
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)-mediated genome engineering and related technologies have revolutionized biotechnology over the last decade by enhancing the efficiency of sophisticated biological systems. Cas12a (Cpf1) is an RNA-guided endonuclease associated to the CRISPR adaptive immune system found in many prokaryotes. Contrary to its more prominent counterpart Cas9, Cas12a recognizes A/T rich DNA sequences and is able to process its corresponding guide RNA directly, rendering it a versatile tool for multiplex genome editing efforts and other applications in biotechnology. While Cas12a has been extensively used in eukaryotic cell systems, microbial applications are still limited. In this review, we highlight the mechanistic and functional differences between Cas12a and Cas9 and focus on recent advances of applications using Cas12a in bacterial hosts. Furthermore, we discuss advantages as well as current challenges and give a future outlook for this promising alternative CRISPR-Cas system for bacterial genome editing and beyond.
Key points
• Cas12a is a powerful tool for genome engineering and transcriptional perturbation
• Cas12a causes less toxic side effects in bacteria than Cas9
• Self-processing of crRNA arrays facilitates multiplexing approaches
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In basic research and industrial biotechnology, genetic engineering is essential for genomic and metabolic manipulation to direct microorganisms towards production of specific valuable products. For this, accessibility of the genome in combination with a highly efficient molecular tool are essential factors for the generation of versatile and robust chassis organisms. In the past decades, vast research has been directed on the development of techniques, which improve genome editing and gene regulation in various microorganisms. The breakthrough by the discovery of the CRISPR-Cas technology has shed light on the adaptive immune system of prokaryotes, and since then opened up tremendous opportunities for targeted genetic engineering approaches in pro- and eukaryotes (Jinek et al. 2012). CRISPR (clustered regularly interspaced short palindromic repeats) is an RNA-guided adaptive defense mechanism in bacteria and archaea that protects them against invasion of viruses and foreign genetic materials (Barrangou et al. 2007; Jinek et al. 2012; Makarova et al. 2020). Native CRISPR-Cas systems have been detected in more than 85% of analyzed archaeal genomes and 40% of bacterial genomes. Today, updated classification of the CRISPR-Cas systems comprises 2 classes, 6 types, and 33 subtypes. Among all CRISPR systems that have been discovered yet, Cas9 from Streptococcus pyogenes is still by far the most studied and well characterized. Based on the architecture of the genomic loci, it is classified as a Class 2 type II-A CRISPR system for its single, large effector Cas9 protein (Makarova et al. 2020). Throughout the years, many studies have been investigating the activity and potential applications of Cas9 for genetic engineering purposes (Adli 2018; Pickar-Oliver and Gersbach 2019).
The more recently discovered Cas12a, which belongs to the Class 2 type V-A CRISPR system, has garnered a lot of attention for its attractive features and potential applications (Zetsche et al. 2015). Despite coming from the same class and postulated to have evolved similarly, Cas9 and Cas12a hold some intrinsic differences which may distinguish their practical applications (Mohanraju et al. 2016; Koonin et al. 2017; Swarts and Jinek 2018). First, both of them recognize different protospacer adjacent motifs (PAM) and have different mechanisms to perform the double strand break. Cas9 recognizes the PAM sequence 5′-NGG, cleaves at proximal position of the PAM, and generates blunt end double-strand breaks (Deltcheva et al. 2011; Jinek et al. 2012). In contrast, Cas12a recognizes T-rich sequence 5′-TTTV and cleaves at distal position of the PAM, generating a staggered double strand break (Zetsche et al. 2015; Kim et al. 2017; Swiat et al. 2017). Second, looking at the domain architectures, Cas9 has two nuclease domains (RuvC and HNH) which each cleaves one strand of the double-strand DNA (dsDNA) (Gasiunas et al. 2012). Contrarily, Cas12a has only a RuvC-like domain and lacks the HNH domain, but is still able to generate a dsDNA break (Zetsche et al. 2015). Third, both types use different crRNA as well as processing mechanism to generate mature crRNA. In the case of Cas9, it needs both tracrRNA and crRNA to allow Cas9 binding to the target sequence. As Cas9 does not possess RNase activity, maturation of crRNA is dependent on the activity of the endoribonuclease RNase III (Deltcheva et al. 2011). In contrast, Cas12a only needs crRNA and does not require the additional tracrRNA. Due to the additional RNase activity, Cas12a can process maturation of the crRNA arrays itself and is therefore independent of other RNase activity (Zetsche et al. 2015; Fonfara et al. 2016). This feature makes Cas12a superior for multiplexing of different targets (Table 1).
Recently, Cas12a has emerged as a reliable genetic tool and attractive alternative to Cas9 (Paul and Montoya 2020). Many studies have investigated its activity in eukaryotes including plants and human cells. However, application of Cas12a in prokaryotes is still limited and it needs more investigations to explore its full potential. In this minireview, we will focus on the current different applications of Cas12a in bacteria. We will discuss challenges and obstacles and give an outlook on what can be expected for future utilization of Cas12a as a robust prokaryotic genetic engineering tool.
