
Abstract
The potential of Saccharomyces cerevisiae for biocatalytic whole-cell transamination was investigated using the kinetic resolution of racemic 1-phenylethylamine (1-PEA) to (R)-1-PEA as a model reaction. As native yeast do not possess any ω-transaminase activity for the reaction, a recombinant yeast biocatalyst was constructed by overexpressing the gene coding for vanillin aminotransferase from Capsicum chinense. The yeast-based biocatalyst could use glucose as the sole co-substrate for the supply of amine acceptor via cell metabolism. In addition, the biocatalyst was functional without addition of the co-factor pyridoxal-5′-phosphate (PLP), which can be explained by a high inherent cellular capacity to sustain PLP-dependent reactions in living cells. In contrast, external PLP supplementation was required when cell viability was low, as it was the case when using pyruvate as a co-substrate. Overall, the results indicate a potential for engineered S. cerevisiae as a biocatalyst for whole-cell transamination and with glucose as the only co-substrate for the supply of amine acceptor and PLP.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chiral amines are important building blocks for the synthesis of bioactive molecules, and efficient and environmentally benign methods are required for their preparation in high yield, productivity, product concentration and enantiomeric purity (Höhne and Bornscheuer 2009). Chemical production of chiral amines with metal catalysts can be cumbersome since the amine group can, on the one hand, bind to metal ions and hamper the reaction (Koszelewski et al. 2011; Rajan et al. 1974) and, on the other hand, lead to heavy metal contamination of the product. Alternatively, chiral amines can be prepared using biocatalysis, for example, with the help of enantioselective ω-transaminases (ω-TAs) (Koszelewski et al. 2010; Mathew and Yun 2012). ω-TAs catalyze the transfer of an amine group from an amine donor to a ketone moiety with pyridoxal-5′-phosphate (PLP) as a co-factor and can be used to prepare chiral amines either via kinetic resolution or through asymmetric synthesis from a prochiral ketone. (S)-selectivity is more common among ω-TAs; however, both (R)- and (S)-selective enzymes have previously been used to convert a broad range of substrates with high efficiency, making this enzyme group a versatile tool for the preparation of various chiral amine molecules (Koszelewski et al. 2010; Tufvesson et al. 2011a).
Transamination reactions may be limited by unfavourable equilibrium and substrate and/or product inhibition. These hurdles have been overcome by addition of excess co-substrates (Savile et al. 2011), in situ product removal (Truppo et al. 2010) and/or coupled enzymatic systems (Höhne et al. 2008; Koszelewski et al. 2008). However, any added complexity to the reaction system leads to an increase in the overall process cost (Tufvesson et al. 2011a). Until now, most processes with ω-transaminases apply purified enzymes or crude cell extract from recombinant Escherichia coli as a biocatalyst (Bea et al. 2011; Kaulmann et al. 2007; Truppo et al. 2009). In the case of kinetic resolution of racemic amines, excess amount of pyruvate has often been used as a reactant (Koszelewski et al. 2010).
Budding yeast Saccharomyces cerevisiae (baker’s yeast) is a eukaryotic microorganism having a broad number of biotechnological applications, including production of biofuels, commodity chemicals (Hong and Nielsen 2012), bioactive compounds (Ro et al. 2006) and biopharmaceuticals (Mollerup et al. 2010). Thus, the feasibility and experience to use yeast in large-scale bioprocessing is well-developed. In organic chemistry, baker’s yeast is known for efficiently catalyzing asymmetric carbonyl reductions for the synthesis of chiral alcohols (D’Arrigo et al. 1997; Johanson et al. 2005; Stewart 2000). S. cerevisiae has been successfully engineered for asymmetric reduction of prochiral ketones (Johanson et al. 2005, 2008), kinetic resolution of racemic diketones (Carlquist et al. 2008, 2009) or asymmetric Baeyer-Villiger oxidation of cyclic ketones for the generation of chiral lactones (Kayser et al. 1998; Stewart 2000).
In the present study, we report on the use of a whole-cell system expressing an ω-transaminase gene originating from Capsicum chinense (Weber et al. 2014) for the kinetic resolution of (rac)-1-phenylethylamine to (R)-1-phenylethylamine. Catalytic activity and cell robustness during the reaction and the use of glucose instead of pyruvate as a co-substrate were evaluated with S. cerevisiae. We provide a proof of concept that cell metabolism can be exploited to produce in vivo the required co-substrate and co-factor of the reaction by only providing glucose in a buffer system with resting cells.
Materials and methods
Chemicals
Acetophenone (ACP), racemic 1-phenylethylamine (1-PEA), (R)-1-PEA, (S)-1-PEA and sodium pyruvate were purchased from Merck (Hohenbrunn, Germany); (R)-1-phenylethanol and (S)-1-phenylethanol from Sigma-Aldrich (Steinheim, Germany); PLP from AppliChem (Darmstadt, Germany); and all other chemicals from VWR (Leuven, Belgium).
