
Abstract
In clostridia, n-butanol production from carbohydrates at yields of up to 76% of the theoretical maximum and at titers of up to 13 g/L has been reported. However, in Escherichia coli, several groups have reported butyric acid or butanol production from recombinant expression of clostridial genes, at much lower titers and yields. To pinpoint deficient steps in the recombinant pathway, we developed an analytical procedure for the determination of intracellular pools of key pathway intermediates and applied the technique to the analysis of three sets of E. coli strains expressing various combinations of butyrate biosynthesis genes. Low expression levels of the hbd-encoded S-3-hydroxybutyryl-CoA dehydrogenase were insufficient to convert acetyl-CoA to 3-hydroxybutyryl-CoA, indicating that hbd was a rate-limiting step in the production of butyryl-CoA. Increasing hbd expression alleviated this bottleneck, but in resulting strains, our pool size measurements and thermodynamic analysis showed that the reaction step catalyzed by the bcd-encoded butyryl-CoA dehydrogenase was rate-limiting. E. coli strains expressing both hbd and ptb-buk produced crotonic acid as a byproduct, but this byproduct was not observed with expression of related genes from non-clostridial organisms. Our thermodynamic interpretation of pool size measurements is applicable to the analysis of other metabolic pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
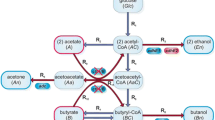
Butyric acid is a saturated C4 fatty acid and intermediate in the clostridial biosynthesis of butanol. As such, an efficient means to produce it from glucose or other biomass-derived carbohydrates could enable efficient biofuels production from renewable biomass. In Clostridium beijerinckii, n-butanol production at yields of up to 74% of the theoretical maximum and at titers of up to 13 g/L has been reported (Ezeji et al. 2004). In Clostridium tyrobutyricum, which ends its fermentation at butyrate and does not produce butanol, butyrate yields of 78% of the theoretical maximum and titers of up to 50 g/L have been reported (Liu et al. 2006). Butyrate biosynthesis in clostridia is believed to proceed by the pathway shown in Fig. 1.

Pathways of butyrate and butanol biosynthesis from acetyl-CoA (AcCoA) in Clostridium acetobutylicum. Also shown is the reaction for the non-clostridial tesB gene product (thioesterase II), which can hydrolyze acyl-CoAs directly to free carboxylic acids in a single step
Because clostridia are more difficult to transform, are sensitive to oxygen, exhibit complex fermentation kinetics, and can form spores, they can be difficult to engineer. Synthetic biology promises to simplify and expedite the design and engineering of metabolic pathways for the bioconversion of various feedstocks to desired fuels and chemicals. Several groups have recently applied synthetic biology approaches to the synthesis of n-butanol in a well-studied, robust host organism such as Escherichia coli (Atsumi et al. 2008; Inui et al. 2008; Nielsen et al. 2009), but always at yields under 15% of the theoretical, and with titers less than 0.7 g/L. Two groups have assayed butanol pathway enzymes in cell extracts of heterologous hosts (Inui et al. 2008; Nielsen et al. 2009) and found that activity levels of the butanoyl-CoA dehydrogenase, encoded by bcd and accompanying electron transfer flavoproteins encoded by etfA and etfB, was either much lower than other pathway enzymes (Inui et al. 2008) or not detectable in cell extracts (Nielsen et al. 2009). However, despite the low or non-measureable activity of the bcd-etfAB encoded butanoyl-CoA dehydrogenase enzyme, bcd and etfAB are required for production of butanol in recombinant E. coli. Butanoyl-CoA dehydrogenase is known to be very oxygen sensitive and yet expression of bcd and etfAB under aerobic conditions led to measureable butanol production in E. coli (Atsumi et al. 2008).
The bcd-etfAB encoded enzyme is thus a prime candidate as a bottleneck in the production of butanol from recombinant E. coli strains expressing the clostridial butanol biosynthesis genes, but bottlenecks may also occur in other reaction steps, and the role of oxygen is yet unclear. To bear further light on these questions, we measured acyl-CoA and related pool sizes in a battery of E. coli strains expressing clostridial genes for butyrate and butanol biosynthesis in a background strain engineered to favor butyrate production and to be an obligate aerobe.
Methods
Strains
E. coli K12 Δpta, E. coli K12 ΔldhA, and E. coli K12 ΔadhE were constructed via the method of Datsenko and Wanner (2000) starting from E. coli K12 MG1655. These genotypes were combined through a series of P1mediated transductions of the corresponding pta::kan, ldhA::kan, and adhE::kan alleles followed by elimination of the kanamycin marker with FLP-mediated recombination by pCP20. The resulting triple Δpta ΔldhA ΔadhE triple knockout was called the pal strain.
