
Abstract
The microbiota associated with vermiculations from karst caves is largely unknown. Vermiculations are enigmatic deposits forming worm-like patterns on cave walls all over the world. They represent a precious focus for geomicrobiological studies aimed at exploring both the microbial life of these ecosystems and the vermiculation genesis. This study comprises the first approach on the microbial communities thriving in Pertosa-Auletta Cave (southern Italy) vermiculations by next-generation sequencing. The most abundant phylum in vermiculations was Proteobacteria, followed by Acidobacteria > Actinobacteria > Nitrospirae > Firmicutes > Planctomycetes > Chloroflexi > Gemmatimonadetes > Bacteroidetes > Latescibacteria. Numerous less-represented taxonomic groups (< 1%), as well as unclassified ones, were also detected. From an ecological point of view, all the groups co-participate in the biogeochemical cycles in these underground environments, mediating oxidation-reduction reactions, promoting host rock dissolution and secondary mineral precipitation, and enriching the matrix in organic matter. Confocal laser scanning microscopy and field emission scanning electron microscopy brought evidence of a strong interaction between the biotic community and the abiotic matrix, supporting the role of microbial communities in the formation process of vermiculations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The hypogean environments are the least known and studied on Earth [1]. Despite the prohibitive abiotic factors (e.g., oligotrophy, total darkness, and high mineral concentrations) for life development, they represent interesting ecological niches, hosting extremophile microorganisms, highly specialized and perfectly adapted to this peculiar ecosystem, showing an unexpected biodiversity within the Bacteria domain and countless novel species [2]. To overcome the limiting factors, microorganisms create mutualistic networks, cooperating in communities and favoring each other’s survival. The autotrophic microorganisms generally draw energy by chemosynthesis, using chemical elements (such as Ca, Mg, Fe, Mn, and S) and organic and inorganic compounds abundant in the host rocks, cave sediments, groundwater, and atmosphere. Concurrently, several microbial groups rely on mixed metabolic pathways (mixotrophy) [3]. In any case, such microbial communities may contribute to the formation of caves, influencing several biogeochemical processes [1, 4,5,6,7]. In particular, they act inducing the precipitation [8, 9] or dissolution of minerals of speleothems and other structures occurring in underground environments, like moonmilk and vermiculation deposits [10, 11]. The genesis of all these examples is, indeed, difficult to be explained only by pure abiotic physicochemical processes [2].
Among cave structures, vermiculations are enigmatic deposits recurring on rock surfaces in caves all over the world [12,13,14], characterized by variable morphologies, colors, and dimensions [15, 16], and generally composed of calcite, associated with quartz, and traces of clay minerals [17]. Recent studies highlighted microbial evidences supporting their biological origin [10, 17,18,19]. Vermiculations can be indeed considered “life hotspots” and a precious support for the studies on cave geomicrobiology. To our knowledge, there are still few studies on their microbial characterization and most of these concern vermiculations from sulfuric acid speleogenetic systems [10, 20, 21].
Aimed at shedding light on the microbial community of vermiculations from the Pertosa-Auletta Cave (Campania, southern Italy) and on its role in their formation, this work represents one of the first microbiological studies of vermicular deposits from a normal epigenic karst system. To this end, molecular biology approaches have been employed. In addition, giving an important contribution to the knowledge of the hidden biological aspects of vermiculations, it represents a key step toward the protection and conservation of these peculiar biosignatures and of the whole cave ecosystem.
Methods
Vermiculation Samplings
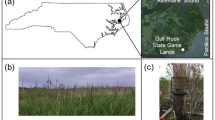
Eleven different points were sampled in the four main branches of the Pertosa-Auletta Cave (Fig. 1), a limestone show cave in southern Italy. Approximately, 2 g of vermiculation deposits was collected. The four branches of the studied karst cave are characterized by various degrees of frequentation, namely (i) Active (A), (ii) Fossil (F), (iii) Paradise (P), and (iv) Tourist (T), where Active indicates the branch still influenced by an active water flow, Fossil identifies inactive conditions of water flow, Paradise is a short piece of the active branch, lit and frequented by humans, and Tourist is the illuminated trail opened to the public for regular visits.