CRISPR-Cas12a as an attractive system for genetic engineering
Also known as Cpf1, Cas12a was initially detected in the genome of Francisella and Prevotella strains (Schunder et al. 2013; Zetsche et al. 2015). Its activity was first demonstrated in Escherichia coli where its facilitation of DNA interference was shown (Zetsche et al. 2015). Since then, multiple studies have exploited the potential of Cas12a homologs from Francisella novicida ATCC 15482 (FnCas12a), Acidaminococcus sp. BV3L6 (AsCas12a), Lachnospiraceae bacterium ND2006 (LbCas12a), and Moraxella bovoculi AAX11_00205 (MbCas12a). Thus far, the Cas12a orthologs are shown to be able to mediate genome editing in human cells (Zetsche et al. 2015; Tóth et al. 2018). In recent years, Cas12a has also been employed for genome editing and gene regulation in bacteria, although the information is very limited compared to its application in eukaryotes (Yao et al. 2018; Adiego-Pérez et al. 2019; Liu et al. 2020). Among the three variants, FnCas12a has been used the most to facilitate genetic engineering in bacteria (Table 2). While some studies reported the activity of LbCas12a in yeast, higher plants, and mammalian cells (Zetsche et al. 2015; Verwaal et al. 2018; Bernabé-Orts et al. 2019; Liu et al. 2019), to the best of our knowledge, it has not been tested in prokaryotes.
Since its discovery, Cas12a has arisen as a potential genetic tool and promising alternative to Cas9. One of the common drawbacks of Cas9 is its toxicity that has been reported in different bacteria (Wendt et al. 2016; Ye et al. 2020). Although the reasons are still poorly understood, it is postulated that, in some bacteria, Cas9 might bind non-specifically to the PAM even without a guide RNA (Jiang et al. 2017; Jones et al. 2017). This will subsequently interfere with gene expression and regulation throughout the genome considering the abundant presence of 5′-NGG site (Cho et al. 2018). Different studies indicate that Cas12a has a less toxic effect, which makes it highly promising for CRISPR-Cas-based genetic engineering in prokaryotes (Jiang et al. 2017). Different toxicity levels of the two Cas proteins is obvious in the cyanobacterium Synechococcus sp. 2973, where promoterless Cas9 is even more toxic than Cas12a expressed under control of a lac promoter (Ungerer and Pakrasi 2016).
Cas12a-mediated genome editing
In bacteria, most common application of CRISPR-Cas system is intended for genome editing. As an RNA-guided endonuclease, similar to Cas9, Cas12a also requires crRNA containing specific spacer sequence to guide the effector module to the target region. In its native system in Francisella novicida U112, mature crRNA consists of 19 nts of a direct repeat followed by 23–25 nts spacer sequence. The nucleotides at position 1–5 represent the “seed” sequence which is essential in establishing stable binding between crRNA and the target strand (Zetsche et al. 2015). It is important that the seed sequence perfectly matches the target strand as mutations in this region will abolish or significantly reduce the nuclease activity of the Cas protein (Semenova et al. 2011; Swarts et al. 2017).
Once the crRNA binds to the target strand, active Cas12a will cleave the target sequence at the distal end of the protospacer (Zetsche et al. 2015), causing a dsDNA break in the chromosome. To maintain genome integrity, the organism has to repair the lethal DSB. Several repair mechanisms have been described so far, like the homology-directed repair (HDR), non-homologous end joining pathway (NHEJ), and alternative-end joining (A-EJ) (Szostak et al. 1983; Chayot et al. 2010; Lieber 2011). While NHEJ is commonly used by eukaryotes, most bacteria primarily rely on HDR to repair the DNA break (Hiom 2009). In bacteria with an active NHEJ pathway like Mycobacterium smegmatis, Cas12a can be utilized for gene disruption studies to investigate its function (Sun et al. 2018a). However, NHEJ-mediated repair often results in random insertions and deletions, which makes it difficult to achieve targeted genome editing. For precise genetic engineering, HDR is often employed to introduce desired modifications to bacterial chromosomes (Ran et al. 2013; Rütering et al. 2017; Schilling et al. 2020a). Utilization of CRISPR-Cas systems greatly reduce the screening effort as the double-strand break caused by the Cas-nuclease is lethal to non-edited cells.
Cas12a has been successfully tested in bacteria from different classes and ecological niches, implying the versatility of the system despite more restrictive PAM sites (Tóth et al. 2020), which results in a ~4.4-fold decreased PAM frequency compared to Cas9 in the genome of E. coli K12. Furthermore, in bacteria where expression of active Cas9 is toxic, e.g., in GC-rich organisms like Corynebacterium glutamicum, Cas12a can successfully facilitate genome editing (Zhao et al. 2020). Nevertheless, it is important to note that different bacteria strains might have different response to the introduced CRISPR-Cas system. In the case of C. glutamicum, it was observed that one out of three tested strains could somehow escape Cas12a cleavage (Jiang et al. 2017).
