
Abstract
Purpose
Human interleukin-21 (IL-21) is a class I cytokine previously reported in clinical studies on immune responsive cancers. Here we report the effects of systemic IL-21 therapy on the immune system in two phase 1 trials with this novel cytokine.
Experimental design
Recombinant IL-21 was administered by intravenous bolus injection at dose levels from 1 to 100 μg/kg using two planned treatment regimens: thrice weekly for 6 weeks (3/week); or once daily for five consecutive days followed by nine dose-free days (5 + 9). The following biomarkers were studied in peripheral blood mononuclear cells (PBMC) during treatment: phosphorylation of STAT3, alterations in the composition of leukocyte subsets, ex vivo cytotoxicity, expression of effector molecules in enriched CD8+ T cells and CD56+ NK cells by quantitative RT-PCR, and gene array profiling of CD8+ T cells.
Results
Effects of IL-21 were observed at all dose levels. In the 5 + 9 regimen IL-21 induced a dose dependent decrease in circulating NK cells and T cells followed by a return to baseline in resting periods. In both CD8+ T cells and CD56+ NK cells we found up-regulation of perforin and granzyme B mRNA. In addition, full transcriptome analysis of CD8+ T cells displayed changes in several transcripts associated with increased cell cycle progression, cellular motility, and immune activation. Finally, cytotoxicity assays showed that IL-21 enhanced the ability of NK cells to kill sensitive targets ex vivo.
Conclusions
IL-21 was biologically active at all dose levels administered with evidence of in vivo NK cell and CD8+ T cell activation.
Similar content being viewed by others


Introduction
IL-21 is a cytokine belonging to the common γ-chain family of cytokines, which also includes IL-2, IL-4, IL-7, IL-9 and IL-15. IL-21 is produced by activated CD4+ T cells and NKT cells and is induced by IL-6 and by a positive autocrine feedback loop [8, 35]. Follicular helper T cells have been shown to be a major source of IL-21 [7]. Similar to the other common γ-chain family of cytokines IL-21 signals through the Janus activated kinase (Jak)/signal transducer and activator of transcription (STAT) pathway, with STAT1 and STAT3 as major targets. In addition signaling through STAT4, STAT6, MAPK and PI3K pathways have been demonstrated [11, 51]. IL-21 promotes activation, cytokine and antigen-dependent proliferation of CD8+ T cells as well as NK cell activation and cytolytic activity [4, 26, 35, 50]. In contrast to IL-2, IL-21 does not promote the proliferation and activity of Treg cells [36] but inhibits the generation of human Treg cells in vitro [25]. Together with IL-6, IL-21 is a main driver of Th17 cell differentiation [22, 23, 34, 52], but depending on the context IL-21 can also promote both Th1 and Th2 responses [42], suggesting that IL-21 may play a role in autoimmune diseases [13, 48]. IL-21 has both proliferative and pro-apoptotic effects on B cells and promotes isotype switching and differentiation of B cells into plasma cells leading to increased immunoglobulin production (reviewed in [24]). In animal models IL-21 enhances anti-tumor immunity through mechanisms that require NK cells and/or CD8+ T cells depending on the model used [4, 27, 30, 40], and IL-21 therapy can lead to sustained tumor-specific CD8+ T cell responses [30], suggesting that IL-21 may be useful as an immunotherapeutic to treat cancer.
Metastatic melanoma (MM) and stage IV renal cell carcinoma (RCC) are two diseases commonly used for testing experimental immunotherapies due to a well described history of responsiveness to immune modulation in these indications [17, 37]. The five-year survival for MM is 5–10% with a median survival of the order of seven months [2, 3]. Recombinant human IL-21 (IL-21) is being developed for clinical use by both ZymoGenetics Inc. and Novo Nordisk A/S. Two phase 1 dose escalation trials of recombinant human IL-21 administered intravenously to MM or RCC patients have recently been conducted showing a favorable safety profile and signs of clinical activity [9, 45]. Due to these positive results, IL-21 is now in phase 2 trials for MM, RCC, and non-Hodgkin’s lymphoma (clinicaltrials.gov identifiers NCT 00336986, NCT00389285, NCT00347971). Here we have investigated the systemic immune effects of IL-21 in blood samples taken from patients in these two phase 1 trials. We have measured alterations in lymphocyte subsets and CD8+ T cell activation markers by flow cytometry, performed transcriptional profiling on enriched CD8+ T cells and carried out ex vivo cytotoxicity assays. Our results confirm that IL-21 at the administered doses was immunologically active in patients with advanced MM and RCC.
Materials and methods
Trial design and patient population
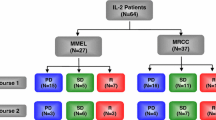
Seventy-two patients were evaluated in two phase 1 dose escalation studies conducted in Australia and the United States. The Australian study was conducted in patients with MM [9], while the US study was conducted in patients with either MM or RCC [45]. Both trials were open-label dose escalation phase 1 studies of recombinant human IL-21 administered by intravenous bolus injection. The primary objective was to assess the safety and tolerability of IL-21 at various dose levels and dosing regimens by determining dose-limiting toxicity (DLT) and estimating a maximum tolerated dose (MTD). Secondary objectives were to: identify a dose response for various biomarkers in the peripheral blood; estimate the minimum biologically effective dose (BED); determine the pharmacokinetics of IL-21; determine if anti-IL-21 antibodies were induced during therapy; and measure effects on tumor size.