Pertosa-Auletta Cave (Campania, southern Italy) karst system; yellow points indicate the collected vermiculations, with the corresponding texture model, in the Active (A, blue), Fossil (F, yellow), Paradise (P, violet), and Tourist (T, green) trails
An accurate description of the study area, as well as on the geochemistry of the vermiculations, is reported in Addesso et al. [17]. In particular, the 11 samples of vermicular deposits, described in detail for their morphology, color, chemical and mineralogical composition [17], showed several shapes as described by Parenzan [15, 22] classification and can be divided into hieroglyphic (A1, A2, A3, F2, T2), dendritic (F1, P1, T1), bubble-like spots (F3), large-leopard spots (F4), and tiger skin (T3). Colors ranged from whitish (A2, F1, T2) to grey (P1, T2) or brown (A1, A3, F1, F3, F4, T3), greenish in P1, probably due to the presence of photoautotrophs [17].
The sampling was performed using disposable and sterile scalpel blades and Eppendorf tubes, carefully avoiding damage to the walls. Stored at 4 °C, the samples were immediately sent to the Instituto de Recursos Naturales y Agrobiologia of Sevilla (IRNAS-CSIC, Spain) and maintained at − 80 °C, until processing.
Molecular Analyses
Total DNA was extracted using FastDNATM Spin Kit for Soil, according to the producer’s protocol (MP Biomedical). The DNA quality was determined by a Nanodrop ND-1000 Spectrophotometer, whereas the amount by a Qubit 2.0 Fluorometer (Invitrogen).
Prokaryotic 16S and eukaryotic 18S rRNA genes were amplified by polymerase chain reaction (PCR), using specific primers: 616F [23] and 1510R [24] for Bacteria, 109F and 915R [25] for Archaea, EukA and EukB [26] for Eukarya, ITS1 and ITS4 [27] for Fungi. PCR reactions were carried out using 0.2-mL PCR tubes with a minimal amount of extracted DNA (from 0.5 to 2.0 μL), pure and diluted to 2 and 5 ng/μL, and 50 μL of Mastermix solution [1 mL = 775 μL H2O(σ), 200 μL of PCR Buffer (BIOLINE) and 5 μL Taq Polymerase (BIOLINE), 10 μL specific primers (Reverse and Forward), 4 μL BSA 10%], employing a FlexCycler (Analytik Jena) and a T100 Thermal Cycler (Bio-Rad). The PCR thermal programs are given in Table S1. The amplified PCR products underwent 1% agarose gel electrophoresis (0.5 M TAE Buffer) for a qualitative analysis. Fingerprints of Archaea and Bacteria communities were obtained by denaturing gradient gel electrophoresis (DGGE) of samples, according to Muyzer et al. [28], using a DCODE™ System (Bio-Rad).
The extracted DNA (with a minimum concentration of ~ 5 ng/μL), after purification by Genomed and Genomic DNA Clean & Concentrator™-10 (Zymo Research), was analyzed by via next-generation sequencing (NGS) targeting the V3–V4 hypervariable region of Prokaryotes 16S rRNA, using Illumina MiSeq 2 × 250 paired end, according to Macrogen (Seoul, Korea) library preparation protocol. Chimeras were identified and removed by means of USEARCH [29]. Resulting reads were processed in Qiime [30], whereas UCLUST [29] was used for the similar sequences assignment to operational taxonomic units (OTUs) by clustering with a 97% similarity threshold. Paired-end reads were merged using FLASH [31]. RDP Release 11 was used as against reference database for taxonomic identification of query sequences. Alpha diversity analysis, including estimation of Chao1, Shannon, Simpson, and Good’s Coverage indices, and rarefaction curves, based on the observed species metric, were performed through Qiime.