Multiple approaches have been established to make Cas12a-based genome editing more robust and fit the experimental design. In the most minimalistic setup, all-in-one plasmid systems are often used. The plasmid carries everything needed to realize the editing: Cas12a, crRNA array, and homology flanks as repair template (Jiang et al. 2017). Two plasmids system where Cas12a and crRNA array are expressed from different plasmids are also well exploited. Depending on the application, the latter is particularly beneficial when coupling CRISPR-Cas with other genetic engineering systems like recombineering (Yan et al. 2017). For this, Cas12a and recombineering genes are combined in one plasmid and used to transform the host strain first. A second plasmid containing the crRNA array is then used to co-transform the host with the oligonucleotides template. Such systems simplify the work when multiple individual targets and mutations are desired, especially in the strains with limited recombination frequencies like mycobacteria (van Kessel and Hatfull 2007). The coupled system is very convenient when aiming for iterative mutagenesis since it skips many laborious cloning steps (Jiang et al. 2017). To date, Cas12a-assisted recombineering has been successfully employed to achieve different point mutations and gene manipulations in E. coli, Yersinia pestis, Mycobacterium smegmatis, C. glutamicum, Zymomonas mobilis, and the halophilic bacterium Halomonas bluephagenesis (Jiang et al. 2017; Yan et al. 2017; Ao et al. 2018; Shen et al. 2019). In C. glutamicum, it was reported that Cas12a-assisted RecET system realized large deletion up to 20 kb with an efficiency of 36.4% (Zhao et al. 2020), slightly higher than Cas9-RecET system with 26.9% (Wang et al. 2018).
Further developments have been made to increase Cas12a-mediated genome editing efficiency. As seen with recombineering, combining Cas12a with other genome editing tools could be beneficial, especially for systems with low efficiency. Zhang et al. (2020) demonstrated that combination of traditional SacB counterselection with CRISPR-Cas12a dramatically increased efficiency of gene insertion and deletion in C. glutamicum (Zhang et al. 2020). Today, Cas12a utilization has gone beyond simple proof-of-principle of genetic manipulations to directed metabolic engineering for production of high-value products such as amino acids, platform chemicals, or polysaccharides (Zhang et al. 2020; Krumbach et al. 2019; Schilling et al. 2020b). Elimination of competing pathways, release of product inhibition, and fine tuning of targeted pathways could be realized by Cas12a-assisted systems (Zhang et al. 2019, 2020; Schilling et al. 2020b).
Cas12a-mediated gene activation and repression
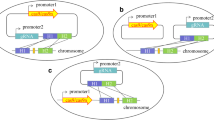
Today, the available CRISPR-Cas systems not only allow gene editing but also gene regulation by means of catalytically inactive DNase-dead Cas (dCas) variants (Fig. 1). For this, dCas can be harnessed for both gene repression by CRISPR interference (CRISPRi) and activation (CRISPRa) and therefore is appealing to tune the level of gene expression. Particularly for targeting essential genes, for which knock-outs would be lethal, knock-down via CRISPRi offers an effective solution to redirect carbon fluxes to desired products. Moreover, utilization of dCas12a can be a quick and straightforward strategy to screen for multiple target genes simultaneously since it only requires the expression of dCas12a and crRNA arrays without the need to supply a homology repair template. Different dCas12a variants have been engineered by introduction of mutations in the RuvC domain (Table 3).

Different modes of action of CRISPR-Cas12-based applications in bacteria. a Staggered double-strand DNA cleavage after binding of crRNA-Cas12a effector complex to the DNA, which can be used to promote homology-directed repair or non-homologous end joining for genome editing efforts. b CRISPRi with catalytically inactive Cas12a variants (dCas12a) can no longer induce DNA cleavage. dCas12a either blocks elongation of transcription acting as a roadblock or prevents binding of the RNA-polymerase to the target promoter site and thereby reduces expression of a gene of interest (GOI). c CRISPRa uses dCas12a fused to a transcriptional activator binding to the upstream (US) region of a target promoter to facilitate the recruitment of RNA-polymerase and thereby enhances expression of a GOI
Compared to gene editing, dCas12a-mediated gene regulation in bacteria was reported less frequently although some studies demonstrated its high efficiency for gene interference (Table 2). Thus far, dCas12a has only been tested in E. coli, Bacillus subtilis, Streptomyces coelicolor, and Paenibacillus polymyxa (Zhang et al. 2017; Li et al. 2018a; Schilling et al. 2020b; Wu et al. 2020). A clear strand bias of the repression efficiency by dCas12a was observed, especially when aiming for interference during transcription elongation. Different studies reported that efficiency of transcriptional perturbation significantly increases when the template strand is targeted (Zhang et al. 2017). In S. coelicolor, repression efficiency of dFnCas12a targeting the template strand can achieve up to 88% whereas it was much less effective when targeting the non-template strand (Li et al. 2018a). Contrarily, dCas9 has shown high preference towards the non-template strand (Tong et al. 2015). In addition, it is also observed that repression efficiency is highest when crRNAs target the site closest to the transcription start site. However, strand bias does not seem to affect dCas12a efficiency when targeting transcription initiation by blocking the promoter region (Zhang et al. 2017). Particularly for transcriptional perturbation, optimal crRNA design is essential to ensure a tight binding of the effector module. Miao et al. (2019) demonstrated that also the PAM sequence and surrounding nucleotides can highly influence the dynamic range of transcriptional perturbation (Miao et al. 2019).
