
Abstract
In all vertebrate animals, CD8+ cytotoxic T lymphocytes (CTLs) are controlled by major histocompatibility complex class I (MHC-I) molecules. These are highly polymorphic peptide receptors selecting and presenting endogenously derived epitopes to circulating CTLs. The polymorphism of the MHC effectively individualizes the immune response of each member of the species. We have recently developed efficient methods to generate recombinant human MHC-I (also known as human leukocyte antigen class I, HLA-I) molecules, accompanying peptide-binding assays and predictors, and HLA tetramers for specific CTL staining and manipulation. This has enabled a complete mapping of all HLA-I specificities (“the Human MHC Project”). Here, we demonstrate that these approaches can be applied to other species. We systematically transferred domains of the frequently expressed swine MHC-I molecule, SLA-1*0401, onto a HLA-I molecule (HLA-A*11:01), thereby generating recombinant human/swine chimeric MHC-I molecules as well as the intact SLA-1*0401 molecule. Biochemical peptide-binding assays and positional scanning combinatorial peptide libraries were used to analyze the peptide-binding motifs of these molecules. A pan-specific predictor of peptide–MHC-I binding, NetMHCpan, which was originally developed to cover the binding specificities of all known HLA-I molecules, was successfully used to predict the specificities of the SLA-1*0401 molecule as well as the porcine/human chimeric MHC-I molecules. These data indicate that it is possible to extend the biochemical and bioinformatics tools of the Human MHC Project to other vertebrate species.
Similar content being viewed by others


Introduction
Major histocompatibility complex class I (MHC-I) molecules are found in all vertebrate animals where they play a crucial role in generating specific cellular immune responses against viruses and other intracellular pathogens. They are highly polymorphic proteins that bind 8–11 amino acid long peptides derived from the intracellular protein metabolism. The resulting heterotrimeric complexes—consisting of the MHC-I heavy chain, the monomorphic light chain, beta-2 microglobulin (β2m), and specifically bound peptides—are translocated to the cell surface where they displayed as target structures for peptide-specific, MHC-I-restricted CTLs. If a peptide of foreign origin is detected, the T cells may become activated and kill the infected target cell.
MHC-I is extremely polymorphic. In humans, more than 3,400 different human leukocyte antigen class I (HLA-I) molecules have been registered (as of January 2011), and this number is currently growing rapidly as more efficient HLA typing techniques are employed worldwide. The polymorphism of the MHC-I molecule is concentrated in and around the peptide-binding groove, where it determines the peptide-binding specificity. Due to this polymorphism, it is highly unlikely that any two individuals will share the same set of HLA-I molecules thereby presenting the same peptides and generating T cell responses of the same specificities—something, that otherwise would give microorganisms a strong evolutionary chance of escape. Rather, this polymorphism can be seen as diversifying peptide presentation thereby individualizing T cell responses and reducing the risk that escape variants of microorganisms might evolve.
In 1999, we proposed that all human MHC specificities should be mapped (“the Human MHC Project”) as a preamble for the application of MHC information and technologies in humans (Buus 1999). Since then, we have developed large-scale tools that are generally applicable towards this goal: production, analysis, prediction and validation of peptide–MHC interactions (Ferre et al. 2003; Harndahl et al. 2009; Hoof et al. 2009; Larsen et al. 2005; Lundegaard et al. 2008; Nielsen et al. 2003, 2007; Ostergaard et al. 2001; Pedersen et al. 1995; Stranzl et al. 2010; Stryhn et al. 1996), and a “one-pot, read-and-mix” HLA-I tetramer technology for specific T cell analysis (Leisner et al. 2008). Here, we demonstrate that many of these tools can be transferred to other vertebrate animals as exemplified by an important livestock animal, the pig. We have successfully generated a recombinant swine leukocyte antigen I (SLA-I) protein, SLA-1*0401, one of the most common SLA molecules of swine (Smith et al. 2005). Using this protein, we have developed the accompanying biochemical peptide-binding assays and demonstrated that the immunoinformatics tools originally developed to cover all HLA-I molecules, despite the evolutionary distance, can be applied to SLA-I molecules. We suggest that the “human MHC project” can be extended to cover other species of interest.
Materials and methods
Peptides and peptide libraries
All peptides were purchased from Schafer-N, Denmark (www.schafer-n.com). Briefly, they were synthesized by standard 9-fluorenylmethyloxycarbonyl (Fmoc) chemistry, purified by reversed-phase high-performance liquid chromatography (to at least >80% purity, frequently 95–99% purity), validated by mass spectrometry, and quantitated by weight.
Positional scanning combinatorial peptide libraries (PSCPL) peptides were synthesized using standard solid-phase Fmoc chemistry on 2-chlorotrityl chloride resins. Briefly, an equimolar mixture of 19 of the common Fmoc amino acids (excluding cysteine) was prepared for each synthesis and used for coupling in 8 positions, whereas a single type of Fmoc amino acid (including cysteine) was used in one position. This position was changed in each synthesis starting with the N-terminus and ending with the C-terminus. In one synthesis, the amino acid pool was used in all nine positions. The library therefore consisted of 20 × 9 + 1 = 181 individual peptide libraries:
-
Twenty PSCPL sublibraries describing position 1: AX8, CX8, DX8, …. YX8
-
Twenty PSCPL sublibraries describing position 2: XAX7, XCX7, XDX7, …. XYX7
-
etc
-
Twenty PSCPL sublibraries describing position 9: X8A, X8C, X8D, …. X8Y
-
A completely random peptide library: X9
X denotes the random incorporation of amino acids from the mixture, whereas the single letter amino acid abbreviation is used to denote identity of the fixed amino acid.
The peptides in each synthesis were cleaved from the resin in trifluoroacetic acid/1,2-ethanedithiol/triisopropylsilane/water 95:2:1:3 v/v/v/v, precipitated in cold diethylether, and extracted with water before desalting on C18 columns, freeze drying, and weighting.
