
Abstract
Optimizing productivity and growth rates of recombinant Chinese hamster ovary (CHO) cells requires insight into the regulation of cellular processes. In this regard, the elucidation of the epigenetic process of DNA methylation, known to influence transcription by a differential occurrence in CpG islands in promoter regions, is increasingly gaining importance. However, DNA methylation has not yet been investigated on a genomic scale in CHO cells and suitable tools have not existed until now. Based on the genomic and transcriptomic CHO data currently available, we developed a customized oligonucleotide microarray covering 19598 CpG islands (89 % of total bioinformatically identified CpG islands) in the CHO genome. We applied our CHO-specific CpG island microarray to investigate the effect of butyrate treatment on differential DNA methylation in CHO cultures in a time-dependent approach. Supplementation of butyrate is known to enhance cell specific productivities in CHO cells and leads to alterations of epigenetic silencing events. Gene ontology clusters regarding, e.g., chromatin modification or DNA repair, were significantly overrepresented 24 h after butyrate addition. Functional classifications furthermore indicated that several major signaling systems such as the Wnt/β-catenin pathway were affected by butyrate treatment. Our novel CHO-specific CpG island microarray will provide valuable information in future studies of cellular processes associated with productivity and product characteristics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
More than 200 biopharmaceutical molecules have been produced by means of mammalian cultivation systems since the first recombinant protein, tissue plasminogen activator (tPA), was approved by the FDA in 1986 (Walsh 2010). About 70 % of these products are made in Chinese hamster ovary (CHO) cells. They account for the production of seven out of the ten top-selling biopharmaceuticals in 2011, comprising, e.g., Rituxan® (Biogen Idec) or Avastin® (Roche) (Huggett and Lähteenmaki 2012). Even though CHO cells have been used extensively over the last three decades, still little is known about the molecular basis for their versatile properties regarding productivity, robustness and adaptability. Addressing this problem more efficiently, the CHO genome and transcriptome were sequenced in 2011 (Becker et al. 2011; Xu et al. 2011). The data obtained in this way now enables researchers to conduct genome-scale analyses investigating regulatory cellular mechanisms, e.g., the identification of miRNA gene loci and the respective pre-miRNA sequences by Hackl et al. (2012).
Among other factors, the transcriptional activity of recombinant genes strongly depends on their integration site within the genome. In many cases such transgenes become rapidly silenced even if they are integrated into active euchromatin (Wurm 2004). Those processes are frequently accompanied by changes in histone modification and DNA methylation (Mutskov and Felsenfeld 2004; Osterlehner et al. 2011). One way to inhibit silencing processes of recombinant genes is the treatment with organic substances such as the short-chain fatty acid (SCFA) butyrate (Kwaks and Otte 2006). There are, however, cytotoxic side effects of butyrate treatment, resulting in growth inhibition and apoptosis (Zhou et al. 2011; Chang et al. 2012). Various approaches to diminish the cytotoxic effects of butyrate treatment have been made, e.g., by combining butyrate treatment with low cultivation temperatures (Chen et al. 2011) or by repression of apoptotic pathways (Sung and Lee 2005; Kim et al. 2009). The exact mechanism of action upon butyrate addition is still unknown, although on the molecular level butyrate was shown to inhibit histone deacetylase (HDAC) activity and induce remodeling of chromatin structures (Boffa et al. 1978), as well as to enhance, e.g., the expression of growth arrest and DNA damage inducible protein (Gadd45) isoforms (Chen et al. 2002). Gadd45 proteins in turn were shown to contribute to DNA demethylation by DNA damage repair (Rai et al. 2008). In this context, the analysis of epigenetic effects — especially on the level of DNA methylation — represents an important step towards gaining further insights into the cellular response to butyrate addition.
In mammals, DNA methylation (the addition of a methyl group to the C5 position of cytosine residues) predominantly takes place in the context of CpG dinucleotides (CpGs) and affects most CpG sites in the genome (Illingworth and Bird 2009). An exception are genomic regions with high CpG frequencies, so-called CpG islands (CGIs), that are in large parts unmethylated and associated with promoter regions of constitutively expressed genes (Ndlovu et al. 2011). Another subgroup of promoter-associated CGIs shows differential methylation between cell types or under certain conditions and can thus be associated with cellular functions (Illingworth and Bird 2009). Changes of promoter methylation often correlate with transcriptional regulation of the associated gene (Ndlovu et al. 2011), whereas the methylation status of intragenic CGIs may affect different processes. It has been shown that many protein-coding genes are expressed by using alternative promoters in a tissue specific manner and thus may be regulated by associated intragenic CGIs (Sun et al. 2011). Methylation of exon–intron boundaries may moreover correlate with alternative splicing processes (Kornblihtt et al. 2013). Finally, intragenic CGIs possibly regulate the expression of non-coding RNAs (ncRNAs) (Sato et al. 2011).
In order to perform genome-wide screenings for differentially methylated CGIs in CHO cells, we developed a microarray covering 19598 promoter-associated and intragenic CGIs. We applied the array to compare butyrate-treated CHO cultures with untreated references over the course of a batch cultivation in order to detect changes in DNA methylation in a time-dependent manner. To our knowledge, this is the first report in literature on a CGI microarray from CHO sequence data and an analysis of the butyrate effect on DNA methylation events in CHO cells.