For expression of the lambda Red expressing electrocompetent cells, an IPTG-inducible variant of the pKD46 plasmid described by Wanner was used. The pal(DE3) derivative of pal was created with the DE3 lysogenization kit (Merck KGaA, Darmstadt, Germany) as per the manufacturer’s directions.
The pal-1e strain was created by evolving E. coli pal pZE21-pab pHATet-CBEH (see below) in MOPS butyrate medium for 130 h after inoculation from an overnight 4 mL culture in LB broth. No noticeable growth occurred until after 110 h, when a population that could grow on butyrate as a sole carbon source. Colonies from this population were streaked from the exponentially growing culture in MOPS butyrate onto LB agar. Two colonies were picked from the resulting plates, grown overnight in LB broth, and re-inoculated into MOPS butyrate where they were found to exhibit a lag phase of less than 1 h. This strain may be a fadR deletion mutant (Campbell et al. 2003) but has not been exhaustively characterized.
Plasmids
The ptb-buk operon was cloned from Clostridium acetobutylicum ATCC 824 genomic DNA using primers “MT ptb-buk Sense” (TCCCCCCGGGGGG AGTTTATAGGGCAAAAGTTTTATAAACATGGGTACTGG) and “MT ptb-buk Anti” (GGGGTACC CCGAGTGTACGACCAGTGATTAAGAGTTTTAA) endowed with XmaI and KpnI restriction sites (emphasized in primer sequences). This PCR product was digested and ligated to KpnI/XmaI digested pZE21-gfp(ASV) (Elowitz and Leibler 2000) in order to create pZE21ptb-buk. The atoB gene was cloned from E. coli K12 MG1655 genomic DNA with primers “CF184 atoB Sense KpnI” (ACTCGGGTACCA TGAAAAATTGTGTCATCGTCAGTG) and “CF185 atoB anti HindIII” (AGAGGAAGCTT TTAATTCAACCGTTCAATCACCATC), digested with HindIII and KpnI, and ligated to the HindIII/KpnI fragment of pZE21-gfp(ASV) to create pZE21-atoB. The promoter-gene PLTetO1-atoB expression cassette from pZE21-atoB was amplified with primers “CF188 P(LTetO1) atoB Sense (AatII)” (GGCTTCCCAACCTTACCAGAGG) and “CF189 P(LTetO1) atoB Anti AatII(XbaI)” (CCTCGACGTCTAGATATTACCGCCTTTGAGTGAGCTG), and the resulting fragment was digested with AatII and ligated into an AatII-digested fragment of pZE21-ptb-buk in order to create pZE21-pab. pRK415-BCS (Nielsen et al. 2009) was digested with SacI and AvrII, and the fragment containing the BCS operon with genes crt, etfAB, bcd, and hbd was ligated to XbaI-digested pHACm (Alper and Stephanopoulos 2007) in order to create pHACm-CBEH. This plasmid carries genes for both tetracycline resistance (from pRK415-BCS) and chloramphenicol resistance (from pHACm). The CmR-conferring cat gene was disrupted in pHACm-CBEH by removing the 38 bp MscI and NcoI linker region and re-ligating the plasmid, in order to form pHATet-CBEH.
Culturing conditions
Batch oxygen-limiting cultures were maintained in 15 mL glass tubes (Bellco Glass, Inc.) stoppered with a butyl rubber septum. The septum was pierced with a 26-gauge or 27-gauge syringe needle. Antibiotics were used as required (Sambrook and Russell 2001).
Anaerobically, E. coli requires bicarbonate for growth at low cell densities (Hornsten 1995). In batch oxygen-limiting cultures without bicarbonate or CO2 supplementation, this requirement results in long lag phases as sufficient bicarbonate must be accumulated by the metabolism of inoculated cells before growth can begin. In continuous cultures, gas sparging can strip away bicarbonate as CO2, stopping growth. To avoid either possibility, MOPS minimal medium (Teknova Inc., Hollister, CA) was supplemented with 10 mM ammonium bicarbonate.
