
Abstract
Background
Multiple mechanisms of immunosuppression have been identified in the tumor microenvironment including regulatory B cells (Breg). Recently, we have shown that Breg suppress T cell function by production of adenosine (ADO). However, the autocrine effect of ADO on B cells and the role of Breg in head and neck cancer remains unclear.
Methods
Blood (n = 42) and tumor tissue (n = 39) of head and neck cancer patients and healthy donors (n = 60) were analyzed by FACS. The effect of ADO on phenotype, intracellular signaling pathways, Ca2+ influx and ADO production was analyzed in Breg and effector B cells (Beff) by FACS, luminescence and mass spectrometry. The blockage of the ADO receptor A2A was analyzed in a murine head and neck cancer model.
Results
ADO-producing Breg were found in tumor tissue and peripheral blood. ADO inhibited the intracellular Bruton’s tyrosine kinase (BTK) and Ca2+ influx only in Beff. The inhibition of BTK by ibrutinib mimicked the effect of ADO, and ibrutinib reduced the production of ADO by downregulation of CD39 in vitro. The inhibition of ADO receptor A2A significantly reduced tumor mass and increased B cell infiltration, in vivo.
Conclusion
Our data demonstrate the presence of a novel ADO-producing Breg population within the tumor microenvironment in mice and humans. A new model is proposed on how ADO-producing Breg can influence the function of Beff cells in healthy donors and cancer patients. Thus, the modulation of the ADO pathway in B cells may serve as a therapeutic approach for cancer patients.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Head and neck squamous cell carcinoma (HNSCC) belongs to the most aggressive cancers worldwide, with a 5-year survival rate less than 50% for advanced cancers [1]. As compared to other tumor entities, HNSCC is regarded as an immunogenic tumor likely to be responsive to immunotherapeutic approaches [2]. Therefore, the approval of the checkpoint inhibitor nivolumab in 2017 has attracted particular attention [3]. However, current immunotherapeutic efforts are mostly focused on Thelper, Tkiller and regulatory T cell (Treg) populations.
Although B cells are a part of the adaptive immune system, they have received little attention in oncological immunotherapy of solid tumors. Recent discoveries demonstrate that B cells have a decisive influence on solid tumor growth and thus can also serve as a therapeutic target. It is now clear that, in addition to antibody production, B cells can also process antigens or acquire immunosuppressive properties. Accordingly, a detailed analysis of defined B-cell populations and their contribution to cancerogenesis is needed.
Until today, it remains unclear whether B cells have pro- or anti-tumorigenic features. In a variety of murine tumor models, B cells have been shown to suppress T cells and increase tumor growth [4, 5]. On the other hand, tumor-infiltrating B cells are associated with a good outcome in patients with lung, breast, ovary or head and neck cancer [6,7,8,9]. In addition, lymphoid structures containing T and B cells have been recently discovered in solid tumor tissue [10]. These structures enable tumor antigen contact, which may support the endogenous anti-tumor immunity [11]. Possibly, these conflicting results on the role of B cells in the tumor environment are due to different B cell populations influencing tumor growth in opposing ways. For example, there are many subsets of regulatory B cell populations (Breg), which are defined by their phenotype and function. Previously, we have described a population of Breg in healthy donors, which express the ectonucleotidases CD39 and CD73 on their surface and produce immunosuppressive adenosine (ADO) [12]. In the peripheral blood of healthy donors, almost 70% of all CD19+ B cells carry both enzymes on their surface.
The importance of ADO in the tumor environment has been known in the past. Solid tumors contain high levels of exogenous immune-suppressive ADO [13, 14], and the inhibition of the adenosine receptor A2B (ADORA2B) signaling leads to a decreased proliferation of human HNSCC cells, in vitro [15]. HNSCC patients with low levels of ADORA2A have a significantly better overall survival rate which correlates with low levels of Treg and a high level of CD8+ T cells [16]. In addition, both a selective ADORA2A inhibitor and a CD73 antibody are currently tested in clinical phase I studies for solid tumors in combination with a PD-1 antibody (NCT02403193/NCT02503774).
The following study reports on the presence of a novel ADO-producing Breg population within the tumor microenvironment (TME) in mice and humans. A new model is proposed on how ADO-producing Breg can influence the function of Beff cells in healthy donors and cancer patients. The discussion includes the modulation of the ADO pathway in B cells as a therapeutic approach for cancer patients.
Materials and methods
Human blood samples
Whole blood samples were obtained from tumor patients (n = 42) and healthy volunteers (n = 60). Peripheral blood mononuclear cells (PBMC) were isolated by Biocoll Separating Solution (Merck, USA) or Pancoll (density 1119 g/ml and 1077 g/ml; PAN Biotech, Germany) and directly used for experiments. Clinicopathological and demographic parameters are listed in Supplementary table S1.
Tumor tissues processing
Human tumor tissue (n = 39) was collected during surgery in NaCl. Minced tissue pieces were collected in RPMI containing 200 IU/ml collagenase I (Worthington, USA) for at least two hours at 37 °C and mashed with a 100 µm EASY strainer (Gibco, USA). The lymphocyte fraction was isolated by Biocoll centrifugation. The purity of the cells was measured by anti-CD45 staining.
FACS antibodies
Antihuman: p-BTK (pY223) PE, CD19 PE-Cy5, IgM PE, CD45 AmCyan and CD45 FITC (Becton Dickinson, USA), CD39 PE-Cy7, CD73 FITC, CD73 eFluor450, CD21 FITC and CD23 APC (eBioscience, USA). Anti-mouse: B220 FITC, CD4 PerCP, CD45 APC-Cy7 (BD-Pharmingen, USA), IgM PE (Southern Biotech, USA) and CD3e eFlour450, CD39 PE-Cy7, IgD FITC, IgM APC (eBioscience, USA), IgD APC-Cy7, CD73 PacificBlue, CD19 PE-Cy7 and CD8a PE (BioLegend, USA). All human mAbs were titrated using normal PBMC to establish optimal dilution.