Material and methods
Cell culture and cultivation process analysis
Anti IL-8-antibody-producing CHO DP-12 cells (clone #1934, ATCC CRL-12445) were cultivated in chemically defined, animal component-free medium CHOMACS® CD (Miltenyi Biotec, Bergisch Gladbach, Germany) supplemented with 6 mM l-glutamine (Fagron GmbH & Co. KG, Barsbüttel, Germany) and 200 nM methotrexate (MTX; Wyeth, Madison, NJ, USA). Insulin-like growth factor was added to the media in a concentration of 0.1 mg/l (Long R3 IGF-I; Sigma-Aldrich, Munich, Germany). For sample preparation, cells were cultivated in 250-ml shaker flasks (Corning Inc., Corning, New York, USA) with a culture volume of 70 ml and a seeding cell density of 3×105 cells/ml. Eight cultures were prepared from one preculture. Culture flasks were kept at 37 °C, 80 % humidity and 5 % CO2 on a shaking platform at 185 rpm (50 mm diameter rotation). Three days after inoculation, 3 mM butyric acid (Sigma-Aldrich) was added to four cultures. The other four were kept as control cultures without butyrate addition. Before (0 h), 1 (24 h) and 2 (48 h) days upon butyrate addition, 3×106 cells per culture were harvested for DNA extraction. Measurements of cell densities and viabilities were performed on a Cedex AS20 cell counter (Innovatis AG, Bielefeld, Germany). IgG concentrations were measured with an HPLC method on protein A columns according to the manufacturer´s protocol (Poros A; Applied Biosystems, Foster City, CA, USA).
DNA preparation
Genomic DNA was prepared by standard phenol chloroform isoamylalcohol (PCI) extraction (Sambrook and Russell 2006) and extended proteinase K (Carl Roth, Karlsruhe, Germany) treatment for at least 6 h. Purified DNA was precipitated with isopropanol. The pellets were dissolved in TE buffer (10 mM Tris–HCl, 1 mM EDTA) with gentle shaking for 24 h at 4 °C.
Enrichment of methylated DNA
Genomic DNA was sonicated to an average fragment size of 500 bp using a Branson Sonifier 450-A (Branson Ultrasonics, Danbury, CT, USA). Sonication was carried out in TE buffer for 20 s with 20 % amplitude while cooling the tubes in ice water, and repeated up to 12 times depending on the persistent presence of high molecular DNA on agarose TAE control gels. We used 9 μg of fragmented genomic DNA per culture, treatment and sampling point for enrichment of methylated DNA by magnetic beads coupled MBD2-Fc protein (Epimark Methylated DNA Enrichment Kit; NEB, Ipswich, MA, USA) according to the manufacturer´s protocol. Eight washing steps were used to wash off unbound DNA. After purification with silica columns, the DNA samples from the four replicate cultures of each time point were pooled before microarray analysis.
CpG island microarray analysis
The analysis of differential CGI methylation was carried out using customized CHO-specific oligonucleotide CGI microarrays. Based on the currently available genomic (Xu et al. 2011) and transcriptomic CHO data (Becker et al. 2011) promoter-associated and intragenic CGIs were predicted according to the algorithm of Takai and Jones (2002). Probes were designed with average distances of 500 bp, in regard to the average DNA fragment lengths after sonication. All probes are present at least twice per array, so that experiments with two arrays (including a dye swap) result in 4–6 n per probe. Customized arrays were printed by Agilent Technologies in an 8×60K high-density format.
Enriched DNA samples were labeled with Cyanine 3- and 5-dUTP, respectively, using the SureTag DNA Labeling Kit (Agilent Technologies, Santa Clara, CA, USA) according to the manufacturer's protocol. For each time point, samples of treated and reference cultures were labeled as dye swap pairs. After removal of unincorporated dye and vacuum concentration, the samples were eluted in 6.3 μl TE buffer. Subsequently the Cy3- and Cy5-labeled samples were combined and 29 μl of hybridization mastermix (4.3 μl 10× blocking agent, 21.5 μl 2× hi-rpm buffer (Agilent Technologies), 8 % formamide (Carl Roth), 1.2 μl mouse Cot-1 DNA (Life Technologies, Darmstadt, Germany) were added per tube. The mixture was incubated for 3 min at 95 °C and subsequently for 30 min at 60 °C. Then, 40 μl of each sample were used to hybridize the microarrays by incubation at 67 °C with 20 rpm for 24 h. After washing and drying the slide by centrifugation, the arrays were scanned with a High-Resolution Microarray C Scanner (Agilent Technologies) with a resolution of 3 μm. Data extraction was carried out using the Feature Extraction Software (Agilent Technologies).
We evaluated our microarray data using the open source software platform EMMA2, which has been designed for consistent storage and efficient analysis of microarray data (Dondrup et al. 2009). Normalization was carried out using the LOWESS method (Yang et al. 2002). Normalized data was subjected to Student's t-tests to determine significantly regulated regions with p values below a threshold of 0.05. All p values were corrected based on the method of Benjamini and Hochberg (1995) to account for the multiple testing situation. As we used pooled samples of each four culture replicates in this first experiment, no internal variance within the respective groups was calculated. Gene ontology classifications were carried out using DAVID (Huang et al. 2009) with an EASE score ≤0.01. Genes found to be differentially methylated without butyrate addition (compare with Fig. 3b, c) were removed from the '24 h' and '48 h' datasets before cluster analysis.