Strains
S. cerevisiae strains CEN.PK 113-7D (MAT a MAL2-8 C SUC2) and CEN.PK113-16B (MAT a, leu2-3,112 MAL2-8 C SUC2) were provided by Professor P. Kötter, EUROSCARF collection, Institute for Molecular Biosciences, Goethe University Frankfurt, Germany. E. coli strain DH5α (Life Technologies, Rockville, MD, USA) was used for subcloning. E. coli strain TMB2100 (Weber et al. 2014) overexpressing vanillin aminotransferase (VAMT) gene in the BL21(DE3) background and S. cerevisiae strain TMB4350 (see construction below) were used for whole-cell transamination experiments. Strains were kept as 20 % glycerol stocks at −80 °C and grown on solid media for 1–2 days prior to experiments.
Nucleic acid manipulation
Plasmid DNA was prepared with the GeneJET™ Plasmid Miniprep Kit (Fermentas, Vilnius, Lithuania). Agarose gel DNA extraction was performed using QIAquick® Gel Extraction Kit (Qiagen GmbH, Hilden, Germany). Primers from MWG-Biotech AG (Ebersberg, Germany) and Pfu DNA polymerase and dNTPs from Fermentas (Vilnius, Lithuania) were used for polymerase chain reactions (PCRs). PCR amplification was performed in a GeneAmp PCR System 9700 (Applied Biosystems, Foster City, CA, USA). PCR products were purified with the E.Z.N.A.® Cycle Sequencing Kit (Omega Bio-Tek, Inc., Doraville, GA, USA). Sequencing was performed by MWG-Biotech AG (Ebersberg, Germany). Restriction endonucleases, shrimp alkaline phosphatase and T4 DNA ligase from Fermentas (Vilnius, Lithuania) were used for DNA manipulation.
Transformation
Competent E. coli DH5α cells were prepared and transformed as described elsewhere (Inoue et al. 1990). Transformants were selected on solid lysogeny broth (LB) medium (Ausubel et al. 1995) containing 100 mg/l ampicillin (IBI Shelton Scientific, Inc., Shelton, CT, USA). E. coli strains were grown in liquid LB medium containing 100 mg/l ampicillin for plasmid amplification. Yeast strains were transformed using the lithium acetate method (Gietz and Schiestl 2007), and transformants were selected on yeast nitrogen base (YNB) medium agar plates (6.7 g/l YNB without amino acids, 15 g/l agar, 20 g/l glucose).
Construction of TMB4350
Plasmid pUC57 VAMT containing the ω-transaminase gene from C. chinense (VAMT, Table 1) was digested with the restriction enzymes BamHI and PstI, and the resulting fragment was ligated into YIplac128-HXT7p-PGKt that had been cut with the same restriction enzymes, thus creating plasmid pNW2. Correct orientation of the insert and sequence was verified by restriction analysis and sequencing. pNW2 was cleaved with ClaI within the LEU2 marker gene and subsequently used to transform the haploid laboratory strain CEN.PK113-16B (MAT a leu2-3,112 MAL2-8 C SUC2) which resulted in strain TMB4350 overexpressing the VAMT-encoding gene.
Cell growth
A single colony of E. coli TMB2100 was inoculated in a 0.5-l shake flask containing 50 ml LB medium (Ausubel et al. 1995) supplemented with 100 mg/l ampicillin. Cells were grown overnight (at least 16 h) in an incubator at 37 °C with shaking at 180 rpm. The pre-culture was used to inoculate 250 ml LB medium containing 100 mg/l ampicillin in a 1-l shake flask at an optical density (OD)620 of 0.1, and growth was performed at 37 °C and 180 rpm. Expression of the transaminase gene VAMT was induced by addition of isopropyl-β-d-thiogalactoside (IPTG) to a final concentration of 1 mM when the OD620 of the culture reached 0.6 to 0.8, and incubation was then continued for 24 h at 15 °C and 180 rpm. S. cerevisiae strains were first grown on solid YPG medium (10 g/l yeast extract, 20 g/l peptone, 15 g/l agar, 20 g/l glucose), and then, a single colony was picked with a sterile inoculation loop and transferred to 5 ml liquid YPG medium (10 g/l yeast extract, 20 g/l peptone, 20 g/l glucose). The pre-culture was grown overnight at 30 °C and 180 rpm and used to inoculate a 1-l shake flask containing 100 ml YPG medium at a starting OD620 of 0.1 and kept at 30 °C and 180 rpm for 24 h. Cells were harvested by centrifugation at 4,000 g for 5 min (S. cerevisiae) or 10 min (E. coli) and washed once with 25 ml water.
Growth inhibition
Inhibition of growth by substrate and product was determined in a growth-based microplate assay using liquid YPG medium supplemented with 0–200 mM of ACP or (rac)-1-PEA. S. cerevisiae cells were pre-grown in YPG medium and inoculated at an OD620 of 0.1 in five wells in 96-well microtitre plates for each product/substrate level. Growth was measured after 24 h and used to determine the level of inhibition at different substrate and product concentrations.