In the Australian study the patients were allocated non-randomly in cohorts of two patients into one of two parallel treatment arms: treatment thrice weekly (Monday, Wednesday, Friday) for six weeks (3/week); or three cycles of daily dosing for five days followed by nine days of rest (5 + 9). If there was no symptomatic tumor progression at the week 8 assessment that required treatment by another modality, further treatment with use of the same dose and regimen was offered. In the US study patients were planned to receive two cycles of the 5 + 9 regimen, although for some patients the recovery period of 9 days was prolonged to 16 days to allow recovery from IL-21-induced toxicity. Eligible patients had histologically confirmed surgically incurable metastatic malignant melanoma. The US trial also included patients with metastatic RCC of predominantly clear cell histology. Details on inclusion criteria and disease assessment have been published previously [9]. The MTD was estimated to be 30 μg/kg for both treatment schedules. In the US trial another 30 patients were treated at the MTD to provide further data on safety and efficacy.
All patients provided written informed consent prior to any study-specific procedures. The Australian trial protocol was approved by the Human Research Ethics Committees of the participating institutions and was performed under the Australian Therapeutic Goods Administration Clinical Trials Notification (CTN) scheme. The study sponsor was Novo Nordisk A/S. In the US trial the institutional review boards of participating medical centers approved the protocol and associated materials, and patients gave written informed consent before study-specific procedures began.
Manufacturing of IL-21
The process for production, refolding and purification of the IL-21 molecule as well as analytical methods for assessing purity and potency were developed by ZymoGenetics Inc. Recombinant human IL-21 is expressed in E. coli as the N-terminal methionylated form of the molecule. Manufacturing of the product was carried out in the GMP facilities of Avecia, Ltd., UK.
STAT3 phosphorylation assay
This assay was carried out only in the Australian trial. Blood samples (2 mL) were drawn into lithium heparinized tubes immediately prior to the first IL-21 dosing (pre-sample) and 15 min after IL-21 dosing (post-sample). The pre-sample was split into two tubes. One tube was immediately put on ice and stored at <4°C until further processing to serve as negative/unstimulated control. To the second tube IL-21 was added to a concentration of 10 ng/mL, where after the tube was incubated for 15 min at RT to serve as the positive, patient-specific control. This dose of IL-21 is sufficient to induce maximal STAT3 phosphorylation, although the fact that stimulation was carried out at RT may have resulted in slightly lower STAT3 phosphorylation than would have been observed at 37°C (data not shown). After drawing the post treatment-sample all three samples were fixed by transferring 500 μL from each sample to tubes containing 9 mL FACS Lysing solution (BD Biosciences, San Jose, CA) and stored up to 120 min on ice. During this step samples were transferred to the laboratory at Cancer Trials Australia (Parkville, Victoria, Australia) where the remaining procedure was carried out. The cells were subsequently washed in PBS containing 1% FCS and fixed by incubation in 2% paraformaldehyde for 10 min at 37°C. Cells were washed again and resuspended in 90% methanol to permeabilize cells. At this step cells were frozen and kept at −80°C until further analysis. Cells were washed and stained for 1 h at RT with PE labeled antibody directed against STAT3 (pY705), clone 4, and FITC labeled CD3 antibody, clone UCHT1 (BD Biosciences). Cells were washed again and acquisition was done on a FACSCalibur (BD Biosciences). Data were analyzed using FACSDiva software (BD Biosciences).
Flow cytometry
US trial: Whole blood was collected into 3 mL heparin tubes for FACS analysis from each subject enrolled in the study. Samples were collected prior to dosing on Days 1 and 4 and on Day 10 of both dosing cycles. All samples were shipped overnight at ambient temperature to Esoterix Laboratory Services (East Windsor, NJ), and analyzed within 24 h of receipt. For each sample, 100 μL of whole blood was placed in TruCount tubes (BD Biosciences), and incubated with the appropriate dilutions of antibodies. The following antibodies were used for immunophenotyping: CD4 (clone SK3), CD8 (clone SK1), CD56 (clone NCAM16.2), CD45 (clone 2D1), CD14 (clone MφP9), CD25 (clone 2A3), and CD38 (clone HB7) were from BD Biosciences; CD3 (clone UCHT1) was from Immunotech; CD19 (clone HIB19) was from BD Pharmingen. Red blood cells were lysed using FACS Lysing Solution (BD Biosciences). After processing, Ab-stained samples were analyzed on a FACS Calibur flow cytometer using CellQuest acquisition software (version 3.3, BD Biosciences). Acquired data were analyzed with WinList flow cytometry data analysis software (version 3.0, Verity Software House, Topsham, ME). Responses to IL-21 dosing were assessed by factorial analysis of variance with repeated measures using JMP v5.1 (SAS Institute, Cary, NC).
Australian trial: Prior to IL-21 dosing, 5–10 mL blood were drawn into Lithium Heparin tubes (BD Biosciences) at Day 1 and Day 5. Blood samples were transferred at ambient temperature to Department of Haematology, Royal Melbourne Hospital, Australia and PBMC were prepared by density gradient centrifugation using Multi-Q-prep method (Beckman Coulter, Fullerton, CA) and resuspended in PBS with BSA to a final volume of 3.6 mL. The following list of antibodies was employed for labelling of blood cells prior to flow cytometric analysis; CD3 (clone UCHT1), CD4 (clone SK3), CD8 (clone T8), CD45 (clone J.33) were from Beckman Coulter and CD45RA (clone L48), CCR7 (clone 3D12) and CD62L (clone DREG-56) were from BD Biosciences. Cells were incubated at room temperature for 10 min with the antibodies prior to flow cytometry. After labelling, red blood cells were lysed and fixed. Acquisition was done on a XL-MCL instrument (Beckman Coulter) and data analysis performed with Expo32 software (Beckman Coulter).