Bisulfite sequencing
The efficiency of enrichment of methylated DNA was compared to bisulfite sequencing experiments of promoter regions of a positive (branched chain-amino-acid aminotransferase cytosolic-like, Bcat1, GeneID 100763658) and a negative (β-actin, Actb, GeneID 100689477) control. Bisulfite conversion of 500 ng genomic DNA was carried out using the Epimark Bisulfite Conversion Kit (NEB) according to the manufacturer´s protocol. Specific genomic regions were amplified using Epimark Taq polymerase (NEB) and primers specific for bisulfite converted DNA (Metabion, Martinsried, Germany). A region of the Actb promoter was amplified with primers actb_for (5′-GGTAAGTAGGGATAATAGGTTTAGT-3′) and actb_rev (5′- CCCCCAAAATAAACAAATAC-3′). The Bcat1 promoter was amplified using the primers bcat1_for (5′- GGGTGTTAGGAATTAAATTTTTAATT-3′) and bcat1_rev (5′- CACAATAACTTTCTCTAAAACTCCC-3′). The primer design was carried out using MethPrimer (Li and Dahiya 2002) and PCR products were purified from agarose TAE (Tris-acetate EDTA) gels using the GeneJet Gel Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA). The purified amplicons were cloned into pJet1.2/blunt cloning vector (CloneJet PCR Cloning Kit; Thermo Fisher Scientific), which was subsequently transformed into One Shot® TOP10 Chemically Competent Escherichia coli cells (Invitrogen, Carlsbad, CA, USA). Five clones were randomly chosen and sequenced (Sequencing Core Facility of the Center for Biotechnology, Bielefeld University). The sequences were compared with the original DNA sequence using the BiQ analyzer software (Bock et al. 2005).
Quantitative real-time PCR
The efficiency of methylated DNA enrichment was determined by qPCR using the Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen) on a LightCycler® 480 (Roche, Penzberg, Germany). For Actb quantification the oligonucleotides qActb_for (5′-CCGCGGAGCGGACACTTTCA-3′) and qActb_rev (5′- AGCGGGTCCACCGGTGTCTA-3′) and for Bcat1 quantification the oligonucleotides qBcat1_for (5′- GCAGGGACGCTGTTTGGCCT-3′) and qBcat1_rev (5′-GGCTTTCCAGGGCTCTGCGT-3′) were used.
Results
Design and establishment of a CHO-specific CGI microarray
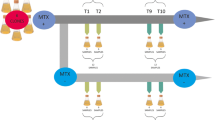
To predict CGIs within the CHO genome, an algorithm according to Takai and Jones (2002) was applied to the currently available genomic and in-house transcriptomic CHO data (Becker et al. 2011; Xu et al. 2011). We identified a total of 43318 CGIs in the CHO genome, 21993 of which could be associated with promoter and intragenic regions, respectively. Our microarray covers 19598 (89 %) of these CGIs with 27446 individual probes within a 60 K high-density format so that each island is represented by at least two probes. This customized microarray was then applied to examine DNA methylation changes upon butyrate addition over time. Figure 1a outlines the experimental procedure. Central to our experiment is the enrichment of methylated DNA by magnetic beads coupled MBD2-Fc protein. To verify the success of the applied method, the enrichment efficiency was controlled and optimized by determining the ratio between two previously identified regions in the CHO genome, one unmethylated (β-actin, Actb) and one methylated (branched chain aminotransferase 1, Bcat1) (Fig. 1b). The quantifications of these positive and negative enrichment controls were performed by real-time PCR (Fig. 1c) and indicate a more than 11,000-fold enrichment of the methylated compared to the unmethylated region. The assessment of an input comparison showed no significant differences between the input fractions (Fig. 1d). Therefore, DNA preparations could be regarded as sufficiently homogenous to allow direct comparison of enriched methylated fractions along with an application of the LOWESS normalization approach.

Experimental design and quality control. a Schematic representation of methylated DNA enrichment combined with the CHO-specific CpG island microarray. b Bisulfite sequencing results of the CGIs of Actb and Bcat1. The black region represents the CGI, the black arrow marks the transcription start site. Black circles represent a methylated CpG position, white circles indicate unmethylated CpG positions. The red arrows indicate the positions of primers used to control the enrichment reaction by quantitative real-time PCR. c Diagram of fold enrichment measured by qPCR. Values were normalized to the input concentration. Enrichment of the methylated region was calculated setting the Actb enrichment to one. d MA plot of the dye swapped input control
In a first application of our microarray the methylated DNA fractions of each four replicate control and butyrate-treated CHO cell cultures per sampling point were pooled, fluorescently labeled and hybridized to microarrays as dye swap pairs. The LOWESS normalized data was subjected to Student's t-tests (adjusted p < 0.05) to identify significantly differentially enriched genomic regions. Genes associated with these regions were searched for Gene Ontology clusters (EASE score <0.01).
Effect of butyrate addition on growth and cell specific IgG productivity
The effect of butyrate on CHO DP-12 cell growth and productivity is shown in Fig. 2. Twenty-four hours after butyrate addition reduced cell growth could already be detected in treated cultures and the viability started to decrease. The butyrate-treated cells reached a maximum of 31×105 cells/ml on day 5 (48 h after butyrate addition). The control cultures continued normal cell growth until a maximum viable cell density of 110×105 cells/ml was reached on day 7. Viabilities as well as viable cell densities of the control cultures started to decrease after day 7. Butyrate treatment resulted in enhanced cell specific productivities on day 5 with a maximum value of 17 pg/cell/day, whereas the control cultures only achieved maximum cell specific productivities of 9 pg/cell/day. This corresponds to a 2-fold increase in IgG productivity upon butyrate addition at this time point.