Stoichiometric modeling of the pal strain
We used a stoichiometric model of E. coli metabolism considered derived from the iJE660 model constructed by Jeremy Edwards and his co-workers (Edwards and Palsson 2000). The model as originally formulated by Edwards et al. included redundant columns (fluxes) in an attempt to model stoichiometrically equivalent reactions. For example, the original model included five duplicate columns for the NADH-driven reduction of L-lactate to lactaldehyde (column indices were 109, 110, 111, 113, and 114). This model was simplified by removing 117 stoichiometrically duplicated fluxes, resulting in a stoichiometric matrix with dimensions 536 × 836. This simplified model was called “iJE660b.” To construct stoichiometric product-growth envelopes, the growth yield was fixed at μ f gDCW/mol glucose, and then fluxes to either succinate or butyrate were minimized and maximized subject to iJE660b and the additional growth = μ f constraint. This procedure was repeated for many values of μ f between zero and the maximum growth rate possible in iJE660b.
Determination of intracellular acyl-CoAs
Our method for the determination of intracellular levels of short-chain acyl-CoA esters is derived from the method reported by Shimazu et al. (2004), but is modified in several aspects. Briefly, in a 1.5-mL Eppendorf tube, a 1-mL aliquot of exponential growth-phase bacterial culture was suspended above 400 μL of bromododecane, which itself was layered above 100 μL of metabolite extraction fluid. Because this cell harvesting method requires a dense, hexane- and bromododecane-immiscible polar, extraction fluid, we formulated an extraction fluid of acetonitrile, 2,2,2-trifluoroethanol, water, and trifluoroacetic acid in a 45:45:9:1 ratio (ρ ∼ 1.08 g/cm3). Samples were centrifuged for 30 s at 18,000×g. Cells were thus quickly centrifuged through the bromododecane layer into the metabolite extraction fluid, where they were rapidly lysed. Trials with GFP-expressing E. coli showed that cells pelleted in bromododecane alone retained fluorescence, but cell debris pelleted in extraction fluid had lost fluorescence after retrieval from the centrifuge. The culture medium and bromododecane supernatants were removed by aspiration, the crude extract was twice washed by the addition of 0.5 mL of hexanes (mixture of isomers) and subsequent vortexing and re-centrifugation. Of the original 100 μL, 70 μL of extract was transferred to a clean microcentrifuge tube in order to separate metabolite extracts from cell debris, and all samples were evaporated to dryness in a vacuum centrifuge (vaccufuge, Eppendorf AG, Hamburg, Germany) at 30 °C. Dried samples were stored indefinitely at −20 °C. This procedure avoids the use of both exotic extractants like Freon and trioctylamine (Shimazu et al. 2004) as well as solid-phase extraction columns (Minkler et al. 2006; Park et al. 2007). Instead of post-column derivatization with bromoacetaldehyde and fluorescent detection of the resulting 1,N 6-ethenoadenine adduct, we used pre-column derivatization with 150 μL of 1 M chloroacetaldehyde in 150 mM sodium citrate buffer (pH = 4.0) and fluorescent detection.
We used two chromatographic methods: one which we called Method A was a simple modification of the protocol of Shimazu “Method 1” (Shimazu et al. 2004), with identical HPLC setup but for a switch of Shimazu’s buffer A with a 50% mixture of (100 mM sodium phosphate, 75 mM sodium acetate, pH 4.5) and 50% 60 mM magnesium chloride, with both buffers amended by addition of 1% v/v unstabilized tetrahydrofuran. The addition of magnesium chloride was necessary for chromatographic resolution of AcCoA and 3HB-CoA, which co-elute in the method originally reported by Shimazu and co-workers. As such, data reported by Shimazu for AcCoA pool sizes in E. coli and Mixococcus xanthus may be overestimates, due to conflation of 3HB-CoA and AcCoA pool sizes. Method A was used for all analyses of Strain Set I.
In a second method we called Method B, buffer A was 5 mM ammonium phosphate/1% v/v THF adjusted to pH 6.5 with phosphoric acid, and buffer B was 99:1 methanol/THF. The column temperature was 60 °C and flow rate was 1.2 mL/min and the gradient regime was: 0% B for 2 min, then from 0% B to 6.25% B by 3 min, from 6.25% B to 14.6% B by 7.5 min, from 14.6% B to 75% B by 13.3 min; a constant flow of 75% B until 15.3 min, to 0% B by 16 min; and a constant flow of 0% B for 2 min. We found this method is preferred for measurement of adenosine phosphates and free CoASH. For all samples analyzed with this method, hexanoyl-CoA was used as an internal standard by 1,000 × dilution of a 10-mM stock of hexanoyl-CoA (Sigma-Aldrich, St. Louis, MO) dissolved in PBS into metabolite extraction fluid, immediately before beginning the extractions. The peak areas of all analytes were normalized to the HexCoA peak size, and the normalized peak area was calibrated against known standards in order to quantify metabolite pool sizes. Method B was used for all analyses of Strain Set II and III.