Surface staining
Cells were stained as previously published [17]. All flow cytometry measurements were taken using a Gallios 10-color flow-cytometer equipped with Kaluza flow cytometry software (both Beckman Coulter, USA). At least 105 cells were acquired for analysis.
Cell separation
CD19+ human B cells were separated from PBMC using the Human B cell Enrichment Kit or the Human B cell Isolation Kit (both stemcell, Canada) according to the manufacturer’s instructions. The purity of the separated cells as monitored by flow cytometry was always ≥ 93%. CD19+ mouse B cells were separated from spleen cells using the Mouse CD19 Positive Selection Kit II, and CD4+ and CD8+ T cells were separated after PE staining with the PE Selection Kit (both stemcell, Canada) according to the manufacturer’s instructions.
B-cell culture
B cells were cultured in 96-well plates (2.5 × 104 cells/well) containing RPMI + 10% FBS superior (Merck, USA), 100 U/ml penicillin and 100 µg/ml streptomycin (Pan Biotech, Germany). B cells were stimulated with IL-4 (1,000 IU/ml, CellGenix, Germany), CD40L (2 µg/ml) and hemagglutinin (423 ng/ml) (both R&D, Canada) and cultured for 6 days (37 °C, 5% CO2). In addition, some cells were treated with ibrutinib (10 µM, Pharmacyclics, USA). FACS analysis of surface markers (CD19, CD39, CD73) was performed before and after stimulation.
Intracellular p-BTK staining
PBMC or isolated CD19+ B cells from healthy volunteers were stimulated with anti-µ-F(ab’)2 (5 µg/ml, Dianova, Germany). Some cells were incubated with ADO (1 mM) (Sigma-Aldrich, USA) for two hours or surface-stained with CD73 eFluor450 before stimulation. The stimulation was stopped by adding 10% formaldehyde solution (AppliChem, Germany). Cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, USA) kept in 50% methanol at − 20 °C for 10 min. Cells were washed and stained with p-BTK (pY223) PE for 45 min. at room temperature before FACS analysis.
Calcium assay
Calcium assays were performed as described previously [18]. For stimulation, anti-µ-F(ab’)2 (5 µg/ml) was added immediately after baseline measurement. The addition of ionomycin was used as a positive control. Calcium flux was measured with the LSR Fortessa using the 355-nm UV laser (Becton Dickinson, USA).
RT-PCR for ADO receptors
RNA was extracted and used for cDNA synthesis as described earlier [19]. Relative quantification of the target gene messenger RNA (mRNA) expression was calculated with the relative quantification in RT-PCR as previously published [20]. The expression levels of target genes were normalized to the levels of six endogenous controls (housekeeping genes RPL13A, β-actin, PBG-D, G6PD, GAPDH and TBP). The target genes of mouse cells were normalized to two housekeeping genes (GAPDH and actin). PCR primers and the respective UPL probes are listed in Supplementary table S2.
ATP hydrolysis by luminescence
ATP measurements in the supernatants of B cells were taken with the ATPlite kit (PerkinElmer, USA) according to the manufacturer’s instructions and as published before [12]. All experiments were performed in technical triplicates.
Mass spectrometry
Supernatants of B cells were centrifuged and boiled for 2 min at 95 °C to inactivate ADO-degrading enzymes and subsequently stored at − 80 °C for subsequent analysis as described before [12]. Purines were measured using liquid chromatography–tandem mass spectrometer by selected reaction monitoring with 13C10 ADO as the internal standard [12, 17]. The experiments were performed in triplicates.
Animal model
Murine squamous carcinoma cells (SCC VII, 5 × 105) were injected into the floor of the mouth of syngeneic C3H/HeJ mice (n = 72 including control groups) as described earlier [21, 22]. The experiments were started with 7–9-week-old male mice. Before tumor induction and 21 days after tumor injection, murine blood was analyzed by an animal blood counter (Scil animal care company GmbH, Germany) and by flow cytometry. On day 21, the tumor was resected and weighed. Tumor-infiltrating lymphocytes (TIL) were isolated and analyzed by flow cytometry as described above. For ADORA2A inhibition, the ADORA2A antagonist SCH-58261 (1 μg/g body weight in PBS; Tocris Bioscience, Great Britain) was injected intraperitoneally starting at day 7 after tumor induction and subsequently every fourth day [23].
Statistical analysis
All data are presented as means of at least three experiments ± SD or as medians with interquartile range. Data were analyzed for Gaussian distribution with Shapiro–Wilk test. One-way and two-way ANOVAs were used for analyzing more than two values for statistical significance. The Wilcoxon matched samples test was used for nonparametric analysis of two mean values. P values ≤ 0.05 were considered to be significant. Statistical analyses were performed using Excel and GraphPad Prism version 6.01. Error bars indicate the SD of the mean data from replicate experiments.
Results
Breg in human HNSCC patients
ADO-producing Breg are defined by the surface expression of the ectonucleotidases, CD39 and CD73. These Breg were found in the peripheral blood of healthy donors (55.4 ± 15.5% of CD19+ B cells, n = 60) and tumor patients (58.8 ± 14.5%, n = 42). In the tumor microenvironment, the frequency of ADO-producing Breg was significantly decreased as compared to the peripheral blood (42.6 ± 16%; p ≤ 0.0001, Fig. 1a). The CD39+CD73+ B cells in the peripheral blood and in the TME are mature follicular B cells (CD23+CD21neg).