Recombinant constructs encoding chimeric and SLA-1*0401 molecules
A synthetic gene encoding a transmembrane-truncated fragment encompassing residues 1 to 275 of human HLA-A*11:01 alpha chain followed by a FXa–BSP–HAT tag (FXa = factor Xa cleavage site comprised of the amino acid sequence IEGR, BSP = biotinylation signal peptide, HAT = histidine affinity tag for purification purposes; see Online Resource 1) had previously been generated and inserted into the pET28 expression plasmid (Novagen) (Ferre et al. 2003). Synthetic genes encoding the corresponding fragments of the SLA-1*0401 alpha chain (α1α2) and α3, respectively, (Sullivan et al. 1997) were purchased from GenScript. To exchange domains and generate chimeric human/swine MHC-I gene constructs, a type II restriction endonuclease-based cloning strategy (SeamLess® Strategene; Cat#214400, Revision#021003a), with modifications, was used. All primers were purchased HPLC-purified from Eurofins MWG Operon (Ebersberg, Germany), and all PCR amplifications were performed in a DNA Engine Dyad PCR instrument (MJ Research, MN, USA). All constructs were validated by DNA sequencing. The following MHC-I heavy chain constructs were made HHH, HHP, HPP, PHP, and PPP, where the first, second, and third letter indicates domains α1 (positions 1–90), α2 (positions 91–181), and α3 (positions 182–275), respectively, and H indicates that the domain is of HLA-A*11:01 origin, whereas P indicates that it is of SLA-1*0401 origin.
Constructs were transformed into DH5α cells, cloned, and sequenced (ABI Prism 3100Avant, Applied Biosystems) (Hansen et al. 2001). Validated constructs of interest were transformed into an Escherichia coli production cell line, BL21(DE3), containing the pACYC184 expression plasmid (Avidity, Denver, USA) containing an isopropyl-β-d-1-thiogalactopyranoside (IPTG)-inducible BirA gene to express biotin-ligase. This leads to almost complete in vivo biotinylation of the desired product (Leisner et al. 2008).
Expression of recombinant proteins
To maintain the pET28-derived plasmids, the media was supplemented with kanamycin (50 μg/ml) throughout the expression cultures. When appropriate, the media was further supplemented with chloroamphenicol (20 μg/ml) to maintain the BirA containing pACYC184 plasmid. E. coli BL21(DE3) cells transformed with appropriate plasmids were grown for 5 h at 30°C, and a 10-ml sample adjusted to OD(600) = 1 was then transferred to a 2-l fed-batch fermentor (LabFors®). To induce protein expression, IPTG (1 mM) was added at OD(600) ∼ 25 and the culture was continued for an additional 3 h at 42°C (for in vivo biotinylation of the product, the induction media was further supplemented with biotin (Sigma #B4501, 125 μg/ml)). Samples were analyzed by reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) before and after IPTG induction. At the end of the induction culture, protease inhibitor (PMSF, 80 μg/l) was added, and cells were lysed in a cell disrupter (Constant Cell Disruptor Systems set at 2,300 bar) and the released inclusion bodies were isolated by centrifugation (Sorval RC6, 20 min, 17,000×g). The inclusion bodies were washed twice in PBS, 0.5% NP-40 (Sigma), and 0.1% deoxycholic acid (Sigma) and extracted into urea–Tris buffer (8 M urea, 25 mM Tris, pH 8.0), and any contaminating DNA was precipitated with streptomycin sulfate (1% w/v).
MHC class I heavy chain purification
The dissolved MHC-I proteins were purified by Ni2+/IDA metal chelating affinity column chromatography followed by Q-Sepharose ion exchange column chromatography, hydrophobic interaction chromatography, and eventually by Superdex-200 size exclusion chromatography. Fractions containing MHC-I heavy chain molecules were identified by A280 absorbance and SDS-PAGE and pooled. Throughout purification and storage, the MHC-I heavy chain proteins were dissolved in 8 M urea to keep them denatured. Note that the MHC-I heavy chain proteins at no time were exposed to reducing conditions. This allowed purification of highly active pre-oxidized moieties as previously described (Ostergaard et al. 2001). Protein concentrations were determined by bicinchoninic acid assay. The degree of biotinylation (usually >95%) was determined by a gel-shift assay (Leisner et al. 2008). The pre-oxidized, denatured proteins were stored at −20°C in Tris-buffered 8 M urea.
Recombinant constructs encoding human and porcine beta-2 microglobulin
Recombinant human β2m was expressed and purified as described elsewhere (Ostergaard et al. 2001), (Ferre et al. 2003). Using a previously reported E. coli codon-optimized gene encoding human β2m as template (Pedersen et al. 1995), a gene encoding porcine β2m was generated by multiple rounds of site-directed mutagenesis (QuikChange® Stratagene, according to the manufacturer’s instructions) (Online Resource 2). Briefly, the genes encoding human or pig β2m were N-terminally fused to a histidine affinity encoding tag (HAT) followed by a restriction enzyme encoding tag (FXa), inserted into the pET28 vector and expressed in inclusion bodies in E. coli. The fusion proteins were extracted into 8 M urea, purified by immobilized metal affinity chromatography (IMAC), and refolded by dilution. The fusion tags were then removed by FXa restriction protease digestion. The liberated intact and native human or pig β2m were purified by IMAC and gel filtration chromatography, analyzed by SDS-PAGE analysis, concentrated, and stored at −20°C until use (Fig. 1).

SDS-PAGE analysis of peak fractions from size exclusion chromatography of porcine β2m (11.4 kDa) after removal of histidine affinity tag (HAT) by FXa protease digestion. Samples were mixed 1:1 in non-reducing SDS sample buffer and loaded onto a 15% polyacrylamide gel
Purification and refolding of recombinant porcine β2m proteins
Porcine β2m was purified in the same way as human β2m (Ostergaard et al. 2001; Ferre et al. 2003). Briefly, the urea-dissolved β2m protein was purified by Ni2+/IDA metal chelating affinity column chromatography, refolded by drop-wise dilution into an excess refolding buffer under stirring (25 mM Tris, 300 mM urea, pH 8.00), and then concentrated (VivaFlow, 10 kDa). The refolded product was purified by Ni2+/IDA metal chelating affinity column chromatography again (this time in aqueous buffer, i.e., without urea). Fractions containing HAT-pβ2m were identified by SDS-PAGE and pooled. Removal of the HAT tag was performed by cleavage with factor Xa restriction protease (FXa) followed by renewed purified by Ni2+/IDA metal chelating affinity and Superdex200 gel filtration column chromatography, concentrated by spin ultrafiltration (10 kDa), mixed 1:1 with glycerol, and stored at −20°C.