Cell growth and IgG production of butyrate-treated and control cultures. Shown are the mean values of each four replicate cultures, error bars represent standard deviations. a Mean viable cell density (continuous lines) and viability (dashed lines). Addition of 3 mM butyrate is marked by a red vertical line, sampling points for microarray analyses are marked by grey arrows. b Mean IgG titer (continuous lines) and cell specific productivity (dashed lines) of butyrate and reference cultures
Butyrate-mediated differential CGI methylation
To investigate genome-wide differential methylation upon butyrate addition, samples from the reference and butyrate-treated cultures had been taken at three time points before (0 h) as well as 1 (24 h) and 2 (48 h) days upon butyrate addition. For each time point, pooled samples of four biological replicates were analyzed. Figure 3a shows MA plots of the combined four replicates of the arrays hybridized with enriched methylated DNA. The plots indicate either hypomethylated (M ≥ 1.0) or hypermethylated (M ≤ −1.0) regions amongst all three time points. The Venn diagrams in Fig. 3b and c illustrate the numbers of genes associated with differentially methylated CGIs and show that the effect of butyrate on DNA methylation was highly dynamic and reversible. We were astonished to find 42 genes to be relatively hypermethylated and 141 genes to be relatively hypomethylated in a comparison between reference and butyrate-treated cultures even before addition of the fatty acid. Similar numbers were seen at the end of the experiment (32 genes affected by relative hypermethylation and 110 genes affected by hypomethylation). A strongly increased number of 1221 genes associated with differentially methylated CGIs was found 24 h after butyrate supplementation, with 342 genes showing relative hypermethylation of corresponding CGIs and 879 genes being associated with relatively hypomethylated genomic regions. The greatest overlap in the sampling points regarding affected genes existed between 24 and 48 h (139 genes in total). Furthermore, 30 genes showed differential methylation both before butyrate treatment and 24 h later. Only ten genes were found to be significantly differentially methylated amongst all three time points, and five differentially methylated genes were both present at 0 and 48 h.

Significantly differentially methylated genes upon butyrate treatment. a MA plots for the indicated sampling points. M values below the threshold of −1.0 (red) represent de novo methylated regions upon butyrate treatment. M values above the threshold of 1.0 (green) represent hypomethylated regions upon butyrate treatment. The blue lines mark for each time point the 95 % and 99 % confidence intervals of all observed M values. The yellow line represents the LOWESS fit. b,c Venn diagrams of genes associated with hypermethylated (b) or hypomethylated (c) CpG islands prior to (0 h), 24 h and 48 h upon treatment of CHO DP-12 cells with 3 mM butyric acid
Gene ontology analyses were carried out in order to gain an overview of affected cellular processes and components (Fig. 4a and b). In the dataset recorded before butyrate treatment, it was found that the GO protein group 'positive regulation of developmental process' was significantly overrepresented. The data of sampling point '24 h' suggest that the genes associated with hypermethylated CGIs play a role in the processes 'response to stimulus', 'phosphorylation' and 'biogenesis', e.g., represented by the more specific functions 'DNA repair', 'response to organic substance' or 'programmed cell death'. Within the group of genes associated with hypomethylated CGIs, the categories 'transcription', 'chromatin', 'signaling', 'immune response', 'developmental process' and 'phosphate metabolism' were frequently affected, e.g., comprising functions such as 'regulation of gene-specific transcription', 'chromatin modification', 'regulation of MAP kinase activity' or 'immune system development'. Another 24 h later (sampling point '48 h'), genes related to 'carbohydrate biosynthetic process' were abundantly associated with hypermethylated CGIs. Hypomethylated genes at the 48 h sampling point could be clustered according to the functional categories 'chromatin modification', 'dendrite morphogenesis', 'post-embryonic development' or 'protein modification by small protein conjugation or removal'.

Gene ontology analysis of genes affected by butyrate-mediated differential methylation of associated CpG islands. a,b Heat maps of significantly enriched GO terms. GO analysis was carried out using DAVID with an EASE score ≤0.01. Genes detected without treatment with 3 mM butyric acid (0 h) were removed from the 24 and 48 h datasets before ontology analysis. Log2 values of fold enrichment are colour-coded according to the legend, n.s. not significantly enriched
Differential methylation of Wnt signaling genes
Within the genes associated with differentially methylated CGIs 24 h upon butyrate addition, genes related to the 'intracellular signaling cascade' were significantly overrepresented. Next to MAPK- and Ca2+-signaling, one of the mainly affected cellular pathways comprised within this group was the Wnt signaling pathway (see KEGG pathway in Fig. 5). Promoter methylation of key elements and target genes of the canonical branch as well as the Wnt/Ca2+ pathway was detected for the following genes: Porcn, Wnt6, Fzd7, Prkx and Rbx1 showed relatively hypermethylated CGIs located in promoter regions. The promoters of Dvl1, Axin, Camk2g, Lrp5, Prkcg, Ppp2r5e, Ppp3cc, Tcf7l1, Tcf7l2 and Tbl1x were detected as relatively hypomethylated. Further examination of the data concerning central β-catenin co-activator proteins showed either relatively hypermethylated (Bcl9l, Pygo2, Med13) or relatively hypomethylated genes (Med1, Trrap, Mll, Mll2). Finally, the Wnt responsive genes Myc and cycD1 were detected as relatively hypermethylated.

Differential methylation of CGIs associated with genes involved in the Wnt signal transduction system 24 h upon butyrate addition. Genes associated with relatively hypomethylated CGIs (green), genes associated with relatively hypermethylated CGIs (red) and genes without significantly differentially methylated CGIs (not coloured). Figure based on KEGG (Kanehisa and Goto 2000)
Discussion
A variety of methods has been developed to conduct genome-scale analyses of DNA methylation, ranging from microarray hybridization techniques to DNA methylation profiling by whole-genome shotgun bisulfite sequencing (WGSBS), the latter yielding genome-wide methylation data in single-base resolution (Bock et al. 2010). However, for economic reasons WGSBS, although providing the greatest genome coverage, is applicable only to a limited number of samples (Laird 2010). Microarray-based techniques enable the analysis of a larger number of samples, but restrict the investigations to a pre-selected number of genomic loci. As we were interested in a comprehensive examination of DNA methylation changes in a medium-throughput format, we developed a customized CHO CGI microarray to simultaneously analyze multiple samples of enriched methylated DNA and detect differentially methylated genomic regions. We applied our microarray to compare butyrate treated with reference CHO cultures in order to analyze the butyrate effect in CHO cells.