Whole-cell transamination
Reactions were performed in magnetically stirred (140 rpm) 100-ml sealed serum flasks using 5 g/l cell dry weight (dw) at 30 °C. The solution contained 20 ml of 100 mM sodium phosphate buffer (pH 7.0), 40 mM pyruvate or 400 mM glucose, 4–75 mM (rac)-1-PEA and 0–1.0 mM PLP.
Cell viability
Cell viability was determined every 24 h during whole-cell transamination by counting colony forming units (CFU/ml) on solid media plates. Samples were diluted in series of tenfold in saline 0.9 % NaCl solution and streaked on solid YPG medium. CFU per millilitre were counted after incubation for 2 days at 30 °C.
Analyses
Growth in shake flasks was monitored by measuring the optical density at a wavelength of 620 nm (OD620) with an Ultrospec 2100pro spectrophotometer (Amersham Biosciences, Sweden). Growth in 96-well microtitre plates was monitored by measuring optical density at a wavelength of 620 nm with a Multiskan Ascent instrument (Thermo Electron Corporation, Vantaa, Finland). Cell dry weight was determined in duplicate by filtering a known volume of cell suspension through nitrocellulose filters (Pall, MI, USA) with 0.2 or 0.45 μM pore size for E. coli and S. cerevisiae, respectively. Filters were washed with distilled water, dried in a microwave oven at 350 W for 8 min, cooled in a desiccator and weighed. Glucose, pyruvate, glycerol, acetate, succinate and ethanol were determined by high-performance liquid chromatography (HPLC) using an Aminex HPX-87H column (Bio-Rad, Richmond, CA, USA) with a RID-10A refractive index detector (Shimadzu, Kyoto, Japan) at 30 °C with 5 mM H2SO4 as a mobile phase and at a flow rate of 0.6 ml/min. Conversion and stereoisomeric purity of 1-PEA were determined using HPLC (Waters Binary HPLC Pump 1525, UV/Vis detector 2489, Autosampler 2707) equipped with a Daicel Chiralcel OD-H column (4.6 × 25 mm, 5 μm) using 85:15 heptane/isopropanol with 0.1 % butylamine as the eluent at a flow rate of 1 ml/min and at a room temperature of 22 °C. The following retention times were obtained: (R)-1-PEA, 5.6 min; (S)-1-PEA, 6.8 min; (R)-1-phenylethanol, 4.9 min; (S)-1-phenylethanol, 5.3 min; and ACP, 4.6 min. The compounds were determined as the peak area at a wavelength of 210 nm.
Results
Transaminase activity in recombinant E. coli and S. cerevisiae
Kinetic resolution of (rac)-1-PEA to (R)-1-PEA was used as a model reaction (Fig. 1) to investigate the potential of recombinant S. cerevisiae as a whole-cell biocatalyst for the preparation of chiral amines. In previous studies, whole-cell transamination has traditionally been performed with recombinant E. coli overexpressing selective ω-TAs, and the biocatalyst has been considered as an “enzyme bag”. To relate the performance of the recombinant yeast system with an E. coli-based system (Weber et al. 2014), the catalytic activity and stability of both biocatalysts were determined in a reaction buffer supplemented with pyruvate and PLP. Adding both components ensured that any difference in catalytic activity did not arise from different abilities of the two species to generate amine acceptors from a fermentable carbon source.

Reaction scheme of kinetic resolution of (rac)-1-phenylethylamine to (R)-1-phenylethylamine with glucose as a co-substrate
As endogenous transaminase activity towards (rac)-1-PEA might decrease the yield of kinetic resolution, non-engineered E. coli and S. cerevisiae lacking the VAMT enzyme were first investigated for background activity. However, no transaminase activity towards (rac)-1-PEA was detected (data not shown), thereby demonstrating that both hosts were suitable for whole-cell transamination of (rac)-1-PEA. Overexpression of the VAMT gene in E. coli and S. cerevisiae led to a selective conversion of (S)-1-PEA (Fig. 2a, b) with initial specific reaction rates 0.022 and 0.002 mmol/g dw/h, respectively. ACP was the only product for both whole-cell systems, and no formation of 1-phenylethanol from the activity of endogenous ketone reductases could be observed (data not shown). The conversion of (S)-1-PEA completely stopped after 24 h with the E. coli biocatalyst, resulting in a final conversion of only 24 % and an enantiomeric excess (ee) of 33 % after 96 h (Fig. 1a). In contrast, the S. cerevisiae biocatalyst remained active for approximately 200 h, and (S)-1-PEA was nearly completely consumed, leading to a final conversion of 45 % and ee of 83 % (Fig. 2b).