SDS-PAGE analysis
Protein samples were mixed 1:1 in SDS sample buffer (4% SDS, 17.4% glycerol, 0.003% bromophenol blue, 0.125 M Tris, 8 mM IAA (iodoacetamide)) with or without reducing agent (2-mercaptoethanol) as indicated, boiled for 3 min, spun at 20,000×g for 1 min, and loaded onto a 12% or 15% running gel with a 5% stacking gel. Gels were run at 180 V, 40 mA for 50 min.
Peptide–MHC class I interaction measured by radioassay and spun column chromatography
A HLA-A*11:01-binding peptide, KVFPYALINK (non-natural consensus sequence A3CON1 (Sette et al. 1994)), was radiolabeled with iodine (125I) using a chloramine-T procedure (Hunter and Greenwood 1962). Dose titrations of MHC-I heavy chains (HHH or HHP) were diluted into refolding buffer (Tris–maleate–PBS) and mixed with β2m (human or porcine) and radiolabeled peptide, and incubated at 18°C overnight. Then binding of radiolabeled peptide to MHC-I was determined in duplicate by Sephadex™ G50 spun column gel chromatography as previously described (Buus et al. 1995). MHC bound peptide eluted in the excluded volume, whereas free peptide was retained on the microcolumn. Both fractions were counted by gamma spectroscopy, and the fraction peptide bound was calculated as excluded radioactivity divided by total radioactivity.
To examine the affinity of the interaction, increasing concentrations of unlabeled competitor peptide were added. When conducted under limiting concentrations of MHC-I molecule, the concentration of competitor peptide needed to effect 50% inhibition of the interaction, the IC50, is an approximation of the affinity of the interaction between MHC-I and the competitor peptide.
Peptide–MHC class I interaction measured by an enzyme-linked immunosorbent assay
Peptide–MHC-I interaction was also measured in a modified version of a previously described enzyme-linked immunosorbent assay (ELISA) (Sylvester-Hvid et al. 2002). Briefly, denatured biotinylated recombinant MHC-I heavy chains were diluted into a renaturation buffer containing β2m and graded concentrations of the peptide to be tested and incubated at 18°C for 48 h allowing equilibrium to be reached. We have previously demonstrated that denatured MHC molecules can de novo fold efficiently, however, only in the presence of appropriate peptide. The concentration of peptide–MHC complexes generated was measured in a quantitative sandwich ELISA (using streptavidin as capture layer and the monoclonal anti-β2m antibody, BBM1, as detection layer) and plotted against the concentration of peptide offered (Sylvester-Hvid et al. 2002). A prefolded, biotinylated FLPSDYFPSV/HLA-A*02:01 (Kast et al. 1994) complex was used as standard. Because the effective concentration of MHC (3–5 nM) used in these assays is below the equilibrium dissociation constant (K D) of most high-affinity peptide–MHC interactions, the peptide concentration, ED50, leading to half-saturation of the MHC is a reasonable approximation of the affinity of the interaction.
Using peptide libraries to perform an unbiased analysis of MHC specificity
The experimental strategy of PSCPL has previously been described (Stryhn et al. 1996). The construction of the sublibraries and the ELISA-driven quantitative measurements of MHC interaction are as given above.
Briefly, the relative binding (RB) affinity of each PSCPL sublibrary was determined as RB (PSCPL) = ED50 (X9) / ED50 (PSCPL) (where ED50 is the concentration needed to half-saturate a low concentration of MHC-I molecules) and normalized so that the sum of the RB values of the 20 naturally occurring amino acids equals 20 (since peptides with a given amino acid in a given position are 20 times more frequent in the corresponding PSCPL sublibrary than in the completely random X9 library). A RB value above 2 was considered as the corresponding position and amino acid being favored, whereas a RB value below 0.5 was considered as being unfavorable (these thresholds represent the 95% confidence intervals). An anchor position (AP) value was calculated by the equation ∑(RB − 1)2. A primary anchor position is characterized by one or few amino acids being strongly preferred and many amino acids being unacceptable. We have arbitrarily defined anchor residues as having an AP value above 15 (Lamberth et al. 2008). The peptide–SLA-I*0401 binding activity of each sublibrary was determined using previously published biochemical binding assay (ELISA) (Sylvester-Hvid et al. 2002) (with the modifications described above).
Sequence logos
Sequences logos describing the predicted binding motif for each MHC molecule were calculated as described by Rapin et al. (2010). In short, the binding affinity for a set of 1,000,000 random natural 9mer peptides was predicted using the NetMHCpan method, and the 1% strongest binding peptides were selected for construction of a position-specific scoring matrix (PSSM). The PSSM was constructed as previously described (Nielsen et al. 2004) including pseudo-count correction for low counts. Next, sequence logos were generated from the amino acid frequencies identified in the PSSM construction. For each position, the frequency of all 20 amino acids is displayed as a stack of letters. The total height of the stack represents the sequence conservation (the information content), while the individual height of the symbols relates to the relative frequency of that particular symbol at that position. Letter shown upside-down are underrepresented compared to the background (for details see Rapin et al. (2010)).
MHC distance trees
MHC distance trees were derived from correlations between predicted binding affinities. For each allelic MHC-I molecule, the binding affinity was predicted for a set of 200,000 random natural peptides using the NetMHCpan method. Next, the distance between any two alleles was defined, as D = 1 − PCC, where PCC is the Pearson correlation between the subset of peptides within the superset of top 10% best binding peptides for each allele. In this measure, two molecules that share a similar binding specificity will have a distance close to 0 whereas two molecules with non-overlapping binding specificities would have a distance close to 2. Using bootstrap, 100 such distance trees were generated, and branch bootstrap values and the consensus tree were calculated.
Results
Generation of chimeric MHC class I molecules
We have previously generated highly active, recombinant human MHC-I (HLA-I) molecules and accompanying high-throughput assays and bioinformatics prediction resources. Here, we transfer the underlying approaches to an important domesticated livestock animal, the pig, and its MHC system, the SLAs. MHC-I molecules are composed of a unique and highly variable distal peptide-binding platform consisting of the alpha 1 (α1) and alpha 2 (α2) domains of the MHC-I heavy chain (HC) and a much more conserved proximal immunoglobulin-like membrane attaching stalk consisting of the alpha 3 (α 3) domain of the HC non-covalently associated with the soluble MHC-I light chain (β2m).