Besides CRISPRi, dCas12a can also be employed for activation of gene expression by linking it to a transcription activator domain. Upon dCas12a binding to the target region, the activator domain facilitates recruitment of RNA polymerase leading to higher expression levels of the gene of interest. Gene activation facilitated by the dCas12a has been well explored in mammalian cells (Campa et al. 2019; Kleinstiver et al. 2019). However, its exploitation in bacteria is very limited and has only been demonstrated in B. subtilis and P. polymyxa (Schilling et al. 2020b; Wu et al. 2020). These studies demonstrated that linking dCas12a to transcription activation domain like RemA or SoxS resulted in higher expression levels of the target genes. Contrary to eukaryotic organisms, for which CRISPRa is primarily based on chromatin rearrangements (Gilbert et al. 2013), for bacterial applications, the activator domain needs to be positioned in a precise distance to the promoter region to activate transcription (Dong et al. 2018). Currently, empirical testing of different crRNAs is required to optimize the dynamic output. However, with an increasing number of studies, it will be possible to develop clear design rule sets for different bacterial promoters to enable efficient experimental design a priori. Interestingly, both studies mentioned above also explored the potential of simultaneous activation and repression by positioning the dCas12a either adjacent to the promoter region to activate transcription initiation, or within the gene to block transcription elongation. These findings once more display the versatility of CRISPR-Cas12a systems, especially when multiple gene targeting is desired.
Multiplex Genome Editing and Regulation
While Cas12a is of importance for bacterial strains in which Cas9 expression shows toxic effects, its simplicity for multiplex targeting remains the most attractive property of Cas12a. To realize multiplex targeting, the spacers-containing crRNAs can either be delivered individually in separate plasmids or in form of a crRNAs array. Nonetheless, it has been reported that supplying the crRNAs in one array is as efficient as supplying them individually (Ao et al. 2018). Therefore, the latter strategy is often used for its simplicity, making use of Cas12a ability to self-process the maturation of crRNAs. It is remarkable to observe that the order of crRNA generally does not affect editing and repression efficiencies (Zhang et al. 2017), although there are exceptions for some specific genes or genomic areas (Li et al. 2018a).
Despite the great potential, Cas12a-based multiplexing has only been investigated in few bacteria: E. coli, B. subtilis, Clostridium difficile, S. coelicolor, and P. polymyxa (Zhang et al. 2017; Ao et al. 2018; Hong et al. 2018; Li et al. 2018a; Schilling et al. 2020b; Wu et al. 2020). Nevertheless, the studies demonstrated the functionality of Cas12a multiplexing with reasonably high efficiency. In bacteria, the highest degree of multiplexing that has been investigated thus far was regulation of four genes in E. coli and P. polymyxa (Zhang et al. 2017; Schilling et al. 2020b). While efficiency of transcriptional perturbation is usually not heavily influenced by an increasing number of targets, efficacy of genome editing via homology-directed repair can decrease (Zhang et al. 2017; Li et al. 2018a). Multiplexing of two gene deletions in C. difficile resulted in an efficiency of 25% which was significantly lower than the efficiency of targeted single gene deletion (Hong et al. 2018). In E. coli, where single-site chromosomal integration showed an efficiency close to 100%, it dropped to 40% and 20% when two and three loci were targeted for simultaneous integrations (Ao et al. 2018). In contrast, Li et al reported 75% efficiency of simultaneous knock-out of two genes in S. coelicolor (Li et al. 2018a).
Optimization of Cas12a activity
There are different strategies that can be employed to achieve higher activity of Cas12a in the desired bacterial host. An important aspect is to ensure adequate expression of Cas12a. Since each organism has distinct codon usage preference (Quax et al. 2015), it is essential that the heterologously expressed Cas12a can be translated at an appropriate level. With decreasing cost of gene synthesis, nowadays, codon-optimized Cas12a is a common starting point in establishing the system especially in bacterial strains where its activity has not yet been investigated. Codon optimization is beneficial to increase the pool of mature Cas12a which could lead to higher efficiency (Ao et al. 2018). Due to its relatively low toxicity, constitutive expression of Cas12a is generally not an issue. In fact, it is preferable in some cases where inducible expression could not provide sufficient efficiency (Li et al. 2018a).
Various studies also investigated different possibilities to enhance the activity of Cas12a. It is reported that engineered AsCas12a variant with E174R/S542R/K548R mutations has twofold higher editing efficiency in human cells than the wild-type variant (Kleinstiver et al. 2019). Furthermore, several variants which recognized non-canonical PAM sites have been designed, which extend the genomic region that can be targeted by the nuclease. It has been demonstrated that AsCas12a carrying the mutations S542R/K607R and S542R/K548V/N552R shows altered PAM recognition to TYCV and TATV, respectively, with improved activities when tested in vitro and in human cells (Gao et al. 2017). When the corresponding mutations are introduced to FnCas12a, LbCas12a, and MbCas12a, the new variants are able to facilitate efficient genome editing with altered PAM recognition (Zhong et al. 2018; Tóth et al. 2020). Based on the observation on how these mutations can be applied to different Cas12a homologs and eukaryotic host organisms, it is reasonable to hypothesize that it will also be relevant for application in bacteria. Engineered Cas12a variants are substantial extensions to currently available bacterial genetic tools, increasing the efficiency of the nucleases or broadening the repertoire of possible PAM to engineer otherwise inaccessible targets or minimize off targeting effects (Kleinstiver et al. 2019). However, most engineered variants of Cas12a have been exclusively tested in eukaryotic organisms and increased efficacy in bacteria remains to be investigated.
Future outlook
First characterized in 2015, Cas12a has emerged as a promising genetic tool and many studies have exploited its potential since then. With the rapidly growing research, there will be several improvements that we can anticipate in the upcoming years which will boost the use of Cas12a for bacterial genome engineering.