The concentrations of acyl-CoAs were calculated from linear (R 2 > 0.9) standard curves obtained from stock solutions (AcCoA, ATP, ADP, AMP, and CoASH) or from spiking known amounts of acyl-CoA into wild-type E. coli K12 metabolite extracts (all other acyl-CoAs). The uncertainty in measured pool sizes was the root-mean-square of the standard error between duplicate or triplicate measurements and the standard error associated with the linear regression. This latter error term was dominant for low pool size measurements (<100 pmol/(A600·mL).
Calculation of ΔIPTGΔG′
The strains used in this study express butyrate or butanol biosynthesis genes from IPTG-inducible promoters. If the induction of gene expression with IPTG changes the pool sizes of pathway metabolites, the free energy change associated with any given pathway step will also change. The difference between the induced state of the pathway and uninduced state of the pathway is shown in Eq. 1, where Q indicates the measured, possibly non-equilibrated reaction quotient \( \prod\limits_i {{{\left( {{C_i}} \right)}^{{\nu_i}}}} \), where C i is the concentration of reactant or product i and ν i is the stoichiometric coefficient of the reactant or product in the biochemical reaction of interest and K′ is the biochemical equilibrium constant (Alberty 2005). In the calculation of ΔIPTGΔG′, the thermodynamic equilibrium constant K′ cancels out, and thus uncertainty in ΔG′ stemming from uncertainty in ln K′ cancels out. To the extent that assumed physiological parameters like the NADH/NAD ratio or the energy charge stay constant after induction, the uncertainty in these values will also cancel out. The prime mark on K′ and ΔG′ indicates a biochemical standard state, where the pH, rather than the absolute number of moles of H+ is taken as the independent variable (Alberty 2005).
Results
Design of the pal strain and a stoichiometric driving force for butyrate production
In order to balance reducing equivalents, the native metabolism of E. coli ferments glucose to produce acetate, ethanol, and lactate, all of which could conceivably compete with butyrate production. To test the effects of removing these pathways on stoichiometrically possible space for butyrate production, the genes pta, adhE, and ldhA, responsible for the fermentative production of acetate, ethanol, and lactate, respectively, were deleted from the constraint-based stoichiometric model of E. coli metabolism (Edwards and Palsson 2000). In stoichiometric models not including clostridial butyrate biosynthesis reactions, these deletions led to a stoichiometric correlation between biomass formation and succinate production, as shown in Fig. 2a. In the Δpta, ΔadhE, and ΔldhA strain model (hereafter, the pal strain/model), growth without any succinate production was possible only up to biomass yields on glucose of 0.003 gDCW/1 mmol glucose, or about 0.017 gDCW/g glucose. This is about ten times lower than usual anaerobic biomass yields on glucose for E. coli (Hempfling and Mainzer 1975). Introduction of the clostridial butyrate biosynthesis pathway reactions shown in Fig. 1 into the stoichiometric model of E. coli pal metabolism caused a shift in the growth-optimal outlet for reducing equivalents, as shown in Fig. 2b. When the butyrate pathway was made available, growth yields exceed 0.094 gDCW/g glucose at a butyrate yield on glucose of only 0.1 mol/mol. The maximum biomass yield of the pal triple knockout model was the same as the (butyrate-producing) wild-type strain model, 0.032 gDCW/mmol glucose, and it occurred at a butyrate yield of 7.5 mol butyrate/10 mol glucose.

Stoichiometrically feasible region for the anaerobic biosynthesis of a succinate and E. coli cell biomass from glucose and carbon dioxide and b butyric acid and E. coli cell biomass from glucose as the sole carbon source. Panel c shows the data in b from an alternate visual perspective for clarity
Thus, although the E. coli metabolic network has alternate routes for clearing glycolysis-generated reducing equivalents under anaerobic conditions (e.g., the production of succinate), introduction of butyrate production provides a more facile route for anaerobic biomass production, at least stoichiometrically. To ensure that the activity of clostridial butyrate biosynthesis enzymes expressed in E. coli was not obscured or outcompeted by native E. coli metabolism, the pal strain background was used for all measurements of the pool sizes of butyrate pathway intermediates. An unexpected property of the pal strain was obligate aerobicity (see Supplemental Information), which allowed for growth under strict oxygen limitation, where the rate of oxygen to supply to the culture limits the rate of growth, a similar to condition previously shown to improve performance of the butanol fermentations (Atsumi et al. 2008).