A priori, the establishment of recombinant SLA molecules is complicated by the lack of validated reagents. Any failure could therefore be caused either by real technical problems in generating SLA molecules, or merely by a lack of information about strong peptide binders to the SLA in question. To reduce this uncertainty, we decided to migrate from human to pig MHC-I in a step-wise manner and generate an intermediary chimeric MHC-I molecule composed of a well-known human peptide-binding platform attached to a SLA stalk, which might allow us to assess whether we could generate a functional SLA stalk consisting of SLA-1*0401 α3 HC and pig β2m. To this end, we used the α1α2 domains of the HLA-A*11:01 molecule, which we expected should be able to bind a known high-affinity HLA-A*11:01-binding peptide (KVFPYALINK). This peptide could be 125I radiolabeled and used in a very robust peptide-binding assay testing whether the human stalk could be replaced with the corresponding SLA stalk. Once that had been successfully established, the entire SLA-1*0401 molecule would be constructed and tested.
We have previously expressed and purified the extracellular segment spanning positions 1–275 of the human HLA-A*11:01 in a denatured and pre-oxidized version that rapidly refold and bind appropriate target peptides (Ostergaard et al. 2001; Ferre et al. 2003). Codon-optimized genes encoding the corresponding segments of SLA-1*0401 (α1α2) and SLA-1*0401 (α3) were constructed as described in the “Materials and methods” section and used to replace the HLA-A*11:01 gene segment in the above construct generating a new construct allowing for the expression of SLA-1*0401. For the generation of HLA-A*11:01/SLA-1*0401 chimeras, the genes encoding α1 (spanning positions 1–90), α2 (spanning positions 91–181), and/or α3 (spanning positions 182–275) domains of HLA-A*11:01 and SLA-1*0401 were exchanged using SeamLess and touch-down cloning strategies. Genes encoding the extracellular segments 1–275 of the above natural or chimeric MHC-I molecules were C-terminally fused to a biotinylation tag (as indicated for SLA-1*0401 in Online Resource 1), inserted into pET28, and expressed in inclusion bodies in E. coli (Fig. 2 shows SDS-PAGE of lysates of recombinant E. coli before and 3 h after IPTG induction). The fusion proteins were extracted into 8 M urea (without any reducing agents), purified by ion exchange, hydrophobic and gel filtration chromatography (all conducted in 8 M urea, without any reducing agents) (Fig. 3 shows SDS-PAGE of the purified SLA-1*0401 after gel filtration), concentrated, and stored in urea at −20°C.

SDS-PAGE analysis of cell lysates of SLA-1*0401 expression in E. coli before and after induction with IPTG. Samples were mixed 1:1 in a reducing SDS sample buffer and loaded onto a 12% polyacrylamide gel

SDS-PAGE analysis of size exclusion chromatography peak fractions from SLA-1*0401 purification. Fraction numbers are shown above each lane. Samples were mixed 1:1 in non-reducing SDS sample buffer and loaded onto a 12% polyacrylamide gel. Lanes were loaded with different volumes, indicated below each lane, to avoid overloading. An arrow indicates the band representing purified SLA-1*0401-BSP-HAT heavy chain (36,675 Da)
Testing a chimeric molecule consisting of a SLA-1*0401 stalk and a HLA-A*11:01 peptide-binding platform—comparing human versus porcine β2m
To test the proximal immunoglobulin-like membrane attaching SLA stalk, we generated recombinant porcine β2m and a chimeric human/porcine MHC-I heavy chain molecule where the α1α2 were derived from the human HLA-A*11:01, and the α3 was derived from the porcine SLA-1*0401. Since this construct contains the entire peptide-binding platform of HLA-A*11:01, we reasoned that the binding of the HLA-A*11:01 restricted peptide, KVFPYALINK, could be used as a functional readout of the refolding, activity, and assembly of the entire chimeric molecule including the porcine SLA stalk. For comparison, we tested the supportive capacity of human β2m and folding ability of the entirely human HLA-A*11:01. A total of four combinations could therefore be tested: porcine or human β2m in combination with either HHP or HHH (where the first letter indicates the origin of the α1 domain (Human HLA-A*11:01 or Porcine SLA-1*0401), the second letter the origin of the α2 domain, and the third letter the origin of the α3 domain). A concentration–titration of heavy chain was added to a fixed excess concentration (3 μM) of β2m and a fixed trace concentration (23 nM) of radiolabeled peptide. As shown in Figs. 4 and 5, the four combinations gave almost the same heavy chain dose titration with a half-saturation occurring around 1–2 nM heavy chain. Porcine β2m supported folding of the chimeric (HHP) α chain slightly better than it supported folding of the human (HHH) α chain. Human β2m supported folding of HHP and HHH equally well. Thus, a recombinant SLA stalk can fold and support peptide binding of the peptide-binding platform. These results also suggest that human β2m can support folding and peptide binding of porcine MHC-I heavy chain molecules.

HLA-A*11:01 complex formation with a known HLA-A*11:01-binding peptide (KVFPYALINK) using either human (open squares) or porcine (filled circles) β2m. The affinity of HLA-A*11:01 to porcine and human β2m was determined as 2.11 nM (95% confidence interval, 1.96 to 2.27) and 1.07 nM (95% confidence interval, 0.871 to 1.32), respectively

MHC-I complex formation of the chimeric class I molecule HHP (HLA-A*11:01 (α1α2), SLA-1*0401 (α3)) with a known HLA-A*11:01-binding peptide (KVFPYALINK), and human (open squares) versus porcine β2m (filled circles). The affinity of HHP to porcine and human β2m was determined as 0.774 nM (95% confidence interval, 0.630 to 0.951) and 1.19 nM (95% confidence interval, 0.958 to 1.47), respectively
Using a positional scanning combinatorial peptide library approach to perform an unbiased analysis of the specificity of SLA-1*0401 and human–pig chimeric MHC class I molecules
Using human β2m to support folding, the recombinant SLA-1*0401 and human–pig chimeric MHC-I molecule were tested for peptide binding. We have previously described how PSCPL can be used to perform an unbiased analysis of MHC-I molecules (Stryhn et al. 1996). A PSCPL consists of 20 sublibraries for each position where one of each of the 20 natural amino acids have been locked and all other positions contain random amino acids. Analyzing how much of each PSCPL sublibrary is needed to support MHC-I folding (see examples in Fig. 6) and comparing each sublibrary with a completely random library, the effect of any amino acid in any position can be examined and expressed as a RB value. Further, an AP value calculated as the sum of squared deviations of RB values for each position can be used to identify the most prominent anchor position (see “Materials and methods” for calculations). Thus, the specificity of a nonamer binding MHC-I molecule can be analyzed comprehensively with 9 × 20 + 1 completely random library = 181 sublibraries (Stryhn et al. 1996).