As often seen in biological systems, there exist antagonistic mechanisms to keep the balance of the natural condition. Recently, it was described that some proteins can act as natural inhibitor of Cas nucleases (Pawluk et al. 2018). Although it is rarely used in practical applications up to now, the so-called anti-CRISPR (Acr) protein represents an appealing approach for various future applications. Understanding of Acr is especially of importance when working with bacterial strains that encode endogenous CRISPR-Cas system, since many of these bacteria also encode native acr genes. For example, Listeria monocytogenes encodes acr for Cas9. Consequently, it severely inhibits commonly used SpCas9 (Marino et al. 2020). Since many Acr proteins inhibit only one specific subtype, it will be interesting to see if it is a feasible approach to use Acr to suppress the native CRISPR system, while at the same time introducing another type of CRISPR-Cas system to facilitate genetic engineering. Furthermore, Acr can also be used to achieve programmable CRISPR-Cas activity at a specific time to alleviate the toxicity of Cas proteins which may result in higher transformation and editing efficiencies (Marino et al. 2020).
To broaden Cas12a application, it will also be interesting to analyze its utilization as a highly efficient base editing tool in bacteria. As described for Cas9, fusing the dead or nickase variant with a cytidine deaminase protein could direct the conversion of cytosine to thymidine within a particular editing window (Komor et al. 2016; Zheng et al. 2018). Application of Cas12a for base editing thus far has only been described for mammalian cells by means of dLbCas12a-cytidine deaminase fusion protein (Li et al. 2018b), where utilization of optimized cytidine deaminases greatly improved the base editing efficiency (Chen et al. 2020). Applying the system into bacteria would be an attractive strategy to achieve C → T-targeted point mutations or pursuit mutagenesis purposes to generate various mutant strains.
Finally, we also anticipate the development of other Cas12a variants including the nickase which only induces ssDNA breaks, while still triggering the repair mechanism. The mutated variants will particularly be of interest for applications in bacteria which are deficient of the dsDNA break repair mechanism (Song et al. 2017). To our knowledge, no Cas12a nickase has been developed so far, although a preliminary study reported that the R1226A mutation of AsCas12a showed nickase activity in vitro (Yamano et al. 2016). This variant will certainly be a beneficial add-on for extended applications of Cas12a.
References
Adiego-Pérez B, Randazzo P, Daran JM, Verwaal R, Roubos JA, Daran-Lapujade P, Van Der Oost J (2019) Multiplex genome editing of microorganisms using CRISPR-Cas. FEMS Microbiol Lett 366:1–19. https://doi.org/10.1093/femsle/fnz086
Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9:9. https://doi.org/10.1038/s41467-018-04252-2
Ao X, Yao Y, Li T, Yang TT, Dong X, Zheng ZT, Chen GQ, Wu Q, Guo Y (2018) A multiplex genome editing method for Escherichia coli based on CRISPR-Cas12a. Front Microbiol 9:1–13. https://doi.org/10.3389/fmicb.2018.02307
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science (80- ) 315:1709–1712. https://doi.org/10.1126/science.1138140
Bernabé-Orts JM, Casas-Rodrigo I, Minguet EG, Landolfi V, Garcia-Carpintero V, Gianoglio S, Vázquez-Vilar M, Granell A, Orzaez D (2019) Assessment of Cas12a-mediated gene editing efficiency in plants. Plant Biotechnol J 17:1971–1984. https://doi.org/10.1111/pbi.13113
Campa CC, Weisbach NR, Santinha AJ, Incarnato D, Platt RJ (2019) Multiplexed genome engineering by Cas12a and CRISPR arrays encoded on single transcripts. Nat Methods 16:887–893. https://doi.org/10.1038/s41592-019-0508-6
Chayot R, Montagne B, Mazel D, Ricchetti M (2010) An end-joining repair mechanism in Escherichia coli. Proc Natl Acad Sci U S A 107:2141–2146. https://doi.org/10.1073/pnas.0906355107
Chen S, Jia Y, Liu Z, Shan H, Chen M, Yu H, Lai L, Li Z (2020) Robustly improved base editing efficiency of Cpf1 base editor using optimized cytidine deaminases. Cell Discov 6:4–7. https://doi.org/10.1038/s41421-020-00195-5
Cho S, Choe D, Lee E, Kim SC, Palsson B, Cho BK (2018) High-level dCas9 expression induces abnormal cell morphology in Escherichia coli. ACS Synth Biol 7:1085–1094. https://doi.org/10.1021/acssynbio.7b00462
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607. https://doi.org/10.1038/nature09886
Dong C, Fontana J, Patel A, Carothers JM, Zalatan JG (2018) Synthetic CRISPR-Cas gene activators for transcriptional reprogramming in bacteria. Nat Commun 9:2489. https://doi.org/10.1038/s41467-018-04901-6