The graphs relative to molecular analysis data were elaborated in the R 3.6.0 programming environment [32]. The barplots, showing the relative abundances at phylum, class, and order levels for each sample, with associated dendrograms explaining the similarities among the samples, were created using “ggplots2”, “dendextend”, and “RColorBrewer” packages. Pearson’s correlation coefficients (r values) were obtained using cor function to evaluate associations (for α = 0.05) between geochemical characteristics and microbial phyla as well as among biological properties of the analyzed vermiculations. Non-metric multidimensional scaling (NMDS) analysis, with superimposition of confidence ellipses for branches (α = 0.05), and principal component analysis (PCA) were performed using meta.mds function, based on Euclidean distance metric, and prcomp function, respectively, both from “vegan” package.
Microscopy
The nucleic acids of the whole cells were visualized using the specific SYBR Green fluorescent dye (1:100 dilution), on samples not handled further, under an Olympus FluoView FV1000 confocal laser scanning microscope, and the 488-nm excitation laser line with emission signal being collected at 510–530 nm. Images were analyzed with the FluoView 2.1 software (Olympus). FESEM images were acquired using FEI Teneo (Thermo Fisher, MA, USA). To this end, samples were prepared as reported in Addesso et al. [17]. In particular, they were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4) at 4 °C for 2 h and washed thrice in cacodylate buffer. Subsequently, they were treated with 1% osmium tetroxide for 1 h at 4 °C and dehydrated by subsequent dilution series in ethanol and acetone finishing with 100% acetone before drying. The samples were dried in a EM CPD 300 (Leica Microsystem, Wetzlar, Germany) critical point drying device at 34.5 °C. Finally, samples were mounted on SEM stubs and sputter-coated with gold (5–10 nm).
Results
All the 11 studied vermiculations, developing on limestone substratum (except A1 and A3, in the Active branch, which were growing at the interface between limestone host rock and bat guano crusts), showed a considerable biological diversity.
Taxonomic Composition of Microbial Community
The preliminary qualitative analysis on the DNA extracted from vermiculations gave positive results for Prokaryotes and negative results for Eukaryotes. Online Resource 1 displays the archaeal (a) and bacterial (b) 16S rRNA gene-DGGE profiles of the sampled vermiculations. NGS analysis of 16S rRNA gene identified archaeal and bacterial taxa. Archaea were scarcely represented (Table 1). At the phylum level, Thaumarchaeota was characterized in all the vermiculations, with a relative abundance varying between 0.01 and 0.07%: Woesearchaeota was present in all the samples (0.01–0.04%), except for A2, P1, and T3, whereas Euryarchaeota was detected in F3 (0.01%) and P1 (0.03%). Moreover, unclassified Archaea were found in A3, F1, F2, F3, P1, T1, and T2 in percentages ranging from 0.01 and 0.09% (Table 1).
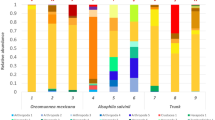
Bacteria composed almost the entire extracted DNA (Fig. 2). The major phylum in the total bacterial community was Proteobacteria (41.3–54.8%), followed by Acidobacteria (7.1–16.8%) > Actinobacteria (1.9–33.8%) > Nitrospirae (2.8–13.3%) > Firmicutes (1.5–6.6%) > Planctomycetes (2.0–4.2%) > Chloroflexi (0.9–2.7%) > Gemmatimonadetes (0.6–1.7%) > Bacteroidetes (0.04–1.7%) > Latescibacteria (0.2–1.3%). NGS analysis highlighted the presence of a very copious group of unclassified phyla with percentage ranging from 6.2 and 19.3%. Other 16 phyla were less represented (< 1%). The microbial abundances were very similar in all the vermiculations, except P1, dominated by Actinobacteria (33.8%) in addition to Proteobacteria (41.3%) (Fig. 2a). The most abundant classes within the Proteobacteria phylum were as follows: Gamma- (19.3–35.8%) > Beta- (6.3–17.4%) > Alpha- (4.6–7.2%) > Delta- (3.3–5.9%) (Fig. 2b). At the order level (Fig. 2c), Gammaproteobacteria was mainly represented by an ample unclassified group (17.7–33.1%) and by Xanthomonadales (< 2.3%), whereas Alphaproteobacteria included the Rhizobiales (1.7–5.1%) and Rhodospirillales (1.1–3.6%) orders. Nitrospirales > Actinomycetales > Thermoanaerobacterales > Planctomycetales > Gemmatimonadales > Gaiellales > Anaerolineales were also identified with an abundance below 5.9%. Numerous unclassified groups were present at the order level, increasing considerably in the subsequent taxonomic levels.