PSCPL position 9 sublibrary analysis of the SLA-1*0401 peptide-binding motif. The amino acids valine, serine, and threonine are disfavored in position 9, evident by a decrease in affinity compared to the reference peptide (X9), whereas the large and bulky amino acids tryptophan and tyrosine are favored as seen by an increase in affinity compared to the reference peptide
Here, this approach was used to perform a complete experimental analysis of SLA-1*0401 and a limited analysis of the chimeric HPP and PHP molecules. A nonamer PSCPL analysis of SLA-1*0401 can be seen in Table 1. AP values identified positions 9, 3, and 2 (in that order of importance) as the anchor positions of SLA-1*0401. In position 9, the amino acid preferences were dominated by the large and bulky aromatic tyrosine (Y), tryptophane (W), and phenylalanine (F), all having RB values above 4 (Table 1). In the almost equally important position 3, preferences for negatively charged amino acids glutamic acid (E) and aspartic acid (D) were observed. In the lesser important position 2, the most preferred amino acids were the hydrophobic amino acids valine (V), isoleucine (I), and leucine (L), followed by the polar amino acids threonine (T) and serine (S).
Finally, a very limited PSCPL analysis was performed for the two chimeric human HLA-A*11:01/porcine SLA-1*0401 MHC-I molecules, HPP and PHP (Table 2). For both chimeric molecules, it could be demonstrated that position 9 is a strong anchor position. The positively charged amino acids, arginine (R) and lysine (K), were preferred in position 9 of the chimeric HPP molecule, whereas the aromatic amino acid, tyrosine (Y), was exclusively preferred in position 9 of the chimeric PHP molecule.
The positively charged amino acids, arginine (R) and lysine (K), were preferred in position 9 of the chimeric HPP molecule similar to the position 9 specificity of the HLA-A*11:01 molecule. In contrast, the aromatic amino acid tyrosine (Y) was preferred in position 9 of the chimeric PHP molecule similar to the position 9 specificity of the SLA-1*0401 molecule.
Using NetMHCpan to predict peptides that bind to SLA-1*0401 or to human–pig chimeric MHC class I molecules
Our recently described neural network-driven bioinformatics predictor, NetMHCpan (version 2.0), has been trained on about 88,000 peptide-binding data points representing more than 80 different MHC-I molecules (primarily HLA-A and HLA-B molecules). We have previously shown that NetMHCpan is an efficient tool to identify peptides that bind to HLA molecules where no prior data exist (Nielsen et al. 2007) and demonstrated that NetMHCpan can be extended to MHC-I molecules of other speciesFootnote 1 (Hoof et al. 2009). We applied NetMHCpan to our peptide repository of about 10,000 peptides, which over the past decade have been selected to scan infectious agents (e.g., SARS and influenza, Sylvester-Hvid et al. 2004; Wang et al. 2010), improve coverage of MHC-I specificities (e.g., Buus et al. 2003; Christensen et al. 2003), etc. We extracted 29 peptides as predicted binders to either the SLA-1*0401, the HPP, or the PHP human/porcine chimeric class I molecules (some of the peptides were predicted to bind to two or even all three of these molecules). All these peptide–MHC-I combinations were tested for binding (Table 3); 13 of 14 peptides bound to the SLA-1*0401 molecule with an affinity (IC50 value) better than 500 nM (6 with an affinity less than 50 nM); all 13 peptides tested on the PHP molecule were strong binders with IC50 values below 50 nM; and 3 of 12 peptides tested on the HPP molecule bound with an affinity better than 500 nM. Of the 39 peptide–MHC-I combinations tested, 20 (51%) were found to be good binders, 9 (23%) were average binders, and 10 (26%) did not bind well (Table 3). This is in stark contrast to the 0.5% frequency of binders among randomly selected peptides (Yewdell and Bennink 1999).
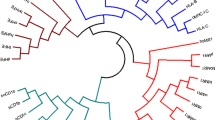
Next, the NetMHCpan method was used to generate PSSMs and sequence logos from the corresponding amino acid frequencies as described by Nielsen et al. (2004). For each position, the frequencies of all 20 amino acids were displayed as a stack of letters showing the sequence conservation/information content (the height of the entire stack) and the relative frequency of amino acids (the height of the individual amino acids). Figure 7 shows a specificity tree clustering of the SLA-1*0401 molecule compared to prevalent representatives of the 12 common HLA supertypes that NetMHCpan originally intended to cover (Lund et al. 2004). By this token, SLA-1*0401 most closely resembles that of HLA-A*01:01.