The stability of production cell lines can be impaired by recombinant gene loss or malfunctioning transcriptional events (Beckmann et al. 2012; He et al. 2012) as well as epigenetic silencing of recombinant genes e.g., affecting the CMV promoter (Kim et al. 2011; Osterlehner et al. 2011). Supplementation of the SCFA butyrate leads to alterations of epigenetic silencing events and can be used to enhance cell specific productivities in CHO cells (Jiang and Sharfstein 2008; Boffa et al. 1978; Gorman et al. 1983; Chen et al. 2002). This is, among other yet unknown effects, due to the inhibitory influence of butyrate on HDACs (Davie 2003). In this context, the analysis of DNA methylation is particularly important to gain further insight into the epigenetic response to butyrate addition. With our work, we are now able to show that butyrate-induced inhibition of growth and increased cell specific productivities in anti-IL8 producing CHO DP-12 cultures is accompanied by significant and reversible changes in DNA methylation.
Butyrate causes methylation of genes related to several functional categories in a time-dependent manner
The analysis of differentially methylated regions showed 1221 genes to be affected 24 h upon butyrate addition. Gene ontology analyses were carried out to obtain an overview of involved cellular processes and components. One overrepresented group of affected genes 24 h upon butyrate addition was related to the 'intracellular signaling cascade'. A closer view showed involvement of pathways such as the MAPK, Ca2+ and the canonical Wnt/β-catenin signaling system. Butyrate-mediated effects on these signal transduction pathways have been reported before in human cell lines (Yang et al. 2001; Zhang et al. 2010) and, among others, may lead to the observed apoptotic events and the negative impact on cell cycle progression. A butyrate-mediated regulation of apoptosis, gene expression, cell cycle, transport, growth and proliferation was shown on other cellular levels and in other cell types as well (Wu et al. 2012; Yee et al. 2008; Chang et al. 2012; Zhou et al. 2011; Li and Li 2006). The presence of components of 'neuron development', 'synaptic transmission' or 'dendrite morphogenesis' was striking, too. Though butyrate is able to induce differentiation of stem cells into neural cells (Yao et al. 2010), an involvement of such specific factors in butyrate-treated CHO cells has not been reported yet. Another significantly abundant group of ontologies refers to 'immune system development'. Several publications report an anti-inflammatory effect of SCFAs, e.g., on human monocytes (Säemann et al. 2000).
Interestingly, 48 h upon butyrate supplementation 91 % of the observed regulations at sampling point '24 h' were not detectable anymore. In order to investigate a possible underlying mechanism more closely, the array data was subjected to an analysis regarding regulatory components of the DNA methylation machinery. The genes Dnmt3a and Uhrf1, coding for major mediators of de novo and maintenance methylation (Bostick et al. 2007; Cedar and Bergman 2009), turned out to be relatively hypomethylated at the 24 h sampling point. Furthermore, the genes Gadd45g and Apobec2, coding for mediators of DNA demethylation (Barreto et al. 2007; Rai et al. 2008) showed changes in their promoter methylation. Strikingly, functional components of the chromatin modifying machinery were especially affected at sampling point '48 h'. As mentioned before, butyrate is known to inhibit HDAC activity (Boffa et al. 1978) and to have an effect on chromatin modification (Costa 2008). However, initially we did not expect such highly dynamic changes on the epigenetic level in CHO cells upon butyrate treatment. Also, we were surprised to see a number of differentially methylated genes among the cultures prior to butyrate treatment that were removed from the '24 h' and '48 h' datasets before further data analysis. These numbers and also the ratios between the respective sampling points remain constant even after applying the most stringent statistical parameters. There are several possible explanations for this finding. On the one hand, a statistically relevant occurrence of differentially methylated genes could of course be caused by a biased enrichment reaction or uneven labeling of enriched methylated DNA. On the other hand it is known that within cell populations or even between individual cells a certain degree of noise can occur within regulatory networks (Chalancon et al. 2012). It was shown that switching between states of DNA methylation can contribute to this noise (Lim and van Oudenaarden 2007). The differentially methylated genes detected before butyrate treatment may represent this effect, which could be further magnified by asynchronous cell growth or minimal chronological differences in sampling. Interestingly, it is known that cultured cells can behave very differently over the course of a production process even if they were treated in the same way (Le et al. 2012). The reason for this differential behavior has not been found yet. However, since in this first application of our novel CHO CGI microarray, the two groups of four replicates for reference and butyrate-treated cultures were pooled after the enrichment of methylated DNA for microarray analysis, the variance within this group cannot be calculated yet. Current experiments address the variance between identically treated cultures and will furthermore allow an estimation of putative biased experimental steps.