Kinetic resolution of 4 mM (rac)-1-PEA with a E. coli overexpressing VAMT and b S. cerevisiae overexpressing VAMT as whole-cell biocatalysts. Reactions were run in sodium phosphate buffer containing 40 mM pyruvate and 0.1 mM PLP. Full triangles indicate conversion, and open squares indicate enantiomeric excess
Tolerance of S. cerevisiae to substrate and product
In order for the whole-cell transamination system to be functional with addition of glucose as the sole source for amine acceptor, yeast metabolism must be active throughout the transamination reaction. It is thus important to know the level at which the substrate and product inhibits metabolism. The concentration boundaries of the bioprocess were estimated by determining the substrate and product inhibition of cell growth for the prototrophic lab strain CEN.PK113-7D and the yeast biocatalyst TMB4350 (Fig. 3a, b). In the control strain without overexpression of VAMT, growth was impaired by concentrations above 30 mM ACP. Addition of (rac)-1-PEA, in contrast, led to strong inhibition of growth already above 5 mM, and no growth was observed above 10 mM. The overexpression of VAMT had no significant influence on the inhibitory effect of ACP. With 1-PEA, however, growth was impaired but still observed up to 15 mM, although the standard deviation between biological replicas was too high to conclude any potential difference in substrate tolerance compared to the control strain. The inhibitory effect of 1-PEA suggests that the reaction should be performed below 15 mM (rac)-1-PEA. However, inhibition of growth occurred at a lower concentration than arrest of the reaction for non-growing cells exposed to reaction conditions with glucose as the sole co-substrate. The specific productivity was highest at 25 mM (rac)-1-PEA; however, the reaction was possible up to approximately 50 mM, although at a lower rate (Fig. 3c). Therefore, in the following experiments, 25 mM (rac)-1-PEA and non-growing cells as a biocatalyst were chosen.

Growth of a S. cerevisiae and b S. cerevisiae overexpressing VAMT in microtitre plates containing liquid YPG medium supplemented with (rac)-1-PEA (full squares) or ACP (full triangles) at varying concentrations. c Relative initial reaction rate for kinetic resolution of varying concentration of (rac)-1-PEA using S. cerevisiae overexpressing VAMT in sodium phosphate buffer containing 0.1 mM PLP and 400 mM (70 g/l) glucose as the sole co-substrate
Glucose as a source for amine acceptor
For S. cerevisiae, glucose is the preferred carbon source over other sugars (Johnston 1999), and the metabolic flux to the central metabolite pyruvate is proficient both under aerobic and anaerobic conditions (Nissen et al. 1997). Therefore, the use of glucose as a co-substrate for whole-cell transamination of 25 mM (rac)-1-PEA was evaluated under conditions where PLP was externally supplemented (Fig. 4a). External addition of 0.1 mM PLP was made to assess the ability of S. cerevisiae to generate amine acceptors independently from the capacity of the cell to sustain required PLP levels. The specific reaction rate was approximately 0.013 mmol/g dw/h during the first 48 h, and then the reaction rate rapidly decreased after 72 h, leading to a conversion of about 23 %. The reaction efficiency was similar compared to directly supplying pyruvate in the reaction solution; initial specific reaction rate was 0.008 mmol/g dw/h, and approximately 22 % conversion was reached after 144 h (Fig. 4a).

Kinetic resolution of 25 mM (rac)-1-PEA using S. cerevisiae overexpressing VAMT in sodium phosphate buffer containing 0.1 mM PLP and 400 mM (70 g/l) glucose or 40 mM pyruvate as the sole co-substrate. a Conversion (circles) and enantiomeric excess (squares) for glucose (full symbols) or pyruvate (open symbols) as a co-substrate. b Viability with pyruvate (open squares) and glucose (full squares) as a co-substrate. c Glucose (full circles), ethanol (crosses), glycerol (full triangles), acetate (stars) and pyruvate (full squares)
While the reaction with pyruvate as a co-substrate does not depend on the cells being viable and metabolically active, the viability is a main determinant for high reaction efficiency with glucose as a co-substrate as amine acceptors need to be provided through glucose catabolism. To gain a deeper understanding of the reaction outcome, cell viability was assessed by analyzing CFU on agar plates containing rich medium throughout the reaction (Fig. 4b). It could be concluded that the arrest in reaction was not due to loss in viability, which remained high throughout the experiment (viability = 7.3 log (CFU/ml) after 144 h). With pyruvate on the other hand, the viability significantly decreased (37 times faster death rate), which resulted in a complete loss of viability after 48 h.
With glucose as a co-substrate, the cells were also metabolically well active after the reaction had stopped, as demonstrated by a continuation of glucose consumption until 120 h (Fig. 4c). The specific glucose consumption rate was 0.68 mmol/g dw/h, and the main extracellular metabolites were, in decreasing order, ethanol, glycerol and acetate. Pyruvate was not detected in the reaction solution, as it was expected since it is not a metabolic end product, and it is thus normally not present at high concentration or transferred into the fermentation broth due to a negative molecular charge at cytosolic pH (Postma et al. 1989).