Specificity tree clustering of the SLA-1*0401 molecule compared to prevalent representatives of the 12 common HLA supertypes (Lund et al. 2004). The distance between any two MHC molecules and the consensus tree is calculated as described in “Materials and methods”. All branch points in the tree have bootstrap values of 100%. Sequence logos of the predicted binding specificity are shown for each molecule. In the logo, acidic amino acids [DE] are shown in red, basic amino acids [HKR] in blue, hydrophobic amino acids [ACFILMPVW] in black, and neutral amino acids [GNQSTY] in green. The axis of the LOGOs indicates in all case positions one through nine of the motif, and the y-axis the information content (see Materials and methods)
The limited PSCPL analysis of the chimeric MHC-I molecules revealed strong P9 signals with specificities that seemed to be determined by the origin of the α1 domain: the HPP chimera showed an HLA-A*11:01-like P9 specificity, whereas the PHP chimera showed a SLA-1*0401/HLA-A*01:01-like specificity. Since the NetMHCpan predictor successfully captured these chimeric specificities (see above), we reasoned that the predictor might also be used to perform in silico dissection of these specificities and used the P9 specificity as an example of such an in silico analysis. The NetMHCpan predictor considers a pseudo-sequence consisting of 34 polymorphic positions, which contain residues that are within 4.0 Å of the atoms of bound nonamer peptides (Nielsen et al. 2007). Of the 34 positions of the pseudo-sequence, 10 delineates the P9 binding pocket; however, only 3 of these, positions 74, 77, and 97, differ between SLA-1*0401 and HLA-A*11:01. To explore the effect of these three residues, we performed in silico experiments where we examined single substitutions Y74D, G77D, and S97I (the letter before the position number indicates the SLA-A*0401 single letter residue, whereas the letter after indicates the HLA-A*11:01 residue) as well as the corresponding triple substitution (YGS-DDI). As described above, PSSMs were generated for each of the in silico molecules followed by a specificity tree clustering (including SLA-A*0401, HLA-A*01:01, and HLA-A*11:01). Figure 8 shows this tree along with the sequence logo plots showing the predicted binding specificity of each in silico MHC-I molecule. Albeit the Y74D and G77D single substitutions showing some of the positively charged P9 peptide residue preference of HLA-A*11:01, they still clustered with HLA-A*01:01. In contrast, the in silico (YGS-DDI) triple substitution clustered with the HLA-A*11:01. This suggests that the NetMHCpan method is capable of defining the residues of the F pocket that determine the specificity of position 9.

Comparison of specific in silico mutations of the SLA-1*0401 molecule and comparison with the two HLA molecules: HLA-A*11:01 and HLA-A*01:01. The distance between any two MHC molecules and the consensus tree is calculated as described in “Materials and methods”. All branch points in the tree have bootstrap values of 100%. The SLA-1*0401 mutations are indicated as Y74D, G77D, and S97I, where the letter before the position number indicates the SLA-1*0401 single letter residue and the letter after indicates the HLA-A*11:01 residue. YGS-DDI is the corresponding triple substitution. Sequence logos are calculated and visualized as described in Fig. 7. The axis of the LOGOs indicates in all case positions one through nine of the motif, and the y-axis the information content (see Materials and methods)
Discussion
We have previously suggested that the specificities of the entire human MHC-I system should be solved (“the human MHC”, Buus 1999; Lauemoller et al. 2000). However, due to the extreme polymorphism of the MHCs, any attempt to address the specificity of the entire MHC system is a significant experimental undertaking. During the past decade, we have established a series of technologies to support a general solution of human MHC class I and II specificities. For MHC-I, this includes (1) a highly efficient E. coli expression system for production of recombinant human and mouse MHC-I molecules (both heavy chain and light chain (β2m) molecules) (Pedersen et al. 1995; Ostergaard et al. 2001), (2) a purification system for obtaining the highly active pre-oxidized MHC-I heavy chain species (Ferre et al. 2003), (3) a high-throughput homogenous peptide–MHC-I binding assay for obtaining large data sets on peptide–MHC-I interactions (Harndahl et al. 2009), (4) a positional scanning combinatorial peptide library approach for a robust and unbiased analysis of the specificity of any MHC-I molecule (Stryhn et al. 1996), (5) an immunobioinformatics approach to generate predictors of the peptide–MHC-I interaction, NetMHCpan, that allows predictions to be made for any human MHC-I molecules, HLA-I, even those that have not yet been covered by existing data set (Hoof et al. 2009; Nielsen et al. 2007), and finally (6) we have demonstrated that pre-oxidized MHC-I molecules can be used to generate MHC-I tetramers in a simple “one-pot, mix-and-read” manner (Leisner et al. 2008). Here, we propose that the next goal should be to extend the overall approach to MHC-I molecules of other species of interest. Mouse and rats have been extensively studied in the past, but much less reagents and information have accrued for the MHC-I molecules of other species. Here, we have used an important livestock animal, the pig, as a model system and demonstrated that it indeed is possible to transfer the original human approach to other species.
Before attempting to generate a recombinant version of the entire porcine SLA-1*0401 molecule, we grafted the more conserved membrane-proximal “stalk” (the immunoglobulin-like class I heavy chain α3 and β2m domains) of porcine SLA-1*0401 onto the peptide-binding domain of HLA-A*11:01 generating a chimeric human/porcine MHC-I molecule. This chimeric molecule retained the peptide-binding specificity of the HLA-A*11:01 molecule, and it clearly demonstrated that the recombinant porcine stalk was functional and, by inference, also properly folded. It also suggests that the peptide-binding specificity of the distal domains do not crucially depend upon the identity of the proximal stalk. Further, comparing the ability of human and porcine β2m to support MHC-I complex formation using either a human or a porcine MHC-I stalk, we demonstrated that every combination (porcine β2m/human-α3, porcine β2m/porcine-α3, human β2m/human-α3, and human β2m/porcine-α3) showed almost the same heavy chain dose titration with identical half-saturations. These results illustrate the ability for porcine and human β2m to support complex formation of SLA molecules and vice versa and suggest evolutionary that the stalk is quite conserved.
Next, we generated the entire SLA-1*0401 heavy chain and succeeded in generating complexes using human β2m as the light chain and PCSPL as peptide donors. The latter solved the a priori problem of not knowing which peptides would be needed to support proper folding of SLA-1*0401, and it did so in an unbiased manner. Furthermore, this approach is highly efficient since it readily establishes a complete matrix representing the amino acid preference for each amino acid and each position of a nonamer peptide. The specificity of SLA-1*0401 shows two primary anchors: one in positions 9 with a preference for aromatic amino acids and another in position 3 with a preference for negatively charged amino acids. In addition, the SLA-1*0401 features a secondary anchor in position 2 with hydrophobic or polar amino acid preferences.
