasopressin increases the rate of protein synthesis in isolated perfused adult rat heart via the V1 receptor. Mol Cell Biochem 1999, 195: 93-98. 10.1023/A:1006980517557
Mets B, Michler RE, Delphin ED, Oz MC, Landry DW: Refractory vasodilation after cardiopulmonary bypass for heart transplantation in recipients on combined amiodarone and angiotensin-converting enzyme inhibitor therapy: a role for vasopressin administration. J Cardiothorac Vasc Anesth 1998, 12: 326-329.
Morales D, Madigan J, Cullinane S, Chen J, Heath M, Oz M, Oliver JA, Landry DW: Reversal by vasopressin of intractable hypotension in the late phase of hemorrhagic shock. Circulation 1999, 100: 226-229.
Sharshar T, Carlier R, Blanchard A, Feydy A, Gray F, Paillard M, Raphael JC, Gajdos P, Annane D: Depletion of neurohypophyseal content of vasopressin in septic shock. Crit Care Med 2002, 30: 497-500. 10.1097/00003246-200203000-00001
Avontuur JA, Tutein Nolthenius RP, Buijk SL, Kanhai KJ, Bruining HA: Effect of L-NAME, an inhibitor of nitric oxide synthesis, on cardiopulmonary function in human septic shock. Chest 1998, 113: 1640-1646.
Avontuur JA, Tutein Nolthenius RP, van Bodegom JW, Bruining HA: Prolonged inhibition of nitric oxide synthesis in severe septic shock: a clinical study. Crit Care Med 1998, 26: 660-667. 10.1097/00003246-199804000-00012
Grover R, Zaccardelli D, Colice G, Guntupalli K, Watson D, Vincent JL: An open-label dose escalation study of the nitric oxide synthase inhibitor, N(G)-methyl-L-arginine hydrochloride (546C88), in patients with septic shock. Glaxo Wellcome International Septic Shock Study Group. Crit Care Med 1999, 27: 913-922. 10.1097/00003246-199905000-00025
Cobb JP: Use of nitric oxide synthase inhibitors to treat septic shock: the light has changed from yellow to red. Crit Care Med 1999, 27: 855-856. 10.1097/00003246-199905000-00002
Meersschaert K, Brun L, Gourdin M, Mouren S, Bertrand M, Riou B, Coriat P: Terlipressin-ephedrine versus ephedrine to treat hypotension at the induction of anesthesia in patients chronically treated with angiotensin converting-enzyme inhibitors: a prospective, randomized, double-blinded, crossover study. Anesth Analg 2002, 94: 835-840.
Morales DL, Garrido MJ, Madigan JD, Helman DN, Faber J, Williams MR, Landry DW, Oz MC: A double-blind randomized trial: prophylactic vasopressin reduces hypotension after cardiopulmonary bypass. Ann Thorac Surg 2003, 75: 926-930. 10.1016/S0003-4975(02)04408-9
Efrati O, Barak A, Ben-Abraham R, Modan-Moses D, Berkovitch M, Manisterski Y, Lotan D, Barzilay Z, Paret G: Should vasopressin replace adrenaline for endotracheal drug administration? Crit Care Med 2003, 31: 572-576. 10.1097/01.CCM.0000050441.09207.16
Lindner KH, Dirks B, Strohmenger HU, Prengel AW, Lindner IM, Lurie KG: Randomised comparison of epinephrine and vasopressin in patients with out-of-hospital ventricular fibrillation. Lancet 1997, 349: 535-537. 10.1016/S0140-6736(97)80087-6
Stiell IG, Hebert PC, Wells GA, Vandemheen KL, Tang AS, Higginson LA, Dreyer JF, Clement C, Battram E, Watpool I, Mason S, Klassen T, Weitzman BN: Vasopressin versus epinephrine for inhospital cardiac arrest: a randomised controlled trial. Lancet 2001, 358: 105-109. 10.1016/S0140-6736(01)05328-4
Cooper DJ, Russell JA, Walley KR, Holmes CL, Singer J, Hebert PC, Granton J, Mehta S, Terins T: Vasopressin and septic shock trial (VASST): innovative features and performance. Am J Resp Crit Care Med 2003, 167: A838.
Dunser MW, Mayr AJ, Ulmer H, Ritsch N, Knotzer H, Pajk W, Luckner G, Mutz NJ, Hasibeder WR: The effects of vasopressin on systemic hemodynamics in catecholamine-resistant septic and postcardiotomy shock: a retrospective analysis. Anesth Analg 2001, 93: 7-13.
Tsuneyoshi I, Yamada H, Kakihana Y, Nakamura M, Nakano Y, Boyle WA III: Hemodynamic and metabolic effects of low-dose vasopressin infusions in vasodilatory septic shock. Crit Care Med 2001, 29: 487-493. 10.1097/00003246-200103000-00004
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
None declared.
Rights and permissions
About this article
Cite this article
Holmes, C.L., Landry, D.W. & Granton, J.T. Science Review: Vasopressin and the cardiovascular system part 2 – clinical physiology. Crit Care 8, 15 (2003). https://doi.org/10.1186/cc2338
Published:
DOI: https://doi.org/10.1186/cc2338