PLP availability in S. cerevisiae
Kinetic resolution of 25 mM (rac)-1-PEA was carried out at different concentrations of PLP and with glucose or pyruvate as a co-substrate to evaluate whether sufficient intracellular PLP could be synthesized during biocatalyst production and/or during the reaction to sustain the activity of recombinant transaminase. The specific productivities were calculated for the first 48 h and are summarized together with final conversion and ee in Table 2. Student’s t test was used to statistically assess differences between experiments. Remarkably, the biocatalyst was operational without addition of PLP to the reaction mixture with either glucose or pyruvate as a co-substrate. The initial productivity was ca two times higher with glucose than with pyruvate as a co-substrate, leading to a significantly higher conversion. For glucose, there was no statistical difference in the initial reaction rate between without addition of PLP (0.013 ± 0.002 mmol/g dw/h) and with the addition of 0.1 mM PLP (0.013 ± 0.002 mmol/g dw/h). However, when taking into account the standard deviation from biological replicates, it cannot be excluded that there still may be a small difference in catalytic activity with or without addition of 0.1 mM PLP. By increasing the PLP concentration to 0.2 or 1.0 mM, the initial specific productivity was increased as compared to without PLP. Further, the addition of PLP led to a longer continuation of the reaction and thus a higher final conversion (17 and 25 % final conversion with 0 and 1 mM PLP, respectively with glucose as a co-substrate). The effect of PLP addition was even more pronounced with pyruvate as a co-substrate (7 and 22 % final conversion with 0 and 0.1 mM PLP, respectively).
Discussion
In the present study, we demonstrate that recombinant S. cerevisiae can be functional as a whole-cell catalyst for the kinetic resolution of racemic amines. When compared with isolated enzyme systems, whole-cell biocatalysis only requires one cultivation step for the production of biocatalyst in which all necessary enzymes can be co-expressed, and there is no need for cell disintegration or costly enzyme purification (Tufvesson et al. 2011b). One main additional advantage of the constructed yeast biocatalyst is that the transamination reaction can proceed with glucose as the only co-substrate, i.e. without external addition of pyruvate co-substrate or PLP co-factor.
When comparing recombinant S. cerevisiae with the E. coli biocatalyst, significantly higher initial catalytic activity was observed with E. coli. As buffered systems containing both pyruvate and PLP were used, the difference did not result from different abilities of the two species to generate amine acceptors and PLP from dissimilation of a carbon source. Instead, the difference indicates that the E. coli biocatalyst may have contained a higher level of the recombinant enzyme than the yeast. However, the recombinant enzyme was not visible by SDS-PAGE analysis of cell extracts from any of the two hosts (Supplementary material Fig. S1), and it cannot be excluded that other unknown mechanisms are involved in the observed difference in activity. Another observation specific for the E. coli system was an arrest of the reaction after 24 h, which could partly be a result from enzyme inhibition by the product ACP as has been observed for other ω-TAs (Park and Shin 2013; Shin and Kim 1997). On the other hand, the reaction continued with yeast for ca 200 h, and ACP was the only detectable product from the conversion of (S)-1-PEA. In fact, ACP seemed to be inert to yeast metabolism, and no conversion to 1-phenyethanol or other by-products was observed in separate bioreduction experiments in which ACP and glucose for co-factor regeneration were directly added to the reaction buffer solution containing freshly harvested metabolically active cells (data not shown). The activity of endogenous yeast reductases, such as YMR226c that has previously been reported to convert ACP (Yang et al. 2007), was thus too low for any significant formation of 1-phenylethanol. Inhibition of the enzyme by ACP cannot solely explain the arrest in reaction for E. coli and not for S. cerevisiae, and other causes such as lower cell robustness and/or enzyme degradation under applied reaction conditions may have been involved. A similar trend has previously been observed when the two hosts were compared for whole-cell bioreduction of a xenobiotic ketone, and it was attributed to a drop in E. coli viability at a faster rate than the S. cerevisiae counterpart (Parachin et al. 2009). Altogether, the S. cerevisiae biocatalyst displayed high stability, high selectivity and no by-product formation during whole-cell transamination; however, the catalytic activity needs to be elevated before being relevant in an industrial setting.