An alternative approach to solve the problem of identifying peptides that support folding of MHC-I molecules of so far unknown specificity is to use our recently developed pan-specific predictor, NetMHCpan. The successful use of this predictor to initiate peptide-binding studies was recently demonstrated for HLA-A*3001 (Lamberth et al. 2008). Although originally developed to cover all HLA-A and HLA-B molecules, it has also been shown to extend to MHC-I molecules of other species (Hoof et al. 2009). Here, we demonstrate that the NetMHCpan predictor is capable of extracting MHC-I sequence information across species and correctly relate this to peptide binding even in the absence of any available data for the specific query MHC-I molecule, i.e., the SLA-1*0401 as well as the chimeric HPP (hα1pα2pα3) and PHP (pα1hα2pα3) molecules. It is not clear why binding of the PHP chimera was more efficiently predicted than binding of the HPP chimera. One could speculate that NetMHCpan has not captured the effect of the different positions of the pseudo-sequence equally well and not all positions and pockets (and by inference—not all chimeric molecules) are therefore predicted equally well.
Using the NetMHCpan predictor to cluster SLA-1*0401 and representative molecules of 12 human HLA supertypes according to predicted peptide-binding specificities, the SLA-1*0401 specificity closely resembled that of HLA-A*01:01 (IEDB, http://www.immuneepitope.org/MHCalleleId/142, accessed March 9th 2011). This result was also obvious from an inspection of the PSCPL analysis of the SLA-1*0401. The PSCPL analysis of the P9 specificity of the SLA-1*0401 and the two chimeric molecules suggested that the P9 specificity primarily was determined by the α1 domain. This contention was further strengthened by a NetMHCpan-driven in silico analysis of the residues delineating the F pocket, which interacts with P9. This suggests that NetMHCpan can be used to design and interpret detailed experiments addressing the structure–function relationship of peptide–MHC-I interaction. In the case of SLA-1*0401, NetMHCpan suggests that Y74, G77, and S97 play a prominent role in defining the P9 F pocket. Whereas the NetMHCpan readily captured the P9 anchor residue of SLA-1*0401, it did not capture the P3 anchor (at least not in the 2.4 version). We surmise that this shortcoming is due to insufficient examples of the use of P3 anchors within the currently available peptide–MHC-I binding data. Inspecting the pseudo-sequence of SLA-1*0401 and HLA-A*01:01 vs. HLA-A*11:01 suggests that the presence of an arginine in position 156 might explain the preference for negatively charged amino acid residues in P3. Future NetMHCpan-guided experiments could pointedly address this question, and the resulting data could complement existing data and be used to update and improve the NetMHCpan predictor.
All in all the two complementary approaches, PSCPL and NetMHCpan, agreed on the specificity of the SLA-1*0401 molecule, as well as of the two chimeric MHC-I molecules. Thus, the specificity of SLA-1*0401 appear to be well established. This specificity has successfully been used to search for foot-and-mouth disease virus (FMDV)-specific CTL epitopes in FMDV-vaccinated, SLA-1*0401-positive pigs, and the recombinant SLA-1*0401 molecules have been used to generate corresponding tetramers and stain pig CTLs (Patch et al. 2011). In conclusion, we here present a set of methods that can be used to generate functional recombinant MHC-I molecules, map their specificities and identify MHC-I-restricted epitopes, and eventually generate peptide–MHC-I tetramers for validation of CTL responses. This suite of methods is not only applicable to humans, but potentially to any species of interest.
Notes
A preliminary report of SLA-1*0401 binding was given in Hoof et al. (2009).
References
Buus S (1999) Description and prediction of peptide-MHC binding: the 'human MHC project'. Curr Opin Immunol 11:209–213
Buus S, Stryhn A, Winther K, Kirkby N, Pedersen LO (1995) Receptor–ligand interactions measured by an improved spun column chromatography technique. A high efficiency and high throughput size separation method. Biochim Biophys Acta 1243:453–460
Buus S, Lauemoller SL, Worning P, Kesmir C, Frimurer T, Corbet S, Fomsgaard A, Hilden J, Holm A, Brunak S (2003) Sensitive quantitative predictions of peptide-MHC binding by a 'Query by Committee' artificial neural network approach. Tissue Antigens 62:378–384
Christensen JK, Lamberth K, Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Buus S, Brunak S, Lund O (2003) Selecting informative data for developing peptide–MHC binding predictors using a query by committee approach. Neural Comput 15:2931–2942
Ferre H, Ruffet E, Blicher T, Sylvester-Hvid C, Nielsen LL, Hobley TJ, Thomas OR, Buus S (2003) Purification of correctly oxidized MHC class I heavy-chain molecules under denaturing conditions: a novel strategy exploiting disulfide assisted protein folding. Protein Sci 12:551–559
Hansen NJ, Pedersen LO, Stryhn A, Buus S (2001) High-throughput polymerase chain reaction cleanup in microtiter format. Anal Biochem 296:149–151
Harndahl M, Justesen S, Lamberth K, Roder G, Nielsen M, Buus S (2009) Peptide binding to HLA class I molecules: homogenous, high-throughput screening, and affinity assays. J Biomol Screen 14:173–180
Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 61:1–13
Hunter W, Greenwood F (1962) Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature 194:495–496
Kast WM, Brandt RM, Sidney J, Drijfhout JW, Kubo RT, Grey HM, Melief CJ, Sette A (1994) Role of HLA-A motifs in identification of potential CTL epitopes in human papillomavirus type 16 E6 and E7 proteins. J Immunol 152:3904–3912
Lamberth K, Roder G, Harndahl M, Nielsen M, Lundegaard C, Schafer-Nielsen C, Lund O, Buus S (2008) The peptide-binding specificity of HLA-A*3001 demonstrates membership of the HLA-A3 supertype. Immunogenetics 60:633–643
Larsen MV, Lundegaard C, Lamberth K, Buus S, Brunak S, Lund O, Nielsen M (2005) An integrative approach to CTL epitope prediction: a combined algorithm integrating MHC class I binding, TAP transport efficiency, and proteasomal cleavage predictions. Eur J Immunol 35:2295–2303