Assessment of butyrate pathway activity by CoA pool size measurements in three strain sets
To examine the factors that control performance of the clostridial butyrate biosynthesis pathway in the E. coli pal strain, we measured in three strain sets the pool sizes of the pathway intermediates coenzyme A (CoASH), acetyl-CoA (AcCoA), acetoacetyl-CoA (AcAcCoA), 3-hydroxybutyryl-CoA (3HB-CoA), crotonyl-CoA (CrtCoA), and butyryl-CoA (ButCoA). None of the strain sets produced measurable quantities of butyric acid, but proved to differ markedly in the pool sizes of various pathway intermediates. Strain Set I includes strains expressing clostridial butyrate biosynthesis genes from low- or medium-copy pSC101 and pZE-derived plasmids and medium strength PLTetO1-derived promoters. No strains in Strain Set I produced measureable quantities of butyric acid during growth on minimal glucose medium. In all of these strains (#1–#9 in Table 1), AcCoA was the sole short-chain acyl-CoA detected from growth-phase cultures. The levels of AcCoA were similar in all of the strains, ranging from between 700 and 1,800 pmol/(A600·mL) for strains #1–#5. Strain #1, containing all genes in the clostridial butyrate biosynthesis pathway (Fig. 1), did not have significantly different AcCoA pool sizes than strains #2–#5, “dropout” strains missing one or more pathway genes. Pool sizes were generally lower in strains #6–#9, which were identical to strains #1–#4 except for introduction of a pRARE plasmid which facilitates the translation of genes containing rare codons. Quantitative data are given in Supplemental Table S4.
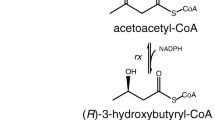
A strain of E. coli with the entire Ralstonia eutropha-derived pathway for polyhydroxybutyrate biosynthesis chromosomally integrated contains an alternate route for the synthesis of 3-hydroxybutyryl-CoA by use of the R. eutropha gene phaB. In contrast to the results with Strain Set I, a clone of this strain transformed with pHATet-CBEH and pZE21-pab (the same butyrate-producing plasmids used in Strain Set I), 3HB-CoA was detectable in cell extracts (Supplemental Table S4). This result suggested that either the expression level of hbd in Strain Set #1 was insufficient for production of the first two pathway intermediates AcAcCoA and 3HB-CoA, or that availability of the redox cofactor NADH was limiting the activity of the hbd-encoded enzyme. In the first case, the bottleneck could be alleviated by increasing the expression of hbd, and in the second, the bottleneck might be eliminated by swapping NADH-requiring hbd for NADPH-requiring 3-hydroxybutyryl-CoA reductases, such as phaB.
Further pool size measurements were made on Strain Set II, which express hbd and other genes for the production of 3-hydroxybutyric acid from monocistronic operons driven by much stronger IPTG-inducible, T7-derived promoters (Tseng et al. 2009). Measurements of intracellular acyl-CoAs in induced cultures of strains from Strain Set II revealed detectable quantities of 3HB-CoA, showing that expression levels of hbd in Strain Set I had been insufficient for measureable intracellular conversion of AcCoA to 3HB-CoA, and that intracellular NADH levels were sufficient for production of 3HB-CoA.
To test the effect of alleviating this bottleneck on the performance of the entire clostridial butanol biosynthesis pathways, pool sizes in a third set of strains was analyzed. This strain set bears plasmids containing the complete clostridial pathway for butanol fermentation (Nielsen et al. 2009), and these plasmids have previously been used to produce small amounts (<600 mg/L) of butanol. In this third strain set, butyryl-CoA was not reliably detected in IPTG-induced cultures (Table 2). In contrast, slight amounts of butyryl-CoA were detected in non-induced strains, but the uncertainty in the level of butyryl-CoA detected under non-inducing conditions was still quite large. Butyryl-CoA levels were too low to be reliably detected in these extracts, as illustrated by the uncertainties given in Table 2 for ButCoA.