Human CD19+ B cells and their BTK phosphorylation in response to ADO. a The expression of the ectonucleotidases CD39 and CD73 measured by FACS in PBMC of healthy donors (n = 60), PBMC of tumor patients (n = 42) and in TIL of HNSCC patients (n = 39). Data are shown as means ± SD. b RNA levels for the four adenosine receptors (ADOR) in isolated peripheral B cells of healthy donors (n = 10) and tumor patients (n = 5). c Human B cells were stimulated with anti-µ-F(ab’)2 and stained for anti-p-BTK (pY223). The experiment was performed with gated B cells (n = 3) and isolated B cells in PBMC (n = 5). d Pre-incubation with ADO induced a decreased BTK phosphorylation in a concentration-dependent manner after stimulation with anti-µ-F(ab’)2 (n = 3). e Incubation with ADO decreased the ability of B cells to phosphorylate BTK in a time-dependent manner. Results are expressed as percentage of phosphorylated BTK shown as mean ± SD (n = 10). f The ability of ADO to decrease BTK phosphorylation in peripheral B cells is comparable in B cells of healthy donors (n = 10) and HNSCC patients (n = 7). Results are expressed as percentage of phosphorylated BTK. The five-minute value was set to 100%. All data are shown as mean ± SD. not significant (ns); p < 0.05 (*); p < 0.01 (**); p < 0.001 (***); p < 0.0001 (****)
Adenosine receptors on B cells
We next aimed to describe the autocrine effect of ADO on B cells. Human B cells express mostly ADORA2A with comparable expression levels in healthy volunteers and tumor patients (Fig. 1b). ADORA2B and ADORA3 showed very low expression levels, while ADORA1 was almost undetectable in human samples.
BTK phosphorylation in B cells
As phosphorylation of the Bruton’s tyrosine kinase (BTK) is a hallmark of B cell activation, we analyzed BTK phosphorylation (pY223) in B cells by FACS. Stimulation with anti-µ-F(ab’)2 induced phosphorylation of BTK in isolated CD19+ B cells as well as in PBMC gated on CD19+ B cells already after 1 min (Fig. 1c). However, gated B cells in PBMC showed a higher variance of BTK phosphorylation. We, therefore, decided to use isolated B cells for further experiments. Treatment of isolated B cells with different ADO concentrations prior to stimulation with anti-µ-F(ab’)2 induced a significant concentration-dependent reduction in the BTK phosphorylation (p ≤ 0.01; Fig. 1d). The effect of ADO was most pronounced, when cells were stimulated with a concentration of 5 µg/mL anti-µ-F(ab’)2, which was applied for further experiments. The inhibition of BTK phosphorylation in B cells by ADO (1 mM) lasted for at least 30 min (Fig. 1e). Peripheral B cells of tumor patients showed a similar response to ADO as compared with B cells of healthy volunteers (p = 0.09, Fig. 1f). These experiments demonstrate the ability of ADO to decrease BTK phosphorylation, which is a key molecule in the B-cell receptor (BCR)-mediated signaling pathway in the B cells of healthy donors as well as HNSCC patients.
ADO decreases BTK phosphorylation exclusively in CD73neg B cells
Next, we investigated whether ADO-mediated downregulation of BTK phosphorylation was restricted to a specific subset of B cells. To this end, B cells obtained from healthy donors were stained with anti-CD73 in order to distinguish CD73+ ADO-producing Breg from CD73neg Beff (Supplementary fig. S1a). In unstimulated Breg as well as Beff, the level of p-BTK was low. Interestingly, basal p-BTK was significantly higher in unstimulated Beff as compared to unstimulated Breg, and ADO treatment significantly decreased basal p-BTK only in unstimulated Beff, but not in unstimulated Breg (Fig. 2a). Accordingly, after stimulation with anti-µ-F(ab’)2, p-BTK was only induced in Beff, but not in Breg, and the effect of stimulation was attenuated by ADO treatment (Fig. 2b). As anti-µ-F(ab’)2 specifically stimulates the µ-chain of the BCR IgM, we examined the expression of IgM on CD73+ Breg and CD73neg Beff cells. Both B cell subtypes expressed comparable IgM levels on their surface and were therefore able to receive the stimulatory BCR signal (Fig. 2c, d). These observations suggest that BTK in CD73+ Breg is less responsive than in CD73neg Beff cells.

Phosphorylation of BTK in B cell subsets. a Unstimulated CD73neg Beff cells showed a significantly higher phosphorylation of BTK than CD73+ Breg two hours after isolation and staining. After pre-treatment with ADO for 2 h, BTK phosphorylation was reduced in CD73neg Beff cells. Data shown as median with interquartile range; paired t-test; n = 5. b Isolated human B cells were first stained for CD73 and then incubated with ADO for two hours. Phosphorylation was measured after stimulation with anti-µ-F(ab’)2. Incubation with ADO reduced BTK phosphorylation in CD73neg Beff cells. Analyzed with a two-way ANOVA; n = 5. c Expression of IgM and CD73 on CD19+ B cells in the density plot of one representative patient. d Frequency of IgM+ cells in the CD73neg Beff and CD73+ Breg subset
ADO decreases the ability of B cells to secrete Ca2+
We observed a clear effect of ADO on BTK phosphorylation in B cells, which in turn is essential for the BCR-mediated Ca2+ flux. In order to further establish this coherency, isolated B cells were treated with ADO or with the BTK inhibitor ibrutinib overnight. Intracellular Ca2+ secretion was measured by FACS by the emission spectrum of Indo-1 after stimulation with anti-µ-F(ab’)2 (Fig. 3a). ADO as well as ibrutinib decreased the ability of B cells to secrete intracellular Ca2+, while ADO further enhanced the ibrutinib-mediated Ca2+ influx inhibition (p ≤ 0.01, Fig. 3b). Peripheral B cells of both healthy donors and cancer patients showed a decrease in Ca2+ influx after treatment with ADO (Fig. 3c). These results suggest that ADO inhibits Ca2+ influx by BTK inhibition in B cells of healthy donors and cancer patients.