Quantitative real-time RT-PCR (qRT-PCR) and microarray analysis
In the Australian trial citrated blood samples were collected from patients on Day 1 (before first IL-21 dose) and on Day 5. CD8+ cells were enriched as previously described [9]; in addition CD56 positive cells were enriched using CD56 Microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) and a magnetic MiniMACS Separation Unit (Miltenyi Biotec). Enriched cells were lysed in RNA extraction buffer and total RNA isolated as previously described. Fifty nanograms of total RNA was used to prepare targets by Two-Cycle Target labeling kit (Affymetrix, Santa Clara, Ca, USA) following the instructions of the manufacturer. Hybridization cocktails were hybridised onto Human Genome U133 Plus 2.0 GeneChips® (Affymetrix) at 45°C for 17 h (60 RPM) in a Hybridization Oven 640 (Affymetrix). GeneChips® were washed and stained in a GeneChip® fluidics station 450 using the fluidics protocol “EukGE-WS2v5_450” (Affymetrix). Chips were scanned in a GeneChip® scanner 3000 (Affymetrix).
For qRT-PCR analyses, cDNA was either prepared from total RNA (granzyme B and perforin-1 analyses) or from each of the IVT cRNA preparations (granzyme A, interferon-γ, CXCR3, and hyaluronan-mediated motility receptor (HMMR) analyses (confirming array results)) using random primers and TaqMan Reverse Transcription reagents (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions. Quantitative PCR was performed in duplicate on each of the cDNA samples (tenfold dilutions of cDNA) using TaqMan PCR core reagents (Applied Biosystems) and the ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems). Primers and FAM-labeled-probes for granzyme B mRNA, granzyme A mRNA, perforin-1 mRNA, interferon-γ mRNA, CXCR3 mRNA, HMMR mRNA, TATA-binding protein mRNA, and 18S rRNA, were ordered as Assays-on-Demand (Applied Biosystems). Probe sequences for these assays were as follows: CXCR3 (ATGGTCCTTGAGGTGAGTGACCACC (assay Hs00171041_m1)), granzyme B (CTTCCTTTAAGGGGGACTCTGGAGG (assay Hs00188051_m1)), granzyme A (TCCTGCTAATTCCTGAAGATGTCTG (assay Hs00196206_m1)), perforin-1 (GAGTGCCGCTTCTACAGTTTCCATG (assay Hs00169473_m1)), interferon-γ (AGAAATATTTTAATGCAGGTCATTC (assay Hs00174143_m1)), HMMR (GCTGAAAGGGAAGGAGGCTGAACTG (assay Hs00234864_m1)), TATA binding protein (TGGGTTTTCCAGCTAAGTTCTTGGA (assay Hs99999910_s1)), and 18S rRNA (TGGAGGGCAAGTCTGGTGCCAGCAG; assay Hs99999901_s1). Data were analyzed using ABI Prism SDS 2.2 software (Applied Biosystems), and expression levels for granzyme A, interferon-γ, CXCR3, and HMMR were normalized to TATA binding protein levels whereas perforin-1 and granzyme B were normalized to 18S rRNA levels.
Cytotoxicity assay
In the Australian trial heparinized blood was collected into CPT tubes (BD Biosciences) from the patients at screening (typically 1–2 weeks before first IL-21 dose) and at study day 1, and day 5 prior to dosing. PBMC were subsequently prepared by centrifugation, frozen in FCS with 10% DMSO and stored for up to one year in liquid nitrogen. Using PBMC from normal donors we have observed that this procedure may decrease cytotoxicity up to 50%. To perform the cytotoxicity assay cells were thawed and tested as effector cells in a standard 4 h 51Cr-release assay using 51Cr-labeled K562 erytholeukemia cells as target cells. For each patient all samples were run together in one assay in order to avoid inter-assay variation. When sufficient cells were available the fraction of NK cells was measured by flow cytometry as described above after thawing of the cells.
Statistical analysis
Microarray data were normalized and gene expression measures derived using the RMA algorithm, and S-PLUS version 7.0 and SAS version 9.1 were used for the statistical analysis. The logged gene expression levels from day 1 and day 5 samples from the 25 patients were fitted to a log dose ANCOVA model assuming that the regulation of a probeset is proportional to the log of the dose. A false discovery rate (FDR) of 1% was used (meaning that the expected percent of false positives is 1%). Clustering and heat-map generation of gene expression data were done using R (http://www.R-project.org). Log fold ratios (day 5 samples compared to day 1 samples (paired samples from individual patients)) were clustered hierarchically by complete linkage and euclidian distance. The Heatplus and geneplotter packages were used for generation of the clustered heatmap whereas GO, GOstats, and Rgraphviz packages were used for GO analysis and construction of the GO-tree (http://www.bioconductor.org). Differentially regulated probesets were also analysed using Ingenuity Pathway Analyses (http://www.ingenuity.com). Immune related genes (Fig. 3b) were identified by filtering genes containing the GO-term “immune regulation” or “immunological synapse”.
Results
Analysis of STAT3 phosphorylation
Upon engagement of IL-21R the primary signal transduction events are the activation of Janus kinase (Jak)1 and Jak3 leading to phosphorylation of STAT1 and STAT3 [1, 41]. As a primary marker of biological activity STAT3 phosphorylation was measured in 29 patients from the Australian trial. Based on in vitro studies we found that the optimal time for measuring STAT3 phosphorylation was 15 min after IL-21 administration as phosphorylated STAT3 (pSTAT3) is subsequently lost (data not shown), probably due to the action of the negative modulator, SOCS1, which has been shown to be induced by IL-21 [14]. We measured pSTAT3 by flow cytometry in whole blood samples drawn immediately before and 15 min after IL-21 dosing. We found that even at the lowest dose level (1 μg/kg) there was maximal induction of pSTAT3 in CD3+ T cells slightly higher than the in vitro positive control (Fig. 1a). In addition pSTAT3 was induced in a fraction of CD3− lymphocytes. Although we did not include markers for B cells and NK cells in these stainings, the non-T cells responding to IL-21 is likely to be a mix of both B cells and NK cells, since in vitro studies using blood from normal donors have shown that pSTAT3 is induced in a fraction of both B cells and NK cells upon IL-21 stimulation (data not shown). IL-21 induced similar levels of STAT3 phosphorylation at all dose levels (Fig. 1b) showing that IL-21 was biologically active even at the lowest dose.