Our HPLC technique also allows determination of intracellular ATP, ADP, and AMP. From these universal energy metabolites, the adenylate energy charge (EC), a ratio of the ATP pool plus one half the ADP pool to total adenine phosphate pool, can be calculated. This ratio is an indicator of the energetic state of the cell. AMP and ADP pool sizes measured in Strain Sets II and III are generally consistent with previously obtained measurements (Shimazu et al. 2004). The extremely low values for EC obtained in strains #11 and #13 after IPTG induction reflect the activity of the tesB gene product: the TesB enzyme does not regenerate ATP during the hydrolysis of 3HB-CoA to form free 3-hydroxybutyric acid; whereas with Ptb and Buk, ATP is regenerated during the two-step formation of 3-hydroxybutyric acid from 3HB-CoA. Pathway intermediates AcAcCoA and CrtCoA were not detected in any of the strain sets we studied.
A thermodynamic framework for relating pool size measurements to pathway activity
Enzymes accelerate the rate at which biochemical reactions reach equilibrium. The addition of IPTG leads to high-level overexpression of pathway enzymes, and thus should tend to push pathway reactions towards equilibrium. This push can be quantified using pool size measurements, if reaction thermodynamic data and pool size measurements are available. Because AcAcCoA and CrtCoA were not detectable in any of the strain sets we studied, we lumped the butyrate biosynthesis pathway into two reactions, the first being synthesis of 3HB-CoA from AcCoA and the second being the synthesis of ButCoA from 3HB-CoA.
Using the pool sizes in Table 2 and relevant thermodynamic parameters from the literature (see Supplemental Information), we estimated the effect of IPTG on equilibration of pathway reactions by computing ΔIPTGΔG′ (see the “Methods” section), as shown in Fig. 3. This figure shows that IPTG increased (made less thermodynamically favorable) the ΔG′ for conversion of AcCoA to 3HB-CoA for two of the 3HB-producing strains (strain #12 and #14), as shown by the positive values for ΔIPTGΔG′. The ΔIPTGΔG′ value for the 3HB-CoA-from-AcCoA reactions for strain #13 was also likely positive, but a value of 0 cannot be excluded because of the slight overlap of the error bar and the ΔIPTGΔG′ = 0 center axis. In strain #11, measurements did not reliably detect intracellular 3HB-CoA, increasing the uncertainty in ΔIPTGΔG′ values.

The change from adding IPTG inducer in measured ΔG′ values for component reactions of the clostridial butyrate or butanol biosynthesis pathway. A positive ΔIPTGΔG′ indicates that induction of pathway genes has filled pool sizes for reaction products (or depleted pool sizes for reactants) of the given reaction, and as such, represents evidence that pathway induction leads to conversion of thermodynamic chemical potential into flux through the given reaction. The graph shows ΔIPTGΔG′ for nine strains numbered according to Table 1. For some strains, induction clearly shifts the 3HB-CoA synthesis reaction (left panel) toward equilibrium. In contrast, no such change is shown for the ButCoA synthesis reaction (right panel), indicating that this pathway step is likely rate-limiting
The failure to detect appreciable levels of butyryl-CoA in Strain Set II and III under either inducing or non-inducing conditions made accurate estimation of ΔIPTGΔG′ fraught with uncertainty. However, it is notable that the estimated ranges for ΔIPTGΔG′ shown for the 3HB-CoA → ButCoA set of pathway reactions all overlap 0. The parent pal strain, with no plasmids or pathway genes, also had estimated ΔIPTGΔG′ values near 0 kJ/mol, as would be expected for a strain with no pathway activity of any type.
Unexpected behavior of ptb and buk
Strain #12 (in the pal background) expressing both hbd and ptb-buk also produced much higher amounts of crotonic acid in the medium supernatant than control strains. Figure 4 shows crotonate production for several strains growing under various conditions in MOPS minimal media. Strain #1, expressing all the genes of the clostridial butyrate biosynthesis pathway at relatively low levels, produced 76 μM crotonic acid during growth on MOPS glucose medium. In MOPS butyrate medium, this strain did not show signs of growth until more than 110 h of culture, after which growth eventually began. Two colonies isolated by streaking onto LB agar after growth in MOPS butyrate showed no lag phase on reinoculation into MOPS butyrate. This mutant evolved from Strain #1, called Strain #1e, produced 10× more crotonate than its parent in growth on MOPS glucose. Crotonic acid production was much lower in Strain #1e growing on MOPS butyrate, where flux through butyrate biosynthetic reactions (Fig. 1) is reversed.