Calcium influx and B cell responses to treatment with the BTK inhibitor ibrutinib. a Isolated B cells were treated overnight with ADO and/or ibrutinib, stained with Indo-1 and stimulated with anti-µ-F(ab’)2 for direct Ca2+ measurement. The overlay plot of the kinetics shows control B cells in black, ADO-treated B cells in light gray and ibrutinib-treated B cells in dark gray of one representative experiment. The results were calculated by the following ratio: intensity indo-1 violet/indo-1 blue. b The Ca2+ influx was significantly reduced by ibrutinib and by the combination of ibrutinib and ADO. AUC = area under the curve as mean ± SD analyzed with one-way ANOVA; median with interquartile range; n = 9. c The data suggest that peripheral B cells of tumor patients (n = 3) show significantly decreased Ca2+ influx after ADO treatment as compared to B cells of healthy donors (n = 9) analyzed with a two-way ANOVA; median with interquartile range. d The MFI of CD39 and CD73 on B cells was measured by FACS before and after treatment with ibrutinib or DMSO as a control. Ibrutinib induced a significant decrease in CD39 expression. Data were analyzed with a two-way ANOVA; median with interquartile range. e B cells were stimulated with CD40L, IL-4 and hemagglutinin and treated with ibrutinib for 6 days. Remaining ATP as measured by luminescence was significantly higher in ibrutinib-treated B cells as compared to control B cells (n = 7). Analyzed by two-way ANOVA; (*) p < 0.05; (**) p < 0.01; (***) p < 0.001; (****) p < 0.0001. f Adenosine concentrations measured by mass spectrometry for B cells of one donor stimulated for 6 days with CD40L, IL-4 and hemagglutinin and treated with ibrutinib. ATP was added at a concentration of 20uM
BTK is essential for the ADO production of B cells
Stimulated B cells were pre-treated with ibrutinib for six days. Next, the expression of the ectonucleotidases was measured by FACS and the hydrolysis of exogenous ATP was measured by luminescence. The expression of CD39 was significantly decreased in ibrutinib-treated B cells as compared to untreated controls (p ≤ 0.01, Fig. 3d). CD73 showed no differences after treatment with ibrutinib (p = 0.8). Additionally, the ability for hydrolysis of exogenous ATP was significantly reduced in ibrutinib-treated B cells as measured by luminescence assay. Experiments of seven representative donors are displayed in Fig. 3e (p ≤ 0.05). In order to confirm these results measured by luminescence, samples of one patient were additionally measured by mass spectrometry. Ibrutinib-treated B cells of this patient expressed less CD39 (MFI 55.7 vs. 31.8). Accordingly, these ibrutinib-treated B cells produced less ADO than the untreated control (Fig. 3f). These results suggest that BTK induces an upregulation of CD39 on B cells, which in turn increases the ability for ADO production.
B cells in murine HNSCC express CD39 and CD73
In order to prove the therapeutic potential of the ADO metabolism, we applied an orthotopic murine tumor model. In tumor-bearing mice, we observed a significant increase in white blood counts (WBC) 21 days after tumor induction, mainly caused by a strong increase in granulocytes and monocytes (Fig. 4a). Additionally, lymphocytes were significantly increased in the blood of tumor-bearing mice due to the increase in T and B cell numbers (Fig. 4b, c). However, the frequency of ADO-producing Breg (CD39+CD73+) remained stable in tumor-bearing mice (Fig. 4d). Also, the percentages of IgM+, IgM+IgD+ or IgD+ B cells were not significantly changed between control mice and tumor-bearing mice (Fig. 4e). Tumor-infiltrating B cells (TIL-B) were detected by expression of CD19 (Fig. 5a). In contrast to peripheral blood, a greater proportion (25–30%) of these B cells expressed CD39 as well as CD73 simultaneously (Figs. 4d and 5b; d21 w/o treatment). TIL-B were more mature as compared to B cells found in the peripheral blood as based on IgD expression (Figs. 4e and 5c).

Cell populations in the peripheral blood of HNSCC-bearing mice. a Leukocytes were determined using animal blood counter (n = 57). b–c Populations of blood lymphocytes and B cells were analyzed by flow cytometry (n = 43/47). d–e The expression profile for CD39/CD73 and IgM/IgD was determined in peripheral B cells. Cell numbers per µl blood were calculated based on WBC. Blood of control and tumor-bearing mice, which were either left untreated or treated with the ADORA2A antagonist SCH-58261, was analyzed at the tumor induction and after 21 days (n = 43/54)

Tumor growth and TIL in HNSCC-bearing mice. HNSCC mice were either left untreated or treated with the ADORA2A antagonist (SCH-58261). a At d21 after tumor induction, tumors were harvested for determination of CD19+ B cell frequency (n = 20). Infiltrating tumor cells were further characterized for b CD39 and CD73 expression (n = 22), as well as c IgM and IgD expression (n = 22). d–e Mice, which were treated with SCH-58261, showed a decrease in tumor weight and in the absolute number of tumor-infiltrating lymphocytes (n = 22/21). f However, the number of tumor-infiltrating B cells per g tumor was decreased (n = 21) and g the number of TIL per g tumor was stable (n = 21)
In our murine tumor model, we observed a high proportion of CD39+CD73+ TIL-B, which have the potential to produce ADO. We, therefore, assumed that increased concentrations of B cell-derived ADO contribute to an immunosuppressive microenvironment and tumor progression. To test this hypothesis, we applied the ADORA2A-inhibitor SCH-58261 in our tumor-bearing mice. ADORA2A is expressed on murine lymphocytes, but not on tumor cells (Supplementary fig. S1b/c). After 21 days, the inhibition of ADORA2A with SCH-58261 significantly reduced tumor growth as well as the absolute number of TIL (Fig. 5d, e). Yet, the number of TIL per tumor weight remained unchanged (Fig. 5f, g). Interestingly, the percentage of TIL-B increased significantly in treated mice (Fig. 5a), while their expression of CD39 / CD73 and their status of maturity (by IgM and IgD expression) remained unchanged (Fig. 5b, c). In contrast, the frequency of peripheral B cells was slightly reduced in treated mice (Fig. 4c), while the expression of CD39 and IgD was upregulated on peripheral B cells (Fig. 4d, e). Of note, the inhibition of ADORA2A induced a significant decrease in the tumor mass even when the antagonistic therapy was started as late as one week after tumor initiation.