Lauemoller SL, Kesmir C, Corbet SL, Fomsgaard A, Holm A, Claesson MH, Brunak S, Buus S (2000) Identifying cytotoxic T cell epitopes from genomic and proteomic information: "the human MHC project". Rev Immunogenet 2:477–491
Leisner C, Loeth N, Lamberth K, Justesen S, Sylvester-Hvid C, Schmidt EG, Claesson M, Buus S, Stryhn A (2008) One-pot, mix-and-read peptide–MHC tetramers. PLoS One 3:e1678
Lund O, Nielsen M, Kesmir C, Petersen AG, Lundegaard C, Worning P, Sylvester-Hvid C, Lamberth K, Roder G, Justesen S, Buus S, Brunak S (2004) Definition of supertypes for HLA molecules using clustering of specificity matrices. Immunogenetics 55:797–810
Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M (2008) NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res 36:W509–W512
Nielsen M, Lundegaard C, Worning P, Lauemoller SL, Lamberth K, Buus S, Brunak S, Lund O (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci 12:1007–1017
Nielsen M, Lundegaard C, Worning P, Hvid CS, Lamberth K, Buus S, Brunak S, Lund O (2004) Improved prediction of MHC class I and class II epitopes using a novel Gibbs sampling approach. Bioinformatics 20:1388–1397
Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Roder G, Peters B, Sette A, Lund O, Buus S (2007) NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2:e796
Ostergaard PL, Nissen MH, Hansen NJ, Nielsen LL, Lauenmoller SL, Blicher T, Nansen A, Sylvester-Hvid C, Thromsen AR, Buus S (2001) Efficient assembly of recombinant major histocompatibility complex class I molecules with preformed disulfide bonds. Eur J Immunol 31:2986–2996
Patch JR, Pedersen LE, Toka FN, Moraes M, Grubman MJ, Nielsen M, Jungersen G, Buus S, Golde WT (2011) Induction of foot-and-mouth disease virus-specific cytotoxic T cell killing by vaccination. Clin Vaccine Immunol 18:280–288
Pedersen LO, Stryhn A, Holter TL, Etzerodt M, Gerwien J, Nissen MH, Thogersen HC, Buus S (1995) The interaction of beta 2-microglobulin (beta 2m) with mouse class I major histocompatibility antigens and its ability to support peptide binding. A comparison of human and mouse beta 2m. Eur J Immunol 25:1609–1616
Rapin N, Hoof I, Lund O, Nielsen M (2010) The MHC motif viewer: a visualization tool for MHC binding motifs. Curr. Protoc. Immunol. Chapter 18:Unit 18.17
Sette A, Sidney J, del Guercio MF, Southwood S, Ruppert J, Dahlberg C, Grey HM, Kubo RT (1994) Peptide binding to the most frequent HLA-A class I alleles measured by quantitative molecular binding assays. Mol Immunol 31:813–822
Smith DM, Lunney JK, Martens GW, Ando A, Lee JH, Ho CS, Schook L, Renard C, Chardon P (2005) Nomenclature for factors of the SLA class-I system, 2004. Tissue Antigens 65:136–149
Stranzl T, Larsen MV, Lundegaard C, Nielsen M (2010) NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics 62:357–368
Stryhn A, Pedersen LO, Romme T, Holm CB, Holm A, Buus S (1996) Peptide binding specificity of major histocompatibility complex class I resolved into an array of apparently independent subspecificities: quantitation by peptide libraries and improved prediction of binding. Eur J Immunol 26:1911–1918
Sullivan JA, Oettinger HF, Sachs DH, Edge AS (1997) Analysis of polymorphism in porcine MHC class I genes: alterations in signals recognized by human cytotoxic lymphocytes. J Immunol 159:2318–2326
Sylvester-Hvid C, Kristensen N, Blicher T, Ferre H, Lauemoller SL, Wolf XA, Lamberth K, Nissen MH, Pedersen LO, Buus S (2002) Establishment of a quantitative ELISA capable of determining peptide–MHC class I interaction. Tissue Antigens 59:251–258
Sylvester-Hvid C, Nielsen M, Lamberth K, Roder G, Justesen S, Lundegaard C, Worning P, Thomadsen H, Lund O, Brunak S, Buus S (2004) SARS CTL vaccine candidates; HLA supertype-, genome-wide scanning and biochemical validation. Tissue Antigens 63:395–400
Wang M, Larsen MV, Nielsen M, Harndahl M, Justesen S, Dziegiel MH, Buus S, Tang ST, Lund O, Claesson MH (2010) HLA class I binding 9mer peptides from influenza A virus induce CD4 T cell responses. PLoS One 5:e10533
Yewdell JW, Bennink JR (1999) Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annu Rev Immunol 17:51–88
Acknowledgments
We thank Lise Lotte Bruun Nielsen, Anne Caroline Schmiegelow, and Iben Sara Pedersen for their expert experimental support. This work was in part supported by the Danish Council for Independent Research, Technology and Production Sciences (274-09-0281) and by the National Institute of Health (NIH) (HHSN266200400025C).
Author information
Authors and Affiliations
Corresponding author
Electronic Supplemental Materials
Below is the link to the electronic supplementary material.
Online Resource 1
Nucleotide sequence used to encode SLA-1*0401 fused to an enzyme restricted cleavage site (Fxa, purple), a biotinylation site (BSP, pink), and a histidine affinity tag (HAT, dark blue) (PDF 62 kb)
Online Resource 2
Primers designed to induce the changes in the DNA coding for human β2m to get the DNA encoding porcine β2m (PDF 72 kb)
Online Resource 3
Comparison of pseudo-sequences for the heavy chain molecules HLA-A*01:01, SLA-1*0401, HLA-A*11:01, HPP (hα1pα2pα3), and PHP (pα1hα2pα3). Orange and purple residue numbers have an impact on binding position 9 and position 3 of the peptide, respectively. Blue residue numbers have an impact on binding position 2 of the peptide, whereas green residue numbers have an impact on binding position 1 of the peptide. Black residues show similarities between the different molecules, whereas red residues illustrate differences when compared to HLA-A*11:01. White residue numbers on a black background indicate a difference in the pseudo-sequence residues between SLA-1*0401 and either HLA-A*01:01 or HLA-A*11:01 or both (PDF 106 kb)
Rights and permissions
About this article
Cite this article
Pedersen, L.E., Harndahl, M., Rasmussen, M. et al. Porcine major histocompatibility complex (MHC) class I molecules and analysis of their peptide-binding specificities. Immunogenetics 63, 821–834 (2011). https://doi.org/10.1007/s00251-011-0555-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00251-011-0555-3