Fonfara I, Richter H, BratoviÄ M, Le Rhun A, Charpentier E (2016) The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 532:517–521. https://doi.org/10.1038/nature17945
Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F (2017) Engineered Cpf1 variants with altered PAM specificities. Nat Biotechnol 35:789–792. https://doi.org/10.1038/nbt.3900
Gasiunas G, Barrangou R, Horvath P, Siksnys V (2012) Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci U S A 109:E2579–E2586. https://doi.org/10.1073/pnas.1208507109
Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, Lim WA, Weissman JS, Qi LS (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154:442–451. https://doi.org/10.1016/j.cell.2013.06.044
Hiom K (2009) DNA repair: common approaches to fixing double-strand breaks. Curr Biol 19:R523–R525. https://doi.org/10.1016/j.cub.2009.06.009
Hong W, Zhang J, Cui G, Wang L, Wang Y (2018) Multiplexed CRISPR-Cpf1-mediated genome editing in Clostridium difficile toward the understanding of pathogenesis of C. difficile infection. ACS Synth Biol 7:1588–1600. https://doi.org/10.1021/acssynbio.8b00087
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y, Wang R, Yang S (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:1–11. https://doi.org/10.1038/ncomms15179
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (80- ) 337:816–821. https://doi.org/10.1126/science.1225829
Jones DL, Leroy P, Unoson C, Fange D, Ćurić V, Lawson MJ, Elf J (2017) Kinetics of dCas9 target search in Escherichia coli. Science (80- ) 357:1420–1424. https://doi.org/10.1126/science.aah7084
Kim HK, Song M, Lee J, Menon AV, Jung S, Kang YM, Choi JW, Woo E, Koh HC, Nam JW, Kim H (2017) In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat Methods 14:153–159. https://doi.org/10.1038/nmeth.4104
Kleinstiver BP, Sousa AA, Walton RT, Tak YE, Hsu JY, Clement K, Welch MM, Horng JE, Malagon-Lopez J, Scarfò I, Maus MV, Pinello L, Aryee MJ, Joung JK (2019) Engineered CRISPR–Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat Biotechnol 37:276–282. https://doi.org/10.1038/s41587-018-0011-0
Knott GJ, Thornton BW, Lobba MJ, Liu JJ, Al-Shayeb B, Watters KE, Doudna JA (2019) Broad-spectrum enzymatic inhibition of CRISPR-Cas12a. Nat Struct Mol Biol 26:315–321. https://doi.org/10.1038/s41594-019-0208-z
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424. https://doi.org/10.1038/nature17946
Koonin EV, Makarova KS, Zhang F (2017) Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol 37:67–78. https://doi.org/10.1016/j.mib.2017.05.008
Krumbach K, Sonntag CK, Eggeling L, Marienhagen J (2019) CRISPR/Cas12a Mediated genome editing to introduce amino acid substitutions into the mechanosensitive channel MscCG of Corynebacterium glutamicum. ACS Synth Biol 8:2726–2734. https://doi.org/10.1021/acssynbio.9b00361
Li L, Wei K, Zheng G, Liu X, Chen S, Jiang W, Lu Y (2018a) CRISPR-Cpf1-assisted multiplex genome editing and transcriptional repression in Streptomyces. Appl Environ Microbiol:84. https://doi.org/10.1128/AEM.00827-18
Li X, Wang Y, Liu Y, Yang B, Wang X, Wei J, Lu Z, Zhang Y, Wu J, Huang X, Yang L, Chen J (2018b) Base editing with a Cpf1–cytidine deaminase fusion. Nat Biotechnol 36:324–327. https://doi.org/10.1038/nbt.4102
Lieber MR (2011) The mechanism of DSB repair by the NHEJ. Annu Rev Biochem 79:181–211. https://doi.org/10.1146/annurev.biochem.052308.093131
Liu P, Luk K, Shin M, Idrizi F, Kwok S, Roscoe B, Mintzer E, Suresh S, Morrison K, Frazão JB, Bolukbasi MF, Ponnienselvan K, Luban J, Zhu LJ, Lawson ND, Wolfe SA (2019) Enhanced Cas12a editing in mammalian cells and zebrafish. Nucleic Acids Res 47:4169–4180. https://doi.org/10.1093/nar/gkz184
Liu Z, Dong H, Cui Y, Cong L, Zhang D (2020) Application of different types of CRISPR/Cas-based systems in bacteria. Microb Cell Factories 19:1–14. https://doi.org/10.1186/s12934-020-01431-z
Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, Moineau S, Mojica FJM, Scott D, Shah SA, Siksnys V, Terns MP, Venclovas Č, White MF, Yakunin AF, Yan W, Zhang F, Garrett RA, Backofen R, van der Oost J, Barrangou R, Koonin EV (2020) Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18:67–83. https://doi.org/10.1038/s41579-019-0299-x
Marino ND, Pinilla-Redondo R, Csörgő B, Bondy-Denomy J (2020) Anti-CRISPR protein applications: natural brakes for CRISPR-Cas technologies. Nat Methods 17:471–479. https://doi.org/10.1038/s41592-020-0771-6
McCarty NS, Graham AE, Studená L, Ledesma-Amaro R (2020) Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat Commun 11:1–13. https://doi.org/10.1038/s41467-020-15053-x