Discussion
Immunosuppression is one of the major tumor escape mechanisms, and ADO is known to play an important role in the generation of an immunosuppressive tumor microenvironment [13, 14, 23,24,25]. Several cell types are capable of producing ADO, including regulatory immune cells, cancer cells as well as non-cellular vesicles, mainly due to the expression and activity of the ectonucleotidases, CD39 and CD73 on their surface [25,26,27]. Although the effect of ADO on T cell effector function has been extensively studied [13], the effect of ADO on B cells is still unknown. At the same time, it becomes evident that B cells may play an important role in the immune response of cancer patients [28]. Recently, we have shown that human peripheral B cells also express CD39 and CD73 on the cell surface and generate large amounts of exogenous ADO. Therefore, these B cells have regulatory characteristics, which enable them to suppress T cell functions, in vitro [12]. In fact, their ability to produce ADO is significantly higher than other immune cells, which are either CD39+CD73neg (e.g., Treg, monocytes) or CD73+CD39neg (e.g., THelper cells, TKiller cells). However, we are well aware that adenosine production is only one functional aspect of tumor-infiltrating B cells. As more than 50% of B cells can produce adenosine, the same cells are probably also capable of secreting cytokines or to process antigens [29]. In view of this hypothesis, we support the idea that tumor-infiltrating B cells may serve as a positive prognostic marker in cancer patients [30, 31]. The correlation between humoral immune responses and different patterns of immune infiltrates in HNSCC patients has been described in detail by Lechner et al. [32]. While humoral immune responses were lower in HPV+ cancer patients, higher humoral immune responses were associated with CD4+ T cell-dominated immune infiltrates.
Our results demonstrate that the frequency of ADO-producing Breg is lower in the TME as compared to the peripheral blood of cancer patients (Fig. 1a). We believe that the frequency is regulated by (I) the selective attraction of different B cell subsets into the TME as well as (II) the influence of the TME on stationary B cells. Nevertheless, even in the TME almost 50% of B cells are capable of producing extracellular ADO by the CD39/CD73 pathway.
Previously, we have demonstrated how chemotherapeutic drugs can influence the ADO production in B cells, in vitro and in vivo [17, 33]. We now describe the presence of ADO-producing Breg in the tumor tissue of HNSCC in human and mice, in which the majority of B cells have a regulatory phenotype (CD39+CD73+). By their continuous production of ADO, Breg help to create an immunosuppressive micromilieu, which is a characteristic feature of many solid tumors [13, 14]. Accordingly, immunosuppression by ADO is regularly regarded as a tumor promoting factor; and solid tumors contain high levels of endogenous immune-suppressive ADO as compared to the normal tissue [5, 13, 14]. In HNSCC patients, low levels of ADORA2A correlate with low frequencies of Treg and a high frequencies of CD8+ T cells, indicating a significantly improved survival rate [16].
The mechanism by which ADO suppresses T cells has been extensively analyzed and includes upregulation of cAMP after ADORA2A activation [16, 24]. Consequently, T-cell proliferation and IFN-γ release are decreased [34]. In contrast to T cells, the knowledge regarding ADO signaling in B cells is very limited. Here, we demonstrate that extracellular ADO downregulates BCR-mediated signaling by decreased phosphorylation of BTK (Bruton’s tyrosine kinase). BTK is a central member of the intracellular signaling cascade after the activation of the common BCR. Mutations along the BTK gene BTK cause X-linked agammaglobulinemia (XLA), characterized by severe defects of the B-cell development and the innate immune system [35]. BTK phosphorylation induces downstream activation of Akt, NF-κB and Ca2+ influx [36, 37], which in turn regulates the activation of pro-inflammatory proteins [38]. Our previously published work has described the influence of ADO on the function of B cells [12]. This includes, e.g., reduced expression of cytokines (IL-6 and IL-8) and reduced proliferation of activated B cells in the presence of ADO.
In the past, other research groups have shown that extracellular ADO induces a reduction in Ca2+ influx in lymphocytes [39]. Our experiments now describe one of the underlying mechanisms. In detail, exogenous ADO decreases phosphorylation of BTK with a consequent decrease in Ca2+ influx in B cells of healthy donors and cancer patients, and this effect is dependent on the ADO receptor A2. In our experiments, the decrease in Ca2+ influx by ADO was further enhanced by the BTK inhibitor ibrutinib, indicating that either ADO or ibrutinib may utilize additional signaling events other than BTK to inhibit the calcium flux in B cells. The BTK inhibitor ibrutinib is well known as a treatment option in chronic lymphocytic leukemia and mantle cell lymphoma, in which ibrutinib silences the downstream pathways of ERK, PI3K, NF-κB and Akt, and induces apoptosis of malignant B cells [40, 41].
The therapeutic potential of ibrutinib in solid tumors is currently being evaluated by a series of research teams including our own group [42,43,44]. However, the prognostic benefit of these molecular changes in patients with solid tumors is still unknown.
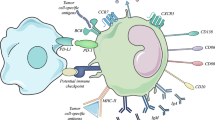
In our experiments, BCR-induced BTK phosphorylation was detectable only in Beff, but not in Breg (Fig. 2b). In contrast, only Breg were able to produce ADO by co-expression of the ectonucleotidases CD39 and CD73. We, therefore, hypothesize that CD73+ Breg are able to suppress BCR signaling in CD73neg Beff by ADO production in the tumor tissue as illustrated in Fig. 6. In addition, our results suggest a negative feedback mechanism in B cells, as ibrutinib decreases the production of extracellular ADO by downregulation of CD39 on B cells.