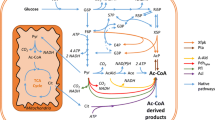
VAMT enzyme from C. chinense can use pyruvate and oxaloacetate as an amine acceptor (Weber et al. 2014), and both are generated as intracellular metabolites from glucose in S. cerevisiae; pyruvate is the end product of glycolysis, whereas oxaloacetate is an intermediate of the Krebs cycle that can also be produced in the cytosol via pyruvate carboxylase (Walker et al. 1991). In practice, however, oxaloacetate is generally not present in the extracellular liquid, whereas low levels of extracellular pyruvate have been detected during aerobic cultivation on glucose (Flikweert et al. 1996). Therefore, the performances of the yeast biocatalyst were only compared between glucose and pyruvate as a co-substrate. In both cases, ACP was generated at similar initial rate, which highlighted the potential of glucose as a co-substrate. The rate of pyruvate synthesis is limited by the glycolytic rate; however, the specific glucose consumption rate was 52 times higher than the specific reaction rate (0.68 and 0.013 mmol/g dw/h, respectively). Pyruvate availability may still be limiting, since it is not an end product of glucose fermentation; consequently, the transaminase competes with other enzymes, namely, pyruvate carboxylase, pyruvate dehydrogenase and pyruvate decarboxylase for pyruvate utilisation (Pronk et al. 1996). More difficult to explain is the discrepancy between reaction continuation with pyruvate as a co-substrate and the significant slowing down of the reaction for glucose, despite a continuation of glucose uptake. It can be speculated that the availability of pyruvate and other intracellular amine acceptors became scarcer at the later stage of the reaction due to a reduced carbon flux to ethanol. However, direct external addition of pyruvate after 72 h had no significant influence on the reaction, and the same final conversion and ee of the product was reached (data not shown). This demonstrates that although pyruvate availability from glucose catabolism may still have partly limited the reaction efficiency, other causes such as degradation/inactivation of VAMT may also explain the reaction arrest. Worth noting is that viability was significantly lower with pyruvate as a co-substrate than with glucose, which may be speculated to be a result of lowering the intracellular pH by the weak acid pyruvate (pKa = 2.49) (Dawson 1968). Uncharged weak acids can diffuse over the plasma membrane (Mira et al. 2010), and once inside the cell, at physiological pH, they dissociate and hydronium ions are formed, decreasing the intracellular pH (Pampulha and Loureiro-Dias 1990). Yeast has an efficient ATP-driven H+ pump for maintaining intracellular pH homeostasis (Verduyn et al. 1990); however, its efficiency under the applied non-growing conditions may be hampered. The negative effect of pyruvate on non-growing cells in a buffer system has, to the best of our knowledge, not been studied previously. Pyruvate can, in fact, be used as the sole carbon source for cell growth (Fendt and Sauer 2010). However, the negative effect on yeast physiology by other weak acids, for example, acetate, has been well-documented previously (Pampulha and Loureiro-Dias 2000).
Another advantage of using glucose as the sole co-substrate was that the yeast biocatalyst was operational without addition of PLP, which suggested that intracellular PLP was available in high amount in the beginning of the reaction. The reaction continued though for a longer time when PLP was added, which may be due to a leakage of intracellular PLP or lack of de novo biosynthesis during the reaction progress. In theory, the amount of PLP should be more than sufficient to sustain the activity of the recombinant transaminase. The initial specific reaction rate was r (S)-1-PEA/X = 0.013 mmol/g dw/h for transamination of (S)-1-PEA during the first 48 h with glucose as the sole co-substrate, which is approximately 100 times less than the theoretical minimum-specific transamination reaction rate capacity in yeast (see Supplementary material for calculation of the theoretical reaction capacity). Proteins constitute 42–60 % of the total yeast biomass composition (Nissen et al. 1997; Verduyn et al. 1991), and 16 out of 20 amino acids require a transaminase for their synthesis either in the amino acid-forming reaction or in the upstream biosynthetic pathway (cf. review by (Ljungdahl and Daignan-Fornier 2012), Supplementary material Table S1). Hence, there is hypothetically a large potential for an increased biocatalytic transaminase activity without the requirement for PLP supplementation. The fact that PLP addition had a more pronounced effect when pyruvate was used as a co-substrate may be linked to the sharp drop in cell viability. This is an advantage of glucose since high cell robustness and physiological fitness under bioprocess conditions are fundamental for fully exploiting the cellular capacity for PLP-dependent reactions. Moreover, the PLP-building blocks glyceraldehyde-3′-phosphate and ribulose-5′-phosphate are intermediates in the glycolysis and the pentose phosphate pathway; thus, an efficient gluconeogenesis is required for their formation from pyruvate (Kanehisa et al. 2012).
The aim of the present study was to explore the possibility of using the metabolic framework of yeast for biocatalytic transamination reactions. Although the productivity and enantioselectivity of the present catalyst are still too low to be relevant in an industrial setting (Tufvesson et al. 2011b), it demonstrates the potential of recombinant yeast as a biocatalyst and the use of glucose as the sole source of amine acceptor, without the requirement of PLP addition. The addition of PLP significantly adds to the process cost, and the glucose-fermenting yeast principle offers a simpler and cheaper bioprocess set-up than traditional systems based on enzyme catalysis. Engineering of process conditions, and of the whole-cell biocatalyst for increased specific activity, and increased space-time yield is the target for future engineering work. This may e.g. be achieved by an elevated ω-TA level in combination with re-constructed pyruvate and/or oxaloacetate dissimilatory reactions. As ACP may be inhibiting for the enzyme, a product removal system may also increase the bioprocess efficiency.