IL-21 induced STAT3 phosphorylation at all dose levels. Whole blood was drawn immediately before the first IL-21 dose and 15 min after dosing. Half of the pre-dose sample was stimulated for 15 min ex vivo with 10 ng/mL IL-21 to provide a positive control. Samples were analyzed by flow cytometry for pSTAT3 in CD3+ T cells. a Representative dot plots of pre-dosing, post-dosing and ex vivo positive control samples. b For each dose level the average pSTAT3 mean fluorescence intensity in CD3+ T cells in percent of the ex vivo positive control samples is shown
Effect on lymphocyte subsets
In the US trial blood samples were drawn on day 1, 4 and 10 during the first and second cycle of IL-21 treatment and three weeks following the final IL-21 dose. These were analyzed by flow cytometry for changes in the number of lymphocyte subsets. The absolute number of lymphocytes decreased in a dose-dependent fashion by trial day 4 but returned to pre-dose levels or slightly higher on day 10 (Fig. 2a). No change was observed from first to second dosing cycle (Fig. 2b). The number of CD4+ and CD8+ T cells, B cells and NK cells all decreased during dosing with the NK cells showing the most profound decrease, whereas monocytes increased (Fig. 2c). On day 10, 5 days after cessation of dosing, monocytes, T cells and NK cells returned to baseline whereas B cell numbers increased slightly beyond baseline levels. Similar results have been obtained from the Australian trial (data not shown). No change in the fraction of activated (CD25+) T cells was seen during dosing (data not shown) but the percent activated T cells increased from 14.9 ± 2.3 pre-dose to 21.4 ± 1.7 three weeks following the final IL-21 dose. Within the B cell population the fraction of naïve B cells, defined as CD19+CD38− decreased during dosing and the fraction of plasma B cells, defined as CD19+CD38HI increased during dosing (Fig. 2d). The plasma cells returned to baseline upon cessation of treatment, whereas the percent naïve B cells remained repressed three weeks following the final IL-21 dose (14.3 ± 1.8% post-dosing compared to 17.9 ± 2.1% pre-dose).

IL-21 induced transient lymphopenia and upregulation of cell adhesion molecules. Absolute number of lymphocytes were measured in the US trial by standard complete blood count analysis and depicted per dose level in cycle 1 (a) or per dose cycle at 30 μg/kg (b) (mean ± SEM). c, d Lymphocyte subsets were measured in the US trial by flow cytometry. c Absolute numbers of T helper cell (CD4+), T effector cell (CD8+), NK cells (CD16/56+), B cells (CD19+) and monocytes (CD14+) at 30 μg/kg cycle 1 (mean ± SEM). d Percent of naïve B cells (CD19+CD38−) and plasma cells (CD19+CD38HI) at 30 μg/kg cycle 1 (mean ± SEM). In the Australian trial CD8+ T cell adhesion markers were measured by flow cytometry. Percentage of CD8+ T cells that express CD62L (e) CCR7 (f) or CD45RA (g) is shown for individual patients in the 5 + 9 regimen. All samples were drawn on the indicated dosing day prior to dosing. As IL-21 administration was initiated on day 1, samples drawn on day 1 represent pre-dose samples
In the Australian trial responses during IL-21 treatment were assessed in peripheral blood mononuclear cells (PBMC) by flow cytometric analysis of cell surface markers on CD8+ T cells. Upon IL-21 treatment, the most predominant changes were an increase at day 5 in the frequency of CD62L and CCR7 positive CD8+ T cells. This increase was seen in a total of 14 out of 14 and 8 out of 10 evaluable subjects in the 5 + 9 regimen for CCR7 and CD62L, respectively (Fig. 2e, f). Moreover, we found an increase in the frequency of naïve (CD45RA+) CD8+ T cells in a total of 11 out of 14 subjects in the 5 + 9 regimen (Fig. 2g). No dose response relationship was observed for any of these parameters. Changes were less prominent in the 3/week regimen (data not shown), which may be due to the fact that patients in this regimen had received only two doses of IL-21 on day 5 compared to four doses in the 5 + 9 regimen, and sampling was done 48 h after the day 3 dose in the 3/week regimen compared to 24 h after the last dose in 5 + 9 regimen. Thus, results are not directly comparable between the regimens.
Full transcriptome analysis on CD8+ cells:
Microarray data from 25 patients from the Australian study were used for identifying IL-21-mediated transcriptional regulations in CD8+ cells. CD8+ cells were isolated from peripheral blood on day 1 prior to the first IL-21 dose and on day 5 prior to dosing and analysed for transcriptional changes. With a FDR of 1%, 6116 probe sets were found to be differentially regulated when the data were fitted to a log dose model. The log dose model assumes that the regulation of a gene is proportional to the log of the dose. Adding the criteria that average gene regulations at least should be twofold in either dose level 4, 5, or 6 (level 4 = 30 μg/kg; level 5 = 50 μg/kg; level 6 = 100 μg/kg), 471 probe sets were identified (Electronic supplementary Table 1).