Adenosine affects the B cell receptor pathway. In B effector cells, binding of the antigen α-µ-F(ab’)2 to the BCR induces Syk and PIP3 activation supported by PI3K signaling. PIP3 recruits BTK, inducing auto-phosphorylation. The activated BTK activates PLCγ2 and IP3, binding to the endoplasmatic reticulum (ER), which secrets Ca2+. On Breg cells, extracellular ADO is produced by hydrolysis of ATP by the ectonucleotidases CD39 and CD73. ADO binds to different ADO receptors, downregulating the auto-phosphorylation of BTK and the Ca2+ influx in CD73neg B cells. In Breg cells, no BTK phosphorylation was found upon binding of the antigen α-µ-F(ab’)2
In knowledge of these molecular mechanisms, we hypothesize that blockade of the adenosine pathway may have a therapeutic potential. Others have previously shown that the inhibition of ADORA2A in mice leads to a delayed growth of HNSCC tumors and enhances the anti-tumor response of CD8+ T cells [16]. Our own murine tumor model confirmed the idea that ADO signaling is a crucial factor contributing to tumor growth.
Other murine tumor studies have shown that the inhibition of ADORA2A decreases the number of T cells in the tumor environment [13] and the metastasis of CD73+ tumors [23]. Our experiments now add to this knowledge by demonstrating that the number of tumor-infiltrating B cells increases during the inhibition of ADORA2A. At the same time, we observed an increased CD39+CD73+ co-expression, when murine tumor-infiltrating B cells were treated with the ADORA2A inhibitor SCH-58261. The current literature describes a CD73+ B-cell subset, which is regularly found in the germinal centers [45]. Others have described that the expression of ectonucleotidases on B cells is dependent on their maturation status [46]. Also, the method of stimulation may have diverse effects on the maturation and expression of ADO-producing ectoenzymes on B cells, in vitro. As treated mice showed a significant decrease in tumor size in our experiments, it remains to be elucidated whether these cells are genuine Breg or whether they belong to a specific mature B cell subset.
In the peripheral blood, the percentage of Breg was low in healthy mice (~ 3%) as compared to healthy humans (~ 60%). However, in tumor-bearing mice, the Breg frequency is increased in the peripheral blood as well as in tumor tissue. In contrast, the majority of human circulating B cells express CD39 and CD73 in healthy donors as well as in HNSCC patients. This observation could be explained by the fact that mice were kept under pathogen-free conditions, while the human immune system is constantly active due to continuous exposure to pathogenic agents. The function of ADO-producing Breg may therefore be the suppression of excessive immune reaction in order to prevent tissue damage or autoimmunity [47].
Conclusion
We demonstrate the presence of a novel ADO-producing Breg population within the tumor microenvironment in mice and humans. A new model is proposed on how ADO-producing Breg can influence the function of Beff cells in healthy donors and cancer patients. Thus, the modulation of the ADO pathway in B cells may serve as a therapeutic approach for cancer patients.
Abbreviations
- ADO:
-
Adenosine
- ADORA:
-
Adenosine receptor A
- BCR:
-
B cell receptor
- Breg :
-
Regulatory B cells
- BTK:
-
Bruton’s tyrosine kinase
- GAPDH:
-
Glyceraldehyde 3-phosphate dehydrogenase
- G6PD:
-
Glucose-6-phosphate dehydrogenase
- HNSCC:
-
Head and neck squamous cell carcinoma
- PBG-D:
-
Porphobilinogen deaminase
- RPL13A:
-
Ribosomal protein L13a
- SCC:
-
Squamous cell carcinoma
- TBP:
-
TATA-box-binding protein
- Treg :
-
Regulatory T cells
References
Kuss I, Hathaway B, Ferris RL, Gooding W, Whiteside TL (2004) Decreased absolute counts of T lymphocyte subsets and their relation to disease in squamous cell carcinoma of the head and neck. Clin Cancer Res Off J Am Assoc Cancer Res 10:3755–3762. https://doi.org/10.1158/1078-0432.ccr-04-0054
Meucci S, Keilholz U, Tinhofer I, Ebner OA (2016) Mutational load and mutational patterns in relation to age in head and neck cancer. Oncotarget 7:69188–69199. https://doi.org/10.18632/oncotarget.11312
Ferris RL, Blumenschein G Jr, Fayette J et al (2016) Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 375:1856–1867. https://doi.org/10.1056/NEJMoa1602252
Gunderson AJ, Kaneda MM, Tsujikawa T et al (2016) Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas cancer. Cancer Discov 6:270–285. https://doi.org/10.1158/2159-8290.cd-15-0827
Hashimoto A, Okada H, Jiang A, Kurosaki M, Greenberg S, Clark EA, Kurosaki T (1998) Involvement of guanosine triphosphatases and phospholipase C-gamma2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J Exp Med 188:1287–1295
Coronella JA, Telleman P, Kingsbury GA, Truong TD, Hays S, Junghans RP (2001) Evidence for an antigen-driven humoral immune response in medullary ductal breast cancer. Cancer Res 61:7889–7899
Distel LV, Fickenscher R, Dietel K et al (2009) Tumour infiltrating lymphocytes in squamous cell carcinoma of the oro- and hypopharynx: prognostic impact may depend on type of treatment and stage of disease. Oral Oncol 45:e167–e174. https://doi.org/10.1016/j.oraloncology.2009.05.640
Milne K, Kobel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, Watson PH, Nelson BH (2009) Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PLoS ONE 4:e6412. https://doi.org/10.1371/journal.pone.0006412
Riemann D, Wenzel K, Schulz T, Hofmann S, Neef H, Lautenschlager C, Langner J (1997) Phenotypic analysis of T lymphocytes isolated from non-small-cell lung cancer. Int Arch Allergy Immunol 114:38–45