Differentially methylated core components of canonical Wnt/β-catenin signaling
In order to prove the ability of our CHO-specific CGI microarray to detect differential methylation within central regulatory networks, canonical Wnt signaling as one of many affected pathways was exemplarily analyzed. Wnt signaling is mediated by the highly conserved family of secreted Wnt glycolipoproteins and directs several developmental processes (MacDonald et al. 2009). We detected butyrate-mediated DNA methylation within the canonical Wnt/β-catenin pathway ranging from members of Wnt biogenesis and signal transduction in the cytoplasm to transcriptional regulation in the nucleus (respective differentially methylated genes in brackets). Porcupine (Porcn), an ER localized membrane-spanning protein, mediates secretion and palmitoylation of Wnt proteins (Wnt6) (Hausmann et al. 2007; Janda et al. 2012). This modification is essential for their interaction with the Frizzled seven-pass transmembrane protein family (Fzd7) and the LDL receptor-related proteins LRP5 and LRP6 (Lrp5) (Janda et al. 2012). The formation of a Wnt–Fzd–LRP complex leads to recruitment of the scaffolding protein Dishevelled (Dvl1), subsequent interaction with the Axin complex (Axin) and thereby stabilization of β-catenin (Logan and Nusse 2004). β-Catenin moves into the nucleus and regulates gene expression of Wnt target genes alongside the transcription factors TCF/LEF (Tcf7l1, Tcf7l2; Hoppler and Kavanagh 2007). The subsequent recruitment of several co-activators (e.g., Med1, Med13, Bcl9l or Pygo2) regulates a plethora of transcriptional events (MacDonald et al. 2009). For example, expression of the Wnt responsive gene c-Myc (Myc) is activated by β-catenin (He et al. 1998). Myc has decisive influence on growth, proliferation and differentiation (Massagué 2004). Its expression was previously shown to be influenced by butyrate (Krupitza et al. 1995; Mariani et al. 2003) and may be accountable for many reported effects of butyrate as well as those we observed in CHO DP-12 cells upon supplementation of the SCFA. Generally, the influence of butyrate on Wnt signaling has been stated before on other cellular levels, e.g., by measuring Wnt activity or Wnt target gene expression upon butyrate treatment (Bordonaro et al. 2008; Shin et al. 2012). However, to our knowledge, this is the first time that such a far-reaching influence of butyrate-mediated DNA methylation changes on the Wnt signal transduction system has been shown.
Perspectives of CHO CGI microarrays
With this work, we present the first CGI microarray that allows the detection of differential DNA methylation in CHO cells on a genomic scale. A comprehensive study conducted on CHO DP-12 cells demonstrates that our newly designed array is applicable to unravel epigenetic events in the course of cultivation processes. In our first experiment the effect of butyrate on DNA methylation unexpectedly turned out to be highly dynamic and reversible. Moreover, signaling pathways such as the MAPK-, Ca2+- and canonical Wnt/β-catenin system were affected by differential DNA methylation. Further investigations will clarify if the observed reversibility of the epigenetic effects is due to a putative feedback mechanism or to process-related circumstances. Eventually, this should lead to identification of central cellular components targeted by butyrate. Considering the different effects of DNA methylation at certain genomic loci, the impact of differential DNA methylation should ideally be examined by combining DNA methylation analysis with transcriptomics, proteomics and metabolomics in order to gain information on multiple cellular levels. This new approach to study genome-wide changes in DNA methylation in dependency on various cultivation characteristics will provide valuable information regarding the optimization of production processes. A promising application is a subsequent target selection for metabolic and epigenetic engineering approaches. This should facilitate the optimization of product titer and quality in recombinant CHO cells and furthermore enable establishment of methods to screen for stably producing cell lines.
References
Barreto G, Schäfer A, Marhold J, Stach D, Swaminathan SK, Handa V, Döderlein G, Maltry N, Wu W, Lyko F, Niehrs C (2007) Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445:671–675. doi:10.1038/nature05515
Becker J, Hackl M, Rupp O, Jakobi T, Schneider J, Szczepanowski R, Bekel T, Borth N, Goesmann A, Grillari J, Kaltschmidt C, Noll T, Pühler A, Tauch A, Brinkrolf K (2011) Unraveling the Chinese hamster ovary cell line transcriptome by next-generation sequencing. J Biotechnol 156:227–235. doi:10.1016/j.jbiotec.2011.09.014
Beckmann TF, Krämer O, Klausing S, Heinrich C, Thüte T, Büntemeyer H, Hoffrogge R, Noll T (2012) Effects of high passage cultivation on CHO cells: a global analysis. Appl Microbiol Biotechnol 94:659–671. doi:10.1007/s00253-011-3806-1
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol 57:289–300. doi:10.2307/2346101
Bock C, Reither S, Mikeska T, Paulsen M, Lengauer T (2005) BiQ Analyzer: visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics 21:4067–4068. doi:10.1093/bioinformatics/bti652
Bock C, Tomazou EM, Brinkman AB, Müller F, Simmer F, Gu H, Jäger N, Gnirke A, Stunnenberg HG, Meissner A (2010) Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat Biotechnol 28:1106–1114. doi:10.1038/nbt.1681