To obtain an overview of the functional roles of the regulated genes, the 471 probe sets were analyzed for statistical overrepresented gene ontology (GO-) terms, describing genes overrepresented in different functional categories. A very clear overrepresentation was found for genes involved in cell cycle (Fig. 3a and ESM Table 2). The six most significant GO-terms are all directly cell cycle related (1:“cell cycle”, 2:“mitotic cell cycle”, 3:“cell division”, 4:“M phase”, 5:“mitosis”, and 6:“M phase of mitotic cell cycle”). The next four GO-terms are also clearly relatable to cell division; 7:“DNA replication”, 8:“DNA metabolism”, 9:“spindle organization and biogenesis”, and 10:“microtubule-based process”. This GO-term analysis clearly suggests an increased proliferation of the CD8+ cells on day 5 compared to day 1. On the gene level this is exemplified by the clear upregulations of cyclin A2, cyclin B1, cyclin B2, cyclin E1, and cyclin E2 with average fold-regulations from ∼4 to ∼13 (when averaging patients from level 4, 5, and 6 (ESM Table 1)). The cyclin up-regulations are part of a regulatory cascade that controls the activity of cyclin-dependent kinase 2 (CDK2), and phosphorylation of downstream targets involved in the initiation of DNA replication (S-phase)[32]. CDK2 and the proliferation markers PCNA (proliferating cell nuclear antigen) and Ki67 antigen (antigen identified by monoclonal antibody Ki-67) were also found to be significantly up-regulated (ESM Table 1). The functional roles of the differentially expressed genes were also analyzed using ingenuity pathway analysis (IPA). In agreement with the GO-term analysis, the top three functions found in IPA were “Cell Cycle”, “DNA Replication, Recombination, and Repair”, and “Cellular Assembly and Organization (e.g, “segregation of chromosomes”, and “formation of mitotic spindle”) (data not shown).

a Gene ontology (GO-) tree of significantly overrepresented GO-terms (Biological Process “BP” terms). Each circle in the tree corresponds to a particular GO term, and the size of the circle indicates the degree of overrepresentation (see also ESM Table 2). Circles close to the root of the tree corresponds to very generic terms whereas circles closer to the end of the branches represent increasingly specific terms. The ten most significantly overrepresented GO terms are: 1 cell cycle, 2 mitotic cell cycle, 3 cell division, 4 M phase, 5 mitosis, 6 M phase of mitotic cell cycle, 7 DNA replication, 8 DNA metabolism, 9 spindle organization and biogenesis, 10 microtubule-based process. b Heatmap of clustered gene regulations (immunorelated genes). Log2-fold regulations (day 5 versus day 1 (pre-dose)) for individual patients enrolled in the “5 + 9” regimen at six different dose levels are shown. Some genes are represented by two or three individual probesets. Average fold regulations (dose levels 30, 50, and 100 μg/kg) are listed for the individual probesets
Quantitative RT-PCR analysis
Although genes involved in immune functions were not among the highest represented GO-terms a separate analysis showed that several genes involved in immune function were regulated by IL-21 (Fig. 3b). Two of the highest up-regulated genes in this list were interferon (IFN)-γ and granzyme B (see appendix a), both of which are classical markers of effector T-cells. Quantitative RT-PCR analysis was performed on selected genes to verify the gene-array data, including also some genes that were less than twofold up-regulated but of potential interest. The quantitative RT-PCR analysis showed significant up-regulation on day 5 versus day 1 of IFN-γ, granzyme A and B, chemokine (C-X-C motif) receptor 3 (CXCR3), and HMMR (Fig. 4). In addition, analysis of granzyme B expression in CD56+ cells, primarily consisting of NK cells, NKT cells and a subset of CD8+ T cells, showed up-regulation of granzyme B in this subset as well.

Quantitative RT-PCR data. a Analysis of granzyme B and perforin mRNA levels in CD8+ and CD56+ cells from patients dosed three times weekly (“3/week”) or five consecutive days of dosing followed by 9 days with no dosing (“5 + 9”). b Analysis of granzyme A (GZMA), interferon-γ (IFN-γ), hyaluronan-mediated motility receptor (HMMR), and CXCR3 mRNA levels in CD8+ cells from patients in the 5 + 9 regimen. Data from day 1 (pre-dose) and day 5 samples are shown. Part of Fig. 4a has previously been published [9]
NK cytotoxicity
To examine whether IL-21 therapy increased the cytolytic activity of circulating NK cells we isolated PBMC in some patients from the Australian trial at two time points prior to IL-21 administration and on trial day 5 and used the PBMC as effectors against K562 target erythroleukemia cells. K562 cells are MHC class I deficient and classical targets of NK cell-mediated cytotoxicity. An increase in target cell killing was observed in post-dosing samples compared to pre-dosing samples in five out of six evaluable patients (Fig. 5). This increase is likely to be underestimated on a per-NK cell basis as the fraction of circulating NK cells was significantly lower on day 5 compared to the pre-dose sample on day 1 (data not shown).

Enhanced NK cytotoxicity towards K562. PBMC isolated at screening, day 1 (pre-dose) and day 5 were frozen immediately and stored in liquid nitrogen. After thawing the PBMC were incubated for 4 h with 51Cr-labeled K562 erythroleukemia cells. For each subject PBMC from all time points were tested in the same assay
Discussion
We have demonstrated that IL-21 has detectable and relevant biological effects on various lymphocyte subsets even at the lowest dose level tested. We found that within the dose range tested (1–100 μg/kg) IL-21 was able to induce maximal STAT3 phosphorylation in T cells from the blood at all dose levels. As previously shown IL-21 therapy resulted in systemically measurable biological effects at the lowest dose level, measured as increased serum sCD25 level and granzyme B expression in CD8+ T cells on study day 5 [9]. Increases in sCD25 and decreases in circulating lymphocytes mediated by IL-21 showed a positive correlation between dose and effect. Furthermore, increases in percent plasma B cells were observed during dosing. This is consistent with in vitro data showing that IL-21 promotes B cell proliferation and differentiation into plasma cells [12, 15, 21]. In vitro IL-21 only promotes plasma cell differentiation in the context of other B cell stimuli such as CD40; therefore, even a small increase in circulating plasma B cells may indicate a pronounced effect of IL-21 on the fraction of B cells that are activated by other stimuli. Interestingly, the gene array analysis showed that expression of the prototypical plasma B cell marker, CD38 was increased in CD8+ cells (Fig. 3b). T cell expression of CD38 is confined to a subset of mature T cells showing an activated phenotype with high ability to secrete cytokines [38]. Thus, IL-21-mediated up-regulation of CD38 on CD8+ cells may be suggestive of T cell activation.