Unexpected activity of the clostridial ptb-buk operons (encoding phosphotransbutyrylase and butyrate kinase) in recombinant E. coli. Crotonic acid is a byproduct of co-expressing both hbd (S-3-hydroxybutyryl-CoA dehydrogenase) and ptb-buk. The expression of butyrate biosynthesis genes (crotonase, crt; hbd; ptb-buk; or the R-3-hydroxybutyryl-CoA dehydrogenase-encoding phaB gene) is shown semi-quantitatively by color. Red indicates omission of a gene, shades of green indicate semiquantitative expression level. Crotonate production does not depend on the presence of crt, correlates with the expression level of hbd, and is much higher when growing on glucose than when growing on butyrate. Strain numbers refer to Table 1
Strain #12 gave 3× more crotonic acid than strain #1. These strains differ in two important ways: (a) hbd is expressed using the high-copy pETDuet1-thi1-hbd plasmid and the strong T7 promoter and (b) crt and thus the crotonase enzyme is not present in strain 12. Thus, the production of crotonate does not depend on the crotonase enzyme or the crt gene. Also, crotonate was produced at much, much lower levels in strains expressing tesB in place of ptb-buk such as 911, 11, or 13, illustrating that crotonate production depends on expression of both hbd and ptb-buk, but not crt. Strains expressing phaB in place of or in addition to hbd also showed very low or indetectable crotonate production. This may be related to the difference in stereochemistry of the 3-hydroxybutyryl-CoA that results from PhaB versus Hbd. That is, dehydration of 3-hydroxybutyryl-CoA to crotonyl-CoA may require the S-enantiomer produced by Hbd. While the inclusion of phaB in addition to hbd should enable the production of both enantiomers, differences in pathway activity could account for the greatly reduced crotonate production in the presence of phaB. Thus, these conditions seemed necessary and sufficient for crotonate production of over 100 μM: (a) co-expression of the hbd and ptb-buk operons, (b) the absence of alternative pathway genes including tesB and phaB, (c) operation of the butyrate pathway in the forward direction as in growth on glucose. The expression of the crt gene was unrelated to crotonate production.
The results shown in Fig. 4 suggest that E. coli strains expressing both hbd and ptb-buk produce crotonate as a minor fermentation product. Crotonate production may involve a side reaction of the ptb-buk system whereby S-3HB-CoA, S-3-hydroxybutyryl phosphate, or S-3-hydroxybutyric acid is converted to crotonate; however, background activities present on the E. coli genome may also be involved. We also observed 3HB production in E. coli pal(DE3) strains expressing both hbd and ptb-buk, but the level of production was not reproducible and varied in duplicate experiments between background levels and those previously measured (Tseng et al. 2009) for E. coli BL21(DE3) strains expressing both tesB and ptb-buk (data not shown).
Discussion
Analysis of short-chain acyl-CoAs and adenosine phosphates in three sets of E. coli strains expressing clostridial genes for the biosynthesis of butyrate and butanol showed that hbd expression leads to observable 3HB-CoA pools only in Strain Set II, in which strong T7 promoters drive the expression of hbd (Tseng et al. 2009). The PLTetO1-derived promoter and low-copy pSC101-derived plasmid used to express hbd in Strain Set #1 did not result in detectable 3HB-CoA. ButCoA was not reliably detected in any of the strain sets, as indicated by the large relative uncertainties given in Table 2.
The ΔIPTGΔG′ for the conversion of AcCoA to 3HB-CoA was positive for all but one of the strains in Strain Set II, showing that induction of ketoacid reductase genes phaB or hbd tended to push the conversion of AcCoA to 3HB-CoA closer to equilibrium. The exception was strain #11 (Table 1), expressing the clostridial hbd gene for reduction of AcAcCoA to 3HB-CoA and the non-energy-conserving tesB gene for conversion of 3HB-CoA to free 3HB. In this strain ΔIPTGΔG′ was negative, indicating that induction may have pushed the AcCoA to 3HB-CoA reaction further away from equilibrium. One possible explanation is that induction boosted the activity of TesB more than Hbd, thus draining away 3HB-CoA and indirectly disequilibrating the AcCoA to 3HB-CoA reaction.
A thermodynamic framework centered on the calculation of ΔIPTGΔG′ revealed that the conversion of 3HB-CoA to ButCoA was not accelerated by induction of the crt or bcd enzymes, even under the oxygen-limiting conditions we studied. This finding is a confirmation of several other independent analyses that also pinpointed bcd as encoding the enzyme whose activity prevents E. coli from reaching its stoichiometric potential (Inui et al. 2008; Nielsen et al. 2009). One possibility is that very stoichiometry of the reaction catalyzed by the proteins coded by the bcd/etfAB genes has been misunderstood (Li et al. 2008), at least in some species of Clostridia. Pool size measurements directly assess the performance of the individual steps of a recombinant pathway step under in vivo conditions, and are thus complementary to other techniques for dissecting the activity of recombinant pathways, such as enzyme assays or feeding studies, which usually involve physiological perturbations. We expect that combining pool size measurements with calculation of ΔinducerΔG′ will be useful for the dissection and analysis of any metabolically engineered pathway whose performance is not yet satisfactory.