Miao C, Zhao H, Qian L, Lou C (2019) Systematically investigating the key features of the DNase deactivated Cpf1 for tunable transcription regulation in prokaryotic cells. Synth Syst Biotechnol 4:1–9. https://doi.org/10.1016/j.synbio.2018.11.002
Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, Van Der Oost J (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science (80- ):353. https://doi.org/10.1126/science.aad5147
Paul B, Montoya G (2020) CRISPR-Cas12a: Functional overview and applications. Biom J 43:8–17. https://doi.org/10.1016/j.bj.2019.10.005
Pawluk A, Davidson AR, Maxwell KL (2018) Anti-CRISPR: discovery, mechanism and function. Nat Rev Microbiol 16:12–17. https://doi.org/10.1038/nrmicro.2017.120
Pickar-Oliver A, Gersbach CA (2019) The next generation of CRISPR–Cas technologies and applications. Nat Rev Mol Cell Biol 20:490–507. https://doi.org/10.1038/s41580-019-0131-5
Quax TEF, Claassens NJ, Söll D, van der Oost J (2015) Codon bias as a means to fine-tune gene expression. Mol Cell 59:149–161. https://doi.org/10.1016/j.molcel.2015.05.035
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308. https://doi.org/10.1038/nprot.2013.143
Rütering M, Cress BF, Schilling M, Rühmann B, Koffas MAG, Sieber V, Schmid J (2017) Tailor-made exopolysaccharides—CRISPR-Cas9 mediated genome editing in Paenibacillus polymyxa. Synth Biol 2:1–12. https://doi.org/10.1093/synbio/ysx007
Schilling C, Ciccone R, Sieber V, Schmid J (2020a) Engineering of the 2,3-butanediol pathway of Paenibacillus polymyxa DSM 365. Metab Eng 61:381–388. https://doi.org/10.1016/j.ymben.2020.07.009
Schilling C, Koffas MAG, Sieber V, Schmid J (2020b) Novel prokaryotic CRISPR-Cas12a-based tool for programmable transcriptional activation and repression. ACS Synth Biol 9:3353–3363. https://doi.org/10.1021/acssynbio.0c00424
Schunder E, Rydzewski K, Grunow R, Heuner K (2013) First indication for a functional CRISPR/Cas system in Francisella tularensis. Int J Med Microbiol 303:51–60. https://doi.org/10.1016/j.ijmm.2012.11.004
Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, Van Der Oost J, Brouns SJJ, Severinov K (2011) Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A 108:10098–10103. https://doi.org/10.1073/pnas.1104144108
Shen W, Zhang J, Geng B, Qiu M, Hu M, Yang Q, Bao W, Xiao Y, Zheng Y, Peng W, Zhang G, Ma L, Yang S (2019) Establishment and application of a CRISPR-Cas12a assisted genome-editing system in Zymomonas mobilis. Microb Cell Factories 18:1–11. https://doi.org/10.1186/s12934-019-1219-5
Song X, Huang H, Xiong Z, Ai L, Yang S (2017) CRISPR-Cas9D10A nickase-assisted genome editing in Lactobacillus casei. Appl Environ Microbiol 83:1–13. https://doi.org/10.1128/AEM.01259-17
Sun B, Yang J, Yang S, Ye RD, Chen D, Jiang Y (2018a) A CRISPR-Cpf1-assisted non-homologous end joining genome editing system of Mycobacterium smegmatis. Biotechnol J 13:1–10. https://doi.org/10.1002/biot.201700588
Sun J, Wang Q, Jiang Y, Wen Z, Yang L, Wu J, Yang S (2018b) Genome editing and transcriptional repression in Pseudomonas putida KT2440 via the type II CRISPR system. Microb Cell Factories 17:1–17. https://doi.org/10.1186/s12934-018-0887-x
Swarts DC, Jinek M (2018) Cas9 versus Cas12a/Cpf1: structure–function comparisons and implications for genome editing. Wiley Interdiscip Rev RNA 9:1–19. https://doi.org/10.1002/wrna.1481
Swarts DC, van der Oost J, Jinek M (2017) Structural basis for guide RNA processing and seed-dependent DNA targeting by CRISPR-Cas12a. Mol Cell 66:221–233.e4. https://doi.org/10.1016/j.molcel.2017.03.016
Swiat MA, Dashko S, Den Ridder M, Wijsman M, Van Der Oost J, Daran JM, Daran-Lapujade P (2017) FnCpf1: a novel and efficient genome editing tool for Saccharomyces cerevisiae. Nucleic Acids Res 45:12585–12598. https://doi.org/10.1093/nar/gkx1007
Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW (1983) The double-strand-break repair model for recombination. Cell 33:25–35. https://doi.org/10.1016/0092-8674(83)90331-8
Tong Y, Charusanti P, Zhang L, Weber T, Lee SY (2015) CRISPR-Cas9 based engineering of actinomycetal genomes. ACS Synth Biol 4:1020–1029. https://doi.org/10.1021/acssynbio.5b00038
Tóth E, Czene BC, Kulcsár PI, Krausz SL, Tálas A, Nyeste A, Varga É, Huszár K, Weinhardt N, Ligeti Z, Borsy A, Fodor E, Welker E (2018) Mb- And FnCpf1 nucleases are active in mammalian cells: activities and PAM preferences of four wild-type Cpf1 nucleases and of their altered PAM specificity variants. Nucleic Acids Res 46:10272–10285. https://doi.org/10.1093/nar/gky815
Tóth E, Varga É, Kulcsár PI, Kocsis-Jutka V, Krausz SL, Nyeste A, Welker Z, Huszár K, Ligeti Z, Tálas A, Welker E (2020) Improved LbCas12a variants with altered PAM specificities further broaden the genome targeting range of Cas12a nucleases. Nucleic Acids Res 48:3722–3733. https://doi.org/10.1093/nar/gkaa110