Dosing with IL-21 had significant effects on a number of circulating leukocyte subsets. In all lymphocyte subsets, including B cells, NK cells, and T cells, the number of circulating cells decreased markedly during dosing, but returned to pre-dose levels or slightly higher 5 days after the last dose. The most profound decrease was observed in NK cells. In contrast, the number of monocytes increased during dosing and also returned to pre-dose levels 5 days following the last dose. We do not consider it likely that these decreases in cells are a result of IL-21 induced apoptosis as there was a fast recovery. Rather we consider it likely that IL-21 therapy results in re-distribution of lymphocytes, in which the total number of these cell types in the body do not necessarily change, but dosing induces the movement of these cells either out of or into circulation. In support of this notion we found that 8 out of 10 and 14 out of 14 evaluable subjects in the 5 + 9 regimen had higher frequency of CD62L- and CCR7-expressing CD8+ T cells, respectively; these surface receptors mediate vascular adhesion and migration of T cells to lymph nodes [39]. The true effect of IL-21 on CD62L and CCR7 may have been underestimated since cells with strong up-regulation would most likely have left the circulation. The gene array and quantitative RT-PCR analysis revealed that the chemokine receptors CCR5, CXCR6 and CXCR3 were up-regulated in CD8+ cells supporting increased migratory properties of these cells. CCR5, CXCR3, and CXCR6 all have “inflammatory” chemokines as ligands [31, 33] and could therefore potentially facilitate the recruitment of effector lymphocytes to sites with tissue injury, inflammation, and/or tumors. Another gene up-regulated by IL-21 treatment was HMMR (Fig. 4b). High concentrations of hyaluronan (hyaluronic acid (HA)) are often found in areas with rapid cell proliferation as in tumors [16], and HMMR has an established role in HA-stimulated chemotaxis of inflammatory cells in vitro [49].
A highly significant overrepresentation of genes involved in cell cycle progression was observed in the GO-term analysis. Up-regulations of CDK2, cyclin A2, cyclin B1, cyclin B2, cyclin E1, and cyclin E2 clearly suggest that at least a fraction of the analyzed CD8+ cells traverse the G0 (resting stage) to G1 phase (pre-replication stage) to the S-phase (replication) transition [32]. These and other regulations indicate a proliferative effect of IL-21 on the CD8+ cells. This is consistent with previous findings that IL-21 can augment proliferation of CD8+ T cells driven by TCR-stimulation or by IL-15 or IL-7 [35, 50]. In vivo IL-15 and IL-7 may be present in sufficient amount to act synergistically with IL-21 to induce proliferation, or a subset of CD8+ T cells may have undergone TCR-driven proliferation enhanced by IL-21 while in the lymphoid system and subsequently returned to the circulation.
A number of highly interesting gene regulations were singled out when specifically searching for immune related genes (Fig. 3b) in the list of differentially expressed probe sets. The increased expression of inducible co-stimulator (ICOS), C1q binding protein, and CD26 in CD8+ cells has previously been observed in T cell activation [5, 18, 44]. Adenosine deaminase (ADA) which can associate with CD26 on the cell surface [20] was also found to be up-regulated following IL-21 dosing (Fig. 3b). When binding to CD26, ADA has been reported to function as a co-stimulatory molecule in T-cell activation [28]. ICOS was also reported to be up-regulated in the sanroque mouse, a mouse with mutation in a RING-type ubiquitin ligase which showed elevated IL-21 levels and development of lupus-like disease [46]. ICOS plays an important costimulatory role on follicular helper T cells, which are a main source of IL-21 [7]. Thus, it is possible that IL-21 has a function in the regulation of ICOS expression on both CD4+ and CD8+ T cells. IL-17A mRNA was not found to be differentially expressed in the CD8+ T cells following IL-21 dosing (data not shown), and was in the present study not examined in T helper cell subsets.
Our data provide several indications that IL-21 administration enhanced immune activation and effector functions. As previously reported, granzyme B and perforin mRNAs were up-regulated in CD8+ cells at day 5 of IL-21 treatment compared to day 1 [9]. In addition we here show that granzyme B mRNA was also up-regulated in CD56+ cells primarily consisting of NK cells. Moreover, granzyme A, IFN-γ and cathepsin-C mRNAs were all up-regulated in CD8+ cells. Perforin and granzymes are crucial proteins in the granule exocytosis pathway used by CD8+ and NK cells to kill target cells [47]. Cathepsin-C is a dipeptidyl peptidase which activates granzyme A and B [29, 43] and we speculate that increased expression of cathepsin-C may lead to increased activation of the CD8+ cells. Of all the regulated genes IFN-γ was most up-regulated. IFN-γ has a broad role in activation of innate and adaptive immune responses and in the protection against viral infections and tumors [19]. The up-regulations of AIM2 and GBP1 could be secondary to the IFN-γ regulation since these genes are known to be up-regulated by IFN-γ [6, 10]. The functional consequences of IL-21 administration were evidenced by increased cytotoxicity against K562 cells in PBMC isolated after IL-21 dosing, an effect that is likely to be mediated by NK cells.
In conclusion, our data suggest that IL-21 stimulates the expansion, motility, and effector functions of CD8+ T-cells and NK cells in vivo, which may lead to enhanced anti-tumor immunity. We hypothesize that homing and migration of cytotoxic T-lymphocytes as well as other lymphocytes are affected by IL-21-mediated regulation of chemokine receptors. All together our data supports the use of IL-21 in the treatment of cancer.