References
Alberty RA (2005) Thermodynamics of biochemical reactions: Wiley-Interscience
Alper H, Stephanopoulos G (2007) Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab Eng 9(3):258–267
Atsumi S, Cann AF, Connor MR, Shen CR, Smith KM, Brynildsen MP, Chou KJY, Hanai T, Liao JC (2008) Metabolic engineering of Escherichia coli for 1-butanol production. Metab Eng 10(6):305–311
Campbell JW, Morgan-Kiss RM, Cronan JE (2003) A new Escherichia coli metabolic competency: growth on fatty acids by a novel anaerobic beta-oxidation pathway. Mol Microbiol 47(3):793–805
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97(12):6640–6645
Edwards JS, Palsson BO (2000) The Escherichia coli MG1655 in silico metabolic genotype: its definition, characteristics, and capabilities. Proc Natl Acad Sci USA 97(10):5528–5533
Elowitz MB, Leibler S (2000) A synthetic oscillatory network of transcriptional regulators. Nature 403(6767):335–338
Ezeji TC, Qureshi N, Blaschek HP (2004) Butanol fermentation research: upstream and downstream manipulations. Chem Rec 4(5):305–314
Hempfling WP, Mainzer SE (1975) Effects of varying the carbon source limiting growth on yield and maintenance characteristics of Escherichia coli in continuous culture. J Bacteriol 123(3):1076–1087
Hornsten EG (1995) On culturing Escherichia coli on a mineral salts medium during anaerobic conditions. Bioprocess Eng 12(3):157–162
Inui M, Suda M, Kimura S, Yasuda K, Suzuki H, Toda H, Yamamoto S, Okino S, Suzuki N, Yukawa H (2008) Expression of Clostridium acetobutylicum butanol synthetic genes in Escherichia coli. Appl Microbiol Biotechnol 77(6):1305–1316
Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK (2008) Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J Bacteriol 190(3):843
Liu X, Zhu Y, Yang ST (2006) Construction and characterization of ack deleted mutant of Clostridium tyrobutyricum for enhanced butyric acid and hydrogen production. Biotechnol Prog 22(5):1265–1275
Lutz R, Bujard H (1997) Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res 25(6):1203–1210
Minkler PE, Kerner J, Kasumov T, Parland W, Hoppel CL (2006) Quantification of malonyl-coenzyme A in tissue specimens by high-performance liquid chromatography/mass spectrometry. Anal Biochem 352(1):24–32
Nielsen DR, Leonard E, Yoon SH, Tseng HC, Yuan C, Prather KL (2009) Engineering alternative butanol production platforms in heterologous bacteria. Metab Eng 11(4–5):262–273
Park JW, Jung WS, Park SR, Park BC, Yoon YJ (2007) Analysis of intracellular short organic acid-coenzyme A esters from actinomycetes using liquid chromatography-electrospray ionization-mass spectrometry. J Mass Spectrom 42(9):1136–1147
Sambrook J, Russell DW (2001) Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press
Shimazu M, Vetcher L, Galazzo JL, Licari P, Santi DV (2004) A sensitive and robust method for quantification of intracellular short-chain coenzyme A esters. Anal Biochem 328(1):51–59
Tseng H-C, Martin CH, Nielsen DR, Prather KLJ (2009) Metabolic engineering of Escherichia coli for the enhanced production of (R)- and (S)-3-hydroxybutyrate. Appl Environ Microbiol
Tyo KEJ, Fischer CR, Simeon F, Stephanopoulos G (2009) Analysis of polyhydroxybutyrate flux limitations by systematic genetic and metabolic perturbations. Metab Eng 12(3):187–195
Acknowledgments
We acknowledge financial support by the DuPont-MIT Alliance, the NSF, the MIT Energy Initiative, Shell Global Solutions (US) Inc., and the NIH Biotechnology Training Program award to CF.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary materials
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Fischer, C.R., Tseng, HC., Tai, M. et al. Assessment of heterologous butyrate and butanol pathway activity by measurement of intracellular pathway intermediates in recombinant Escherichia coli . Appl Microbiol Biotechnol 88, 265–275 (2010). https://doi.org/10.1007/s00253-010-2749-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-010-2749-2