References
Ausubel F, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K (1995) Short protocols in molecular biology, 3rd edn. Wiley, New York
Bea HS, Park HJ, Lee SH, Yun H (2011) Kinetic resolution of aromatic β-amino acids by ω-transaminases. Chem Commun 47:5894–5896. doi:10.1039/C1CC11528F
Carlquist M, Wallentin CJ, Warnmark K, Gorwa-Grauslund MF (2008) Genetically engineered Saccharomyces cerevisiae for kinetic resolution of racemic bicyclo[3.3.1]nonane-2,6-dione. Tetrahedron-Asymmetry 19:2293–2295. doi:10.1016/j.tetasy.2008.09.024
Carlquist M, Olsson C, Bergdahl B, van Niel EWJ, Gorwa-Grauslund MF, Frejd T (2009) Kinetic resolution of racemic 5,6-epoxy-bicyclo[2.2.1]heptane-2-one using genetically engineered Saccharomyces cerevisiae. J Mol Catal B Enzym 58:98–102. doi:10.1016/j.molcatb.2008.11.014
D’Arrigo P, Pedrocchi-Fantoni G, Servi S (1997) Old and new synthetic capacities of baker’s yeast. Adv Appl Microbiol 44:81–123
Dawson RMC (1968) Data for biochemical research, 2nd edn. Clarendon, Oxford
Entian KD, Kötter P (2007) Yeast genetic strain and plasmid collections. Methods Microbiol 36:629–666
Fendt SM, Sauer U (2010) Transcriptional regulation of respiration in yeast metabolizing differently repressive carbon substrates. BMC Syst Biol 4:12. doi:10.1186/1752-0509-4-12
Flikweert MT, Zanden LVD, Janssen WM, Steensma HY, Dijken JPV, Pronk JT (1996) Pyruvate decarboxylase: an indispensable enzyme for growth of Saccharomyces cerevisiae on glucose. Yeast 12:247–257
Gietz RD, Schiestl RH (2007) High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2:31–34
Höhne M, Bornscheuer UT (2009) Biocatalytic routes to optically active amines. Chem Cat Chem 1:42–51
Höhne M, Kühl S, Robins K, Bornscheuer UT (2008) Efficient asymmetric synthesis of chiral amines by combining transaminase and pyruvate decarboxylase. Chem Bio Chem 9:363–365
Hong KK, Nielsen J (2012) Metabolic engineering of Saccharomyces cerevisiae: a key cell factory platform for future biorefineries. Cell Mol Life Sci 69:2671–2690
Inoue H, Nojima H, Okayama H (1990) High efficiency transformation of Escherichia coli with plasmids. Gene 96:23–28
Johanson T, Katz M, Gorwa-Grauslund MF (2005) Strain engineering for stereoselective bioreduction of dicarbonyl compounds by yeast reductases. FEMS Yeast Res 5:513–525
Johanson T, Carlquist M, Olsson C, Rudolf A, Frejd T, Gorwa-Grauslund MF (2008) Reaction and strain engineering for improved stereo-selective whole-cell reduction of a bicyclic diketone. Appl Microbiol Biotechnol 77:1111–1118. doi:10.1007/s00253-007-1240-1
Johnston M (1999) Feasting, fasting and fermenting: glucose sensing in yeast and other cells. Trends Genet 15:29–33
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular datasets. Nucleic Acids Res 40:109–114. doi:10.1093/nar/gkr988
Kaulmann U, Smithies K, Smith MEB, Hailes HC, Ward JM (2007) Substrate spectrum of ω-transaminase from Chromobacterium violaceum DSM30191 and its potential for biocatalysis. Enzym Microbial Tech 41:628–637
Kayser MM, Chen G, Stewart JD (1998) Enantio- and regioselective Baeyer-Villiger oxidations of 2- and 3-substituted cyclopentanones using engineered baker’s yeast. J Org Chem 63:7103–7106
Koszelewski D, Lavandera I, Clay D, Rozzell D, Kroutil W (2008) Asymmetric synthesis of optically pure pharmacologically relevant amines employing ω-transaminases. Adv Synth Catal 350:2761–2766
Koszelewski D, Tauber K, Faber K, Kroutil W (2010) ω-Transaminases for the synthesis of non-racemic α-chiral primary amines. Trends Biotechnol 6:324–332. doi:10.1016/j.tibtech.2010.03.003
Koszelewski D, Grischek B, Glueck SM, Kroutil W, Faber K (2011) Enzymatic racemization of amines catalyzed by enantiocomplementary ω-transaminases. Chemistry 17:378–383
Ljungdahl PO, Daignan-Fornier B (2012) Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae. Genetics 190:885–929. doi:10.1534/genetics.111.133306
Mathew S, Yun H (2012) ω-Transaminases for the production of optically pure amines and unnatural amino acids. J ACS Catal 2:993–1001
Mira N, Teixeira M, Sá-Correia I (2010) Adaptive response and tolerance to weak acids in Saccharomyces cerevisiae: a genome-wide view. Omics 14:525–540. doi:10.1089/omi.2010.0072
Mollerup I, Jensen SW, Larsen P, Schou O, Snel L (2010) Insulin purification Encyclopedia of industrial biotechnology: bioprocess, bioseparation, and cell technology. Novo Nordisk A/S, Bagsvaerd, pp 1–20