References
Asao H, Okuyama C, Kumaki S, Ishii N, Tsuchiya S, Foster D, Sugamura K (2001) Cutting edge: the common gamma-chain is an indispensable subunit of the IL-21 receptor complex. J Immunol 167:1–5
Balch CM, Soong SJ, Gershenwald JE, Thompson JF, Reintgen DS, Cascinelli N, Urist M, McMasters KM, Ross MI, Kirkwood JM, Atkins MB, Thompson JA, Coit DG, Byrd D, Desmond R, Zhang YT, Liu PY, Lyman GH, Morabito A (2001) Prognostic factors analysis of 17,600 melanoma patients: validation of the American Joint Committee on Cancer melanoma staging system. J Clin Oncol 19:3622–3634
Balch CM, Buzaid AC, Soong SJ, Atkins MB, Cascinelli N, Coit DG, Fleming ID, Gershenwald JE, Houghton A Jr., Kirkwood JM, McMasters KM, Mihm MF, Morton DL, Reintgen DS, Ross MI, Sober A, Thompson JA, Thompson JF (2001) Final version of the American joint committee on cancer staging system for cutaneous melanoma. J Clin Oncol 19:3635–3648
Brady J, Hayakawa Y, Smyth MJ, Nutt SL (2004) IL-21 Induces the functional maturation of murine NK Cells. J Immunol 172:2048–2058
Chen A, Gaddipati S, Hong Y, Volkman DJ, Peerschke EI, Ghebrehiwet B (1994) Human T cells express specific binding sites for C1q. Role in T cell activation and proliferation. J Immunol 153:1430–1440
Cheng YS, Colonno RJ, Yin FH (1983) Interferon induction of fibroblast proteins with guanylate binding activity. J Biol Chem 258:7746–7750
Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, Mackay CR (2004) T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol 173:68–78
Coquet JM, Kyparissoudis K, Pellicci DG, Besra G, Berzins SP, Smyth MJ, Godfrey DI (2007) IL-21 Is produced by NKT cells and modulates NKT cell activation and cytokine production. J Immunol 178:2827–2834
Davis ID, Skrumsager BK, Cebon J, Nicholaou T, Barlow J, Møller NPH, Skak K, Lundsgaard D, Frederiksen KS, Thygesen P, McArthur G (2007) An open-label, two-arm, phase I trial of recombinant human interleukin-21 in patients with metastatic melanoma. Clin Cancer Res 13:3630–3636
DeYoung KL, Ray ME, Su YA, Anzick SL, Johnstone RW, Trapani JA, Meltzer PS, Trent JM (1997) Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 15:453–457
di Carlo E, de Totero D, Piazza T, Fabbi M, Ferrini S (2007) Role of IL-21 in immune-regulation and tumor immunotherapy. Cancer Immunol Immunother 56:1323–1334
Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, Leonard WJ, Lipsky PE (2005) IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells. J Immunol 175:7867–7879
Fröhlich A, Marsland BJ, Sonderegger I, Kurrer M, Hodge MR, Harris NL, Kopf M (2007) IL-21 receptor signaling is integral to the development of Th2 effector responses in vivo. Blood 109:2023–2031
Gagnon J, Ramanathan S, Leblanc C, Ilangumaran S (2007) Regulation of IL-21 signaling by suppressor of cytokine signaling-1 (SOCS1) in CD8+ T lymphocytes. Cell Signal 19:806–816
Good KL, Bryant VL, Tangye SG (2006) Kinetics of human B cell behavior and amplification of proliferative responses following stimulation with IL-21. J Immunol 177:5236–5247
Gotte M, Yip GW (2006) Heparanase, hyaluronan, and CD44 in cancers: A breast carcinoma perspective. Cancer Res 66:10233–10237
Guida M, Colucci G (2007) Immunotherapy for metastatic renal cell carcinoma: is it a therapeutic option yet? Ann Oncol 18:149–152
Hutloff A, Dittrich AM, Beler KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA (1999) ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature 397:263–266
Ikeda H, Old LJ, Schreiber RD (2002) The roles of IFN-gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 13:95–109
Kameoka J, Tanaka T, Nojima Y, Schlossman SF, Morimoto C (1993) Direct association of adenosine deaminase with a T cell activation antigen, CD26. Science 261:466–469
Konforte D, Paige CJ (2006) Identification of cellular intermediates and molecular pathways induced by IL-21 in human B cells. J Immunol 177:8381–8392
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK (2007) IL-21 initiates an alternative pathway to induce proinflammatory TH17 cells. Nature 448:484–487
Laurence A, O’Shea JJ (2007) TH-17 differentiation: of mice and men. Nat Immunol 8:903–905
Leonard WJ, Spolski R (2005) Interleukin-21: a modulator of lymphoid proliferation, apoptosis and differentiation. Nat Rev Immunol 5:688–698
Li Y, Yee C (2008) IL-21 mediated Foxp3 suppression leads to enhanced generation of antigen-specific CD8+ cytotoxic T lymphocytes. Blood 111:229–235
Li Y, Bleakley M, Yee C (2005) IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol 175:2261–2269
Ma HL, Whitters MJ, Konz RF, Senices M, Young DA, Grusby MJ, Collins M, Dunussi-Joannopoulos K (2003) IL-21 activates both innate and adaptive immunity to generate potent antitumor responses that require perforin but are independent of IFN-gamma. J Immunol 171:608–615
Martin M, Huguet J, Centelles JJ, Franco R (1995) Expression of ecto-adenosine deaminase and CD26 in human T cells triggered by the TCR-CD3 complex. Possible role of adenosine deaminase as costimulatory molecule. J Immunol 155:4630–4643
McGuire MJ, Lipsky PE, Thiele DL (1993) Generation of active myeloid and lymphoid granule serine proteases requires processing by the granule thiol protease dipeptidyl peptidase I. J Biol Chem 268:2458–2467
Moroz A, Eppolito C, Li Q, Tao J, Clegg CH, Shrikant PA (2004) IL-21 enhances and sustains CD8+ T cell responses to achieve durable tumor immunity: comparative evaluation of IL-2, IL-15, and IL-21. J Immunol 173:900–909