Boffa LC, Vidali G, Mann RS, Allfrey VG (1978) Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem 253:3364–3366
Bordonaro M, Lazarova DL, Sartorelli AC (2008) Butyrate and Wnt signaling: a possible solution to the puzzle of dietary fiber and colon cancer risk? Cell Cycle 7:1178–1183
Bostick M, Kim JK, Estève P-O, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317:1760–1764. doi:10.1126/science.1147939
Cedar H, Bergman Y (2009) Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10:295–304. doi:10.1038/nrg2540
Chalancon G, Ravarani CNJ, Balaji S, Martinez-Arias A, Aravind L, Jothi R, Babu MM (2012) Interplay between gene expression noise and regulatory network architecture. Trends Genet 28:221–232. doi:10.1016/j.tig.2012.01.006
Chang M-C, Tsai Y-L, Chen Y-W, Chan C-P, Huang C-F, Lan W-C, Lin C-C, Lan W-H, Jeng J-H (2012) Butyrate induces reactive oxygen species production and affects cell cycle progression in human gingival fibroblasts. J Periodontal Res 48:66–73. doi:10.1111/j.1600-0765.2012.01504.x
Chen F, Kou T, Fan L, Zhou Y, Ye Z, Zhao L, Tan W-S (2011) The combined effect of sodium butyrate and low culture temperature on the production, sialylation, and biological activity of an antibody produced in CHO cells. Biotechnol Bioprocess Eng 16:1157–1165. doi:10.1007/s12257-011-0069-8
Chen Z, Clark S, Birkeland M, Sung CM, Lago A, Liu R, Kirkpatrick R, Johanson K, Winkler JD, Hu E (2002) Induction and superinduction of growth arrest and DNA damage gene 45 (GADD45) α and β messenger RNAs by histone deacetylase inhibitors trichostatin A (TSA) and butyrate in SW620 human colon carcinoma cells. Cancer Lett 188:127–140
Costa FF (2008) Non-coding RNAs, epigenetics and complexity. Gene 410:9–17. doi:10.1016/j.gene.2007.12.008
Davie JR (2003) Inhibition of histone deacetylase activity by butyrate. J Nutr 133:2485S–2493S
Dondrup M, Albaum SP, Griebel T, Henckel K, Jünemann S, Kahlke T, Kleindt CK, Küster H, Linke B, Mertens D, Mittard-Runte V, Neuweger H, Runte KJ, Tauch A, Tille F, Pühler A, Goesmann A (2009) EMMA 2 — a MAGE-compliant system for the collaborative analysis and integration of microarray data. BMC Bioinforma 10:50. doi:10.1186/1471-2105-10-50
Gorman CM, Howard BH, Reeves R (1983) Expression of recombinant plasmids in mammalian cells is enhanced by sodium butyrate. Nucleic Acids Res 11:7631–7648
Hackl M, Jadhav V, Jakobi T, Rupp O, Brinkrolf K, Goesmann A, Pühler A, Noll T, Borth N, Grillari J (2012) Computational identification of microRNA gene loci and precursor microRNA sequences in CHO cell lines. J Biotechnol 158:151–155. doi:10.1016/j.jbiotec.2012.01.019
Hausmann G, Bänziger C, Basler K (2007) Helping Wingless take flight: how WNT proteins are secreted. Nat Rev Mol Cell Biol 8:331–336. doi:10.1038/nrm2141
He L, Winterrowd C, Kadura I, Frye C (2012) Transgene copy number distribution profiles in recombinant CHO cell lines revealed by single cell analyses. Biotechnol Bioeng 109:1713–1722. doi:10.1002/bit.24428
He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-myc as a target of the APC pathway. Science 281:1509–1512
Hoppler S, Kavanagh CL (2007) Wnt signalling: variety at the core. J Cell Sci 120:385–393. doi:10.1242/jcs.03363
Huang DW, Sherman BT, Zheng X, Yang J, Imamichi T, Stephens R, Lempicki RA (2009) Extracting biological meaning from large gene lists with DAVID. Curr Protoc Bioinformatics Chapter 13:Unit 13.11. doi:10.1002/0471250953.bi1311s27
Huggett B, Lähteenmaki R (2012) Public biotech 2011 — the numbers. Nat Biotech 30:751–757. doi:10.1038/nbt.2320
Illingworth RS, Bird AP (2009) CpG islands — 'a rough guide'. FEBS Lett 583:1713–1720. doi:10.1016/j.febslet.2009.04.012
Janda CY, Waghray D, Levin AM, Thomas C, Garcia KC (2012) Structural basis of Wnt recognition by Frizzled. Science 337:59–64. doi:10.1126/science.1222879
Jiang Z, Sharfstein ST (2008) Sodium butyrate stimulates monoclonal antibody over-expression in CHO cells by improving gene accessibility. Biotechnol Bioeng 100:189–194. doi:10.1002/bit.21726
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. doi:10.1093/nar/28.1.27
Kim M, O'Callaghan PM, Droms KA, James DC (2011) A mechanistic understanding of production instability in CHO cell lines expressing recombinant monoclonal antibodies. Biotechnol Bioeng 108:2434–2446. doi:10.1002/bit.23189
Kim Y-G, Kim JY, Lee GM (2009) Effect of XIAP overexpression on sodium butyrate-induced apoptosis in recombinant Chinese hamster ovary cells producing erythropoietin. J Biotechnol 144:299–303. doi:10.1016/j.jbiotec.2009.09.016
Kornblihtt AR, Schor IE, Alló M, Dujardin G, Petrillo E, Muñoz MJ (2013) Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol 14:153–165. doi:10.1038/nrm3525
Krupitza G, Harant H, Dittrich E, Szekeres T, Huber H, Dittrich C (1995) Sodium butyrate inhibits c-myc splicing and interferes with signal transduction in ovarian carcinoma cells. Carcinogenesis 16:1199–1205
Kwaks THJ, Otte AP (2006) Employing epigenetics to augment the expression of therapeutic proteins in mammalian cells. Trends Biotechnol 24:137–142. doi:10.1016/j.tibtech.2006.01.007
Laird PW (2010) Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet 11:191–203. doi:10.1038/nrg2732
Le H, Kabbur S, Pollastrini L, Sun Z, Mills K, Johnson K, Karypis G, Hu W-S (2012) Multivariate analysis of cell culture bioprocess data—lactate consumption as process indicator. J Biotechnol 162:210–223. doi:10.1016/j.jbiotec.2012.08.021