Ungerer J, Pakrasi HB (2016) Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci Rep 6:1–9. https://doi.org/10.1038/srep39681
van Kessel JC, Hatfull GF (2007) Recombineering in Mycobacterium tuberculosis. Nat Methods 4:147–152. https://doi.org/10.1038/nmeth996
Verwaal R, Buiting-Wiessenhaan N, Dalhuijsen S, Roubos JA (2018) CRISPR/Cpf1 enables fast and simple genome editing of Saccharomyces cerevisiae. Yeast 35:201–211. https://doi.org/10.1002/yea.3278
Wang B, Hu Q, Zhang Y, Shi R, Chai X, Liu Z, Shang X, Zhang Y, Wen T (2018) A RecET-assisted CRISPR-Cas9 genome editing in Corynebacterium glutamicum. Microb Cell Factories 17:1–16. https://doi.org/10.1186/s12934-018-0910-2
Wendt KE, Ungerer J, Cobb RE, Zhao H, Pakrasi HB (2016) CRISPR/Cas9 mediated targeted mutagenesis of the fast growing cyanobacterium Synechococcus elongatus UTEX 2973. Microb Cell Factories 15:1–8. https://doi.org/10.1186/s12934-016-0514-7
Wu Y, Liu Y, Lv X, Li J, Du G, Liu L (2020) CAMERS-B: CRISPR/Cpf1 assisted multiple-genes editing and regulation system for Bacillus subtilis. Biotechnol Bioeng 117:1817–1825. https://doi.org/10.1002/bit.27322
Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y, Fedorova I, Nakane T, Makarova KS, Koonin EV, Ishitani R, Zhang F, Nureki O (2016) Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 165:949–962. https://doi.org/10.1016/j.cell.2016.04.003
Yan M-Y, Yan H, Ren G, Zhao J, Guo X, Sun Y-C (2017) CRISPR-Cas12a-assisted recombineering in bacteria. Appl Environ Microbiol 83:1–13. https://doi.org/10.1128/AEM.00947-17
Yao R, Liu D, Jia X, Zheng Y, Liu W, Xiao Y (2018) CRISPR-Cas9/Cas12a biotechnology and application in bacteria. Synth Syst Biotechnol 3:135–149. https://doi.org/10.1016/j.synbio.2018.09.004
Ye S, Enghiad B, Zhao H, Takano E (2020) Fine-tuning the regulation of Cas9 expression levels for efficient CRISPR-Cas9 mediated recombination in Streptomyces. J Ind Microbiol Biotechnol 47:413–423. https://doi.org/10.1007/s10295-020-02277-5
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, Van Der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771. https://doi.org/10.1016/j.cell.2015.09.038
Zhang Y, Yuan J (2020) CRISPR / Cas12a mediated genome engineering in photosynthetic bacteria. bioRxiv:1–25. https://doi.org/10.1101/2020.10.05.327569
Zhang X, Wang J, Wang J, Cheng Q, Zheng X, Zhao G (2017) Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov 3:1–9. https://doi.org/10.1038/celldisc.2017.18
Zhang J, Hong W, Zong W, Wang P, Wang Y (2018) Markerless genome editing in Clostridium beijerinckii using the CRISPR-Cpf1 system. J Biotechnol 284:27–30. https://doi.org/10.1016/j.jbiotec.2018.07.040
Zhang J, Yang F, Yang Y, Jiang Y, Huo YX (2019) Optimizing a CRISPR-Cpf1-based genome engineering system for Corynebacterium glutamicum. Microb Cell Factories 18:1–13. https://doi.org/10.1186/s12934-019-1109-x
Zhang J, Qian F, Dong F, Wang Q, Yang J, Jiang Y, Yang S (2020) De novo engineering of Corynebacterium glutamicum for l -Proline Production. ACS Synth Biol 9:1897–1906. https://doi.org/10.1021/acssynbio.0c00249
Zhao N, Li L, Luo G, Xie S, Lin Y, Han S, Huang Y, Zheng S (2020) Multiplex gene editing and large DNA fragment deletion by the CRISPR/Cpf1-RecE/T system in Corynebacterium glutamicum. J Ind Microbiol Biotechnol 47:599–608. https://doi.org/10.1007/s10295-020-02304-5
Zheng K, Wang Y, Li N, Jiang FF, Wu CX, Liu F, Chen HC, Liu ZF (2018) Highly efficient base editing in bacteria using a Cas9-cytidine deaminase fusion. Commun Biol 1:1–6. https://doi.org/10.1038/s42003-018-0035-5
Zhong Z, Zhang Y, You Q, Tang X, Ren Q, Liu S, Yang L, Wang Y, Liu X, Liu B, Zhang T, Zheng X, Le Y, Zhang Y, Qi Y (2018) Plant genome editing using FnCpf1 and LbCpf1 nucleases at redefined and altered PAM sites. Mol Plant 11:999–1002. https://doi.org/10.1016/j.molp.2018.03.008
Zhou Y, Liu X, Wu J, Zhao G, Wang J (2020) CRISPR-Cas12a-assisted genome editing in Amycolatopsis mediterranei. Front Bioeng Biotechnol 8:1–9. https://doi.org/10.3389/fbioe.2020.00698
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
MM: literature research, review structure, and writing of the manuscript; CS: literature research and writing of the manuscript. JS: Review structure, writing of the manuscript, and final proofread. All authors read and approved of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Meliawati, M., Schilling, C. & Schmid, J. Recent advances of Cas12a applications in bacteria. Appl Microbiol Biotechnol 105, 2981–2990 (2021). https://doi.org/10.1007/s00253-021-11243-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-021-11243-9