Moser B, Wolf M, Walz A, Loetscher P (2004) Chemokines: multiple levels of leukocyte migration control. Trends immunol 25:75–84
Murray AW (2004) Recycling the cell cycle: cyclins revisited. Cell 116:221–234
Nanki T, Shimaoka T, Hayashida K, Taniguchi K, Yonehara S, Miyasaka N (2005) Pathogenic role of the CXCL16-CXCR6 pathway in rheumatoid arthritis. Arthritis Rheum 52:3004–3014
Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C (2007) Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448:480–483
Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, Schrader S, Burkhead S, Heipel M, Brandt C, Kuijper JL, Kramer J, Conklin D, Presnell SR, Berry J, Shiota F, Bort S, Hambly K, Mudri S, Clegg C, Moore M, Grant FJ, Lofton-Day C, Gilbert T, Rayond F, Ching A, Yao L, Smith D, Webster P, Whitmore T, Maurer M, Kaushansky K, Holly RD, Foster D (2000) Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 408:57–63
Peluso I, Fantini MC, Fina D, Caruso R, Boirivant M, MacDonald TT, Pallone F, Monteleone G (2007) IL-21 counteracts the regulatory T cell-mediated suppression of human CD4+ T lymphocytes. J Immunol 178:732–739
Riker AI, Radfar S, Liu SH, Wang YX, Khong HT (2007) Immunotherapy of melanoma: a critical review of current concepts and future strategies. Expert Opin Biol Ther 7:345–358
Sandoval-Montes C, Santos-Argumedo L (2005) CD38 is expressed selectively during the activation of a subset of mature T cells with reduced proliferation but improved potential to produce cytokines. J Leukoc Biol 77:513–521
Schaerli P, Moser B (2005) Chemokines—control of primary and memory T-cell traffic. Immunol Res 31:57–74
Sondergaard H, Frederiksen KS, Thygesen P, Galsgaard ED, Skak K, Kristjansen PEG, Ødum N, Kragh M (2007) Interleukin 21 therapy increases the density of tumor infiltrating CD8+ T cells and inhibits the growth of syngeneic tumors. Cancer Immunol Immunother 56:1417–1428
Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I, Sareneva T (2003) IL-21 in Synergy with IL-15 or IL-18 Enhances IFN-γ production in human NK and T Cells. J Immunol 170:5464
Strengell M, Sareneva T, Foster D, Julkunen I, Matikainen S (2002) IL-21 up-regulates the expression of genes associated with innate immunity and Th1 response. J Immunol 169:3600–3605
Sutton VR, Waterhouse NJ, Browne KA, Sedelies K, Ciccone A, Anthony D, Koskinen A, Mullbacher A, Trapani JA (2007) Residual active granzyme B in cathepsin C-null lymphocytes is sufficient for perforin-dependent target cell apoptosis. J Cell Biol 176:425–433
Tanaka T, Camerini D, Seed B, Torimoto Y, Dang NH, Kameoka J, Dahlberg HN, Schlossman SF, Morimoto C (1992) Cloning and functional expression of the T cell activation antigen CD26. J Immunol 149:481–486
Thompson JA, Curti BD, Redman BG, Weber JS, Agarwala SS, Sievers EL (2006) Interleukin-21 (IL-21): Tolerability and anti-tumor activity following two 5-day cycles in patients with stage IV melanoma (MM) or renal cell carcinoma (RCC). ASCO Abstract no. 2505
Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, Goodnow CC (2005) A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature 435:452–458
Voskoboinik I, Smyth MJ, Trapani JA (2006) Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol 6:940–952
Wurster AL, Rodgers VL, Satoskar AR, Whitters MJ, Young DA, Collins M, Grusby MJ (2002) Interleukin 21 is a T helper (Th) cell 2 cytokine that specifically inhibits the differentiation of naive Th cells into interferon gamma-producing Th1 cells. J Exp Med 196:969–977
Zaman A, Cui Z, Foley JP, Zhao HJ, Grimm PC, DeLisser HM, Savani RC (2005) Expression and role of the hyaluronan receptor RHAMM in inflammation after bleomycin injury. Am J Respir Cell Mol Biol 33:447–454
Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, Pise-Masison CA, Radonovich MF, Brady JN, Restifo NP, Berzofsky JA, Leonard WJ (2005) Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med 201:139–148
Zeng R, Spolski R, Casas E, Zhu W, Levy DE, Leonard WJ (2007) The molecular basis of IL-21-mediated proliferation. Blood 109:4135–4142
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR (2007) IL-6 programs TH-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8:967–974
Acknowledgments
We acknowledge the invaluable assistance of the staff of the Cancer Trials Australia laboratory. Specific thanks for technical assistance with quantitative RT-PCR and micro-array analyses to Lene Normann Nielsen, to Peter Chapple for technical assistance with flow cytometry, to David Edwards for statistical analyses of the micro-array data, to Kate Lillie for assistance with sample collection and study coordination, and to Bodil Andreasen for technical assistance with cytotoxicity assays.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
K. S. Frederiksen and D. Lundsgaard have contributed equally to this work.
Electronic supplementary material
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Frederiksen, K.S., Lundsgaard, D., Freeman, J.A. et al. IL-21 induces in vivo immune activation of NK cells and CD8+ T cells in patients with metastatic melanoma and renal cell carcinoma. Cancer Immunol Immunother 57, 1439–1449 (2008). https://doi.org/10.1007/s00262-008-0479-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-008-0479-4