Nissen TL, Schulze U, Nielsen J, Villadsen J (1997) Flux distributions in anaerobic, glucose-limited continuous cultures of Saccharomyces cerevisiae. Microbiology 143:203–218
Pampulha ME, Loureiro-Dias MC (1990) Activity of glycolytic enzymes of Saccharomyces cerevisiae in the presence of acetic acid. Appl Microbiol Biotechnol 34:375–380. doi:10.1007/BF00170063
Pampulha ME, Loureiro-Dias MC (2000) Energetics of the effect of acetic acid on growth of Saccharomyces cerevisiae. FEMS Microbiol Lett 184:69–72. doi:10.1111/j.1574-6968.2000.tb08992.x
Parachin NS, Carlquist M, Gorwa-Grauslund M (2009) Comparison of engineered Saccharomyces cerevisiae and engineered Escherichia coli for the production of an optically pure keto alcohol. Appl Microbiol Biotechnol 84:487–497
Park ES, Shin JS (2013) ω-Transaminase from Ochrobactrum anthropi is devoid of substrate and product inhibitions. Appl Environ Microbiol 79:4141–4144. doi:10.1128/AEM.03811-12
Postma E, Verduyn C, Scheffers WA, Dijken JPV (1989) Enzymic analysis of the Crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae. Appl Environ Microbiol 55:468–477
Pronk JT, Steensma HY, Dijken JPV (1996) Pyruvate metabolism in Saccharomyces cerevisiae. Yeast 12:1607–1633
Rajan KS, Davis JM, Colburn RW (1974) Metal chelates in the storage and transport of neurotransmitters. Interactions of copper(2+) ion with ATP and biogenetic amines. J Neurochem 22:137–147
Ro DK, Paradise EM, Ouellet M, Fisher FKJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MCY, Withers ST, Shiba Y, Sarpong R, Keasling JD (2006) Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 440:940–943
Savile CA, Janey JM, Mundorff EC, Moore JC, Tam S, Jarvis WR, Colbeck JC, Krebber A, Fleitz FJ, Brands J, Devine PN, Huisman GW, Hughes GJ (2011) Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329:305–309. doi:10.1126/science.1188934
Shin JS, Kim BG (1997) Kinetic resolution of α-methylbenzylamine with ω-transaminase screened from soil microorganisms: application of a biphasic system to overcome product inhibition. Biotechnol Bioeng 55:348–358. doi:10.1002/(SICI)1097-0290(19970720)55:2<348::AID-BIT12>3.0.CO;2-D
Stewart JD (2000) Organic transformations catalyzed by engineered yeast cells and related systems. Curr Opin Biotechnol 11:363–368
Truppo MD, Turner NJ, Rozzell JD (2009) Efficient kinetic resolution of racemic amines using a transaminase in combination with an amino acid oxidase. Chem Commun 2127-2129
Truppo MD, Rozzell JD, Turner NJ (2010) Efficient production of enantiomerically pure chiral amines at concentrations of 50 g/L using transaminases. Org Process Res Dev 14:234–237
Tufvesson P, Lima-Ramos J, Jensen JS, Al-Haque N, Neto W, Woodley JM (2011a) Process considerations for the asymmetric synthesis of chiral amines using transaminases. Biotechnol Bioeng 108:1479–1493. doi:10.1002/bit.23154
Tufvesson P, Lima-Ramos J, Nordblad M, Woodley JM (2011b) Guidelines and cost analysis for catalyst production in biocatalytic processes. Org Process Res Dev 15:266–274
Verduyn C, Postma E, Scheffers WA, Dijken JPV (1990) Energetics of Saccharomyces cerevisiae in anaerobic glucose-limited chemostat cultures. J Gen Microbiol 136:405–412
Verduyn C, Stouthamer A, Scheffers WA, Dijken JPV (1991) A theoretical evaluation of growth yields of yeasts. Antonie Van Leeuwenhoek 59:49–63
Walker ME, Val DL, Rohde M, Devenish RJ, Wallace JC (1991) Yeast pyruvate carboxylase: identification of two genes encoding isoenzymes. Biochem Biophys Res Commun 176:1210–1217
Weber N, Ismail A, Gorwa-Grauslund M, Carlquist M (2014) Biocatalytic potential of vanillin aminotransferase from Capsicum chinense. Submitted for publication
Yang Y, Zhu S, Piegat TJ, Hua L (2007) Enzymatic ketone reduction: mapping the substrate profile of a short-chain alcohol dehydrogenase (YMR226c) from Saccharomyces cerevisiae. Tetrahedron-Asymmetry 18:1799–1803
Acknowledgments
This work was financed by the Swedish Research Council FORMAS and the Royal Swedish Academy of Agriculture and Forestry.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 149 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Weber, N., Gorwa-Grauslund, M. & Carlquist, M. Exploiting cell metabolism for biocatalytic whole-cell transamination by recombinant Saccharomyces cerevisiae . Appl Microbiol Biotechnol 98, 4615–4624 (2014). https://doi.org/10.1007/s00253-014-5576-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-014-5576-z