Schuler PJ, Laban S, Doescher J, Bullinger L, Hoffmann TK (2017) Novel treatment options in head and neck cancer. Oncol Res Treat 40:342–346. https://doi.org/10.1159/000477254
Germain C, Gnjatic S, Tamzalit F et al (2014) Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. Am J Respir Crit Care Med 189:832–844. https://doi.org/10.1164/rccm.201309-1611OC
Saze Z, Schuler PJ, Hong CS, Cheng D, Jackson EK, Whiteside TL (2013) Adenosine production by human B cells and B cell-mediated suppression of activated T cells. Blood 122:9–18. https://doi.org/10.1182/blood-2013-02-482406
Ohta A, Gorelik E, Prasad SJ et al (2006) A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A 103:13132–13137. https://doi.org/10.1073/pnas.0605251103
Blay J, White TD, Hoskin DW (1997) The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res 57:2602–2605
Kasama H, Sakamoto Y, Kasamatsu A et al (2015) Adenosine A2b receptor promotes progression of human oral cancer. BMC Cancer 15:563. https://doi.org/10.1186/s12885-015-1577-2
Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, Kulkarni AB, Zhang WF, Sun ZJ (2017) Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer 16:99. https://doi.org/10.1186/s12943-017-0665-0
Ziebart A, Huber U, Jeske S, Laban S, Doescher J, Hoffmann TK, Brunner C, Jackson EK, Schuler PJ (2018) The influence of chemotherapy on adenosine-producing B cells in patients with head and neck squamous cell carcinoma. Oncotarget 9:5834–5847. https://doi.org/10.18632/oncotarget.23533
Storch B, Meixlsperger S, Jumaa H (2007) The Ig-alpha ITAM is required for efficient differentiation but not proliferation of pre-B cells. Eur J Immunol 37:252–260. https://doi.org/10.1002/eji.200636667
Elsaesser AF, Schwarz S, Joos H, Koerber L, Brenner RE, Rotter N (2016) Characterization of a migrative subpopulation of adult human nasoseptal chondrocytes with progenitor cell features and their potential for in vivo cartilage regeneration strategies. Cell Biosci 6:11. https://doi.org/10.1186/s13578-016-0078-6
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45
O'Malley BW Jr, Cope KA, Johnson CS, Schwartz MR (1997) A new immunocompetent murine model for oral cancer. Arch Otolaryngol Head Neck Surg 123:20–24
Vahle AK, Kerem A, Ozturk E, Bankfalvi A, Lang S, Brandau S (2012) Optimization of an orthotopic murine model of head and neck squamous cell carcinoma in fully immunocompetent mice–role of toll-like-receptor 4 expressed on host cells. Cancer Lett 317:199–206. https://doi.org/10.1016/j.canlet.2011.11.027
Beavis PA, Divisekera U, Paget C et al (2013) Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci U S A 110:14711–14716. https://doi.org/10.1073/pnas.1308209110
Bastid J, Regairaz A, Bonnefoy N et al (2015) Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res 3:254–265. https://doi.org/10.1158/2326-6066.cir-14-0018
Deaglio S, Dwyer KM, Gao W et al (2007) Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med 204:1257–1265. https://doi.org/10.1084/jem.20062512
Schuler PJ, Saze Z, Hong CS et al (2014) Human CD4+ CD39+ regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73+ exosomes or CD73+ cells. Clin Exp Immunol 177:531–543. https://doi.org/10.1111/cei.12354
Figueiro F, Muller L, Funk S, Jackson EK, Battastini AM, Whiteside TL (2016) Phenotypic and functional characteristics of CD39high human regulatory B cells (Breg). Oncoimmunology 5:e1082703. https://doi.org/10.1080/2162402x.2015.1082703
Biragyn A, Lee-Chang C (2012) A new paradigm for an old story: the role of regulatory B cells in cancer. Front Immunol 3:206. https://doi.org/10.3389/fimmu.2012.00206
Yuseff MI, Pierobon P, Reversat A, Lennon-Dumenil AM (2013) How B cells capture, process and present antigens: a crucial role for cell polarity. Nat Rev Immunol 13:475–486. https://doi.org/10.1038/nri3469
Pretscher D, Distel LV, Grabenbauer GG, Wittlinger M, Buettner M, Niedobitek G (2009) Distribution of immune cells in head and neck cancer: CD8+ T-cells and CD20+ B-cells in metastatic lymph nodes are associated with favourable outcome in patients with oro- and hypopharyngeal carcinoma. BMC Cancer 9:292. https://doi.org/10.1186/1471-2407-9-292
Hladikova K, Koucky V, Boucek J et al (2019) Tumor-infiltrating B cells affect the progression of oropharyngeal squamous cell carcinoma via cell-to-cell interactions with CD8(+) T cells. J Immunother Cancer 7:261. https://doi.org/10.1186/s40425-019-0726-6
Lechner A, Schlosser HA, Thelen M et al (2019) Tumor-associated B cells and humoral immune response in head and neck squamous cell carcinoma. Oncoimmunology 8:1535293. https://doi.org/10.1080/2162402X.2018.1535293
Huber U, Ziebart A, Schwarz S, Jerg M, Laban S, Hoffmann TK, Schuler PJ (2015) Adenosine-producing B cells in patients with head and neck cancer. Oral Oncol 51:e34. https://doi.org/10.1016/j.oraloncology.2015.02.023
Lukashev D, Sitkovsky M, Ohta A (2007) From "Hellstrom Paradox" to anti-adenosinergic cancer immunotherapy. Purinergic Signal 3:129–134. https://doi.org/10.1007/s11302-006-9044-9
Weber ANR, Bittner Z, Liu X, Dang TM, Radsak MP, Brunner C (2017) Bruton's tyrosine kinase: an emerging key player in innate immunity. Front Immunol 8:1454. https://doi.org/10.3389/fimmu.2017.01454
Inabe K, Ishiai M, Scharenberg AM, Freshney N, Downward J, Kurosaki T (2002) Vav3 modulates B cell receptor responses by regulating phosphoinositide 3-kinase activation. J Exp Med 195:189–200