Li L-C, Dahiya R (2002) MethPrimer: designing primers for methylation PCRs. Bioinformatics 18:1427–1431
Li RW, Li C (2006) Butyrate induces profound changes in gene expression related to multiple signal pathways in bovine kidney epithelial cells. BMC Genomics 7:234. doi:10.1186/1471-2164-7-234
Lim HN, van Oudenaarden A (2007) A multistep epigenetic switch enables the stable inheritance of DNA methylation states. Nat Genet 39:269–275. doi:10.1038/ng1956
Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20:781–810. doi:10.1146/annurev.cellbio.20.010403.113126
MacDonald BT, Tamai K, He X (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell 17:9–26. doi:10.1016/j.devcel.2009.06.016
Mariani MR, Carpaneto EM, Ulivi M, Allfrey VG, Boffa LC (2003) Correlation between butyrate-induced histone hyperacetylation turn-over and c-myc expression. J Steroid Biochem Mol Biol 86:167–171. doi:10.1016/S0960-0760(03)00267-X
Massagué J (2004) G1 cell-cycle control and cancer. Nature 432:298–306. doi:10.1038/nature03094
Mutskov V, Felsenfeld G (2004) Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J 23:138–149. doi:10.1038/sj.emboj.7600013
Ndlovu MN, Denis H, Fuks F (2011) Exposing the DNA methylome iceberg. Trends Biochem Sci 36:381–387. doi:10.1016/j.tibs.2011.03.002
Osterlehner A, Simmeth S, Göpfert U (2011) Promoter methylation and transgene copy numbers predict unstable protein production in recombinant Chinese hamster ovary cell lines. Biotechnol Bioeng 108:2670–2681. doi:10.1002/bit.23216
Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR (2008) DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell 135:1201–1212. doi:10.1016/j.cell.2008.11.042
Säemann MD, Böhmig GA, Österreicher CH, Burtscher H, Parolini O, Diakos C, Stöckl J, Hörl WH, Zlabinger GJ (2000) Anti-inflammatory effects of sodium butyrate on human monocytes: potent inhibition of IL-12 and up-regulation of IL-10 production. FASEB J 14:2380–2382. doi:10.1096/fj.00-0359fje
Sambrook J, Russell DW (2006) Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc 2006:pdb.prot4455. doi:10.1101/pdb.prot4455
Sato F, Tsuchiya S, Meltzer SJ, Shimizu K (2011) MicroRNAs and epigenetics. FEBS J 278:1598–1609. doi:10.1111/j.1742-4658.2011.08089.x
Shin H, Kim J-H, Lee YS, Lee YC (2012) Change in gene expression profiles of secreted frizzled-related proteins (SFRPs) by sodium butyrate in gastric cancers: induction of promoter demethylation and histone modification causing inhibition of Wnt signaling. Int J Oncol 40:1533–1542. doi:10.3892/ijo.2012.1327
Sun H, Wu J, Wickramasinghe P, Pal S, Gupta R, Bhattacharyya A, Agosto-Perez FJ, Showe LC, Huang TH-M, Davuluri RV (2011) Genome-wide mapping of RNA Pol-II promoter usage in mouse tissues by ChIP-seq. Nucleic Acids Res 39:190–201. doi:10.1093/nar/gkq775
Sung YH, Lee GM (2005) Enhanced human thrombopoietin production by sodium butyrate addition to serum-free suspension culture of bcl-2-overexpressing CHO cells. Biotechnol Prog 21:50–57. doi:10.1021/bp049892n
Takai D, Jones PA (2002) Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A 99:3740–3745. doi:10.1073/pnas.052410099
Walsh G (2010) Post-translational modifications of protein biopharmaceuticals. Drug Discov Today 15:773–780. doi:10.1016/j.drudis.2010.06.009
Wu S, Li RW, Li W, Li C-J (2012) Transcriptome characterization by RNA-seq unravels the mechanisms of butyrate-induced epigenomic regulation in bovine cells. PloS ONE 7:e36940. doi:10.1371/journal.pone.0036940
Wurm FM (2004) Production of recombinant protein therapeutics in cultivated mammalian cells. Nat Biotech 22:1393–1398. doi:10.1038/nbt1026
Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S, Andersen MR, Neff N, Passarelli B, Koh W, Fan HC, Wang J, Gui Y, Lee KH, Betenbaugh MJ, Quake SR, Famili I, Palsson BO, Wang J (2011) The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat Biotechnol 29:735–741. doi:10.1038/nbt.1932
Yang J, Kawai Y, Hanson RW, Arinze IJ (2001) Sodium butyrate induces transcription from the Gαi2 gene promoter through multiple Sp1 sites in the promoter and by activating the MEK-ERK signal transduction pathway. J Biol Chem 276:25742–25752. doi:10.1074/jbc.M102821200
Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30:e15
Yao X, Zhang J, Huang H, Dai L, Liu Q, Zhang M (2010) Histone deacetylase inhibitor promotes differentiation of embryonic stem cells into neural cells in adherent monoculture. Chin Med J 123:734–738
Yee JC, de Leon GM, Philp RJ, Yap M, Hu W-S (2008) Genomic and proteomic exploration of CHO and hybridoma cells under sodium butyrate treatment. Biotechnol Bioeng 99:1186–1204. doi:10.1002/bit.21665
Zhang Y, Zhou L, Bao YL, Wu Y, Yu CL, Huang YX, Sun Y, Zheng LH, Li YX (2010) Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem-Biol Interact 185:174–181. doi:10.1016/j.cbi.2010.03.035
Zhou Q, Dalgard CL, Wynder C, Doughty ML (2011) Histone deacetylase inhibitors SAHA and sodium butyrate block G1-to-S cell cycle progression in neurosphere formation by adult subventricular cells. BMC Neurosci 12:50. doi:10.1186/1471-2202-12-50
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Wippermann, A., Klausing, S., Rupp, O. et al. Establishment of a CpG island microarray for analyses of genome-wide DNA methylation in Chinese hamster ovary cells. Appl Microbiol Biotechnol 98, 579–589 (2014). https://doi.org/10.1007/s00253-013-5282-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-013-5282-2