Saito K, Tolias KF, Saci A, Koon HB, Humphries LA, Scharenberg A, Rawlings DJ, Kinet JP, Carpenter CL (2003) BTK regulates PtdIns-4,5–P2 synthesis: importance for calcium signaling and PI3K activity. Immunity 19:669–678
Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386:855–358. https://doi.org/10.1038/386855a0
Linnemann C, Schildberg FA, Schurich A et al (2009) Adenosine regulates CD8 T-cell priming by inhibition of membrane-proximal T-cell receptor signalling. Immunology 128:e728–e737. https://doi.org/10.1111/j.1365-2567.2009.03075.x
Chang BY, Francesco M, De Rooij MF et al (2013) Egress of CD19(+)CD5(+) cells into peripheral blood following treatment with the Bruton tyrosine kinase inhibitor ibrutinib in mantle cell lymphoma patients. Blood 122:2412–2424. https://doi.org/10.1182/blood-2013-02-482125
Herman SE, Gordon AL, Hertlein E et al (2011) Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 117:6287–6296. https://doi.org/10.1182/blood-2011-01-328484
Guo W, Liu R, Bhardwaj G et al (2014) Targeting Btk/Etk of prostate cancer cells by a novel dual inhibitor. Cell Death Dis 5:e1409. https://doi.org/10.1038/cddis.2014.343
Wang JD, Chen XY, Ji KW, Tao F (2016) Targeting Btk with ibrutinib inhibit gastric carcinoma cells growth. Am J Transl Res 8:3003–3012
Wei L, Su YK, Lin CM et al (2016) Preclinical investigation of ibrutinib, a Bruton's kinase tyrosine (BTK) inhibitor, in suppressing glioma tumorigenesis and stem cell phenotypes. Oncotarget 7:69961–69975. https://doi.org/10.18632/oncotarget.11572
Conter LJ, Song E, Shlomchik MJ, Tomayko MM (2014) CD73 expression is dynamically regulated in the germinal center and bone marrow plasma cells are diminished in its absence. PLoS ONE 9:e92009. https://doi.org/10.1371/journal.pone.0092009
Barankiewicz J, Dosch HM, Cohen A (1988) Extracellular nucleotide catabolism in human B and T lymphocytes. The source of adenosine production. J Biol Chem 263:7094–7098
van de Veen W, Stanic B, Wirz OF, Jansen K, Globinska A, Akdis M (2016) Role of regulatory B cells in immune tolerance to allergens and beyond. J Allergy Clin Immunol 138:654–665. https://doi.org/10.1016/j.jaci.2016.07.006
Fu KK, Rayner PA, Lam KN (1984) Modification of the effects of continuous low dose rate irradiation by concurrent chemotherapy infusion. Int J Radiat Oncol Biol Phys 10:1473–1478. https://doi.org/10.1016/0360-3016(84)90371-7
Khurana D, Martin EA, Kasperbauer JL, O'Malley BW Jr, Salomao DR, Chen L, Strome SE (2001) Characterization of a spontaneously arising murine squamous cell carcinoma (SCC VII) as a prerequisite for head and neck cancer immunotherapy. Head Neck 23:899–906. https://doi.org/10.1002/hed.1130
Acknowledgements
Open Access funding provided by Projekt DEAL. We thank Katja Hasch, Gabriela Cudek, Monika Jerg and Heidrun Jäger for their support in performing the experiments. Acknowledgements go to the research group of Hassan Jumaa (Institute of Immunology, University Medical Center Ulm, Germany) for their support with the Calcium influx experiments.
Funding
The research was supported by the German Research Foundation (DFG) Grant # SCHU 2536/3 (Schuler PJ), by NIH grants DK068575, DK079307, DK091190, HL109002 (Jackson EK), by the International Graduate School in Molecular Medicine Ulm (Jeske SS) and by the doctoral funding program ‘Experimental Medicine’ of the Medical Faculty of the University of Ulm (Brand M).
Author information
Authors and Affiliations
Contributions
SSJ, MB and AZ performed experiments; JD prepared figures; EKJ performed mass spectrometry experiments; JG and SL provided patient samples; CB, SSJ and PJS designed research and wrote the paper; TKH and JD edited the paper.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and ethical standards.
Human studies were approved by the ethics committee of the University Ulm, Helmholtzstr. 20, 89081 Ulm, Germany (#255/14). The Helsinki Declaration was followed. Mice were kept in groups of 4–5 mice in individually ventilated cages with nesting material under specific pathogen-free conditions in the animal facility at the University of Ulm. All animal experiments were performed in accordance with institutional and governmental regulations in compliance with European rules (2010/63/EU) and were approved by the regional animal ethics committee Regierungspräsidium (Tübingen, Germany), Konrad-Adenauer-Str. 20, 72072 Tübingen, Germany (TVA1270).
Informed consent
Informed consent was obtained in written form from all human donors including the healthy control group before sample collection. Donors consented to the use of their specimens and data for research and for publication.
Animal source
Animals were obtained from Janvier Labs; CS 4105 Le Genest St Isle; 53941 St Berthevin Cedex, France.
Cell line authentication
The murine SCC VII cell line, which derived from a spontaneously arising squamous cell carcinoma of C3H mice, was first described by Fu et al. [48], further characterized [49] and kindly provided by Sven Brandau [22] (University of Essen, Germany). The SCC VII cell line was authenticated in a bioassay, in which 1 × 105 SCC VII cells were injected subcutaneously (back/flank) of fully immunocompetent C3H/HeN mice and tumor growth was observed within 21 days post-inoculation with a size of maximal 1 cm3 as described [22, 48, 49].
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jeske, S.S., Brand, M., Ziebart, A. et al. Adenosine-producing regulatory B cells in head and neck cancer. Cancer Immunol Immunother 69, 1205–1216 (2020). https://doi.org/10.1007/s00262-020-02535-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-020-02535-6