Bacterial composition of vermiculations from Pertosa-Auletta Cave; the barplots show the relative abundances (%) at phylum (a), class (b), and order (c) levels of samples from the Active (A, blue), Fossil (F, yellow), Paradise (P, violet), and Tourist (T, green) branches, with corresponding dendrograms (a, b, c) and PCA analysis (d, e, f)
The dendrograms (Fig. 2), showing similarities and divergences between specimens based on taxon relative abundances, highlighted three groups, keeping enough in the graph representations of all three taxonomic levels. The clustering analysis showed a clear separation of P1, the only sample located in Paradise branch, from the other two groups, closer to each other (Fig. 2a–c). At the phylum level (Fig. 2a), A2, F1, F3, and T2 clustered together from the rest (A1, A3, F2, F4, T1, T3). At the class level (Fig. 2b), F2 grouped with A2, F1, F3, and T2, splitting up from A1, A3, F4, T1, and T3. Lastly, at the order level (Fig. 2c), F1, F3, F4, and T2 assembled a new cluster divided from the remaining samples. Figure 2 also shows the corresponding PCAs based on the total bacterial communities at the phylum (Fig. 2d), class (Fig. 2e), and order (Fig. 2f) levels. Analogous clusters of the dendrograms were also observed in PCA plots. The first (PC1) and the second (PC2) principal components accounted together for 86.04%, 83.81% and 84.73% of the data variance, respectively for phylum, class, and order taxonomic levels.
Microbial Community Richness and Diversity
The rarefaction curve plots, built based on the number of observed microbial groups vs. the number of sequences per sample, for both the four branches and the 11 individual samples, are reported in Online Resource 2 (a and b, respectively). Most curves tended to approach the saturation plateau, reinforcing the sufficiency of sequencing analysis, adequately representative of the investigated communities.
Alpha diversity estimation, using several metrics, is reported in Table 2. The total OTUs generated for each sample ranged from a maximum of 2127 to a minimum of 1323, whereas the average value of Good’s Coverage was 99.78%, indicating that the analysis well covers the microbial diversity in vermiculation samples. Chao1 richness estimator resulted between 1444.8 and 2313.3. Shannon and Simpson diversity indices presented similar estimates among the samples (around 7 and close to 1, respectively), except for P1, which presented the lowest values (5.78 and 0.87, respectively).
Relationships Between Microbial Community and Geochemical Characteristics
Pearson’s correlation coefficients between microbial phyla and geochemical and mineralogical characteristics [17] of each vermiculation are shown in Table 3a. Deferribacteres, Latescibacteria, and Nitrospirae displayed positive correlations (0.74 < r < 0.77; p < 0.01), with organic C, P, and Mo, respectively. Unclassified Archaea, Armatimonadetes, and Ignavibacteriae were negatively correlated (− 0.61 < r < − 0.66; p < 0.05) with S. Chloroflexi were positively related with Ca, Mg, Sr, Ti, V, and Zn (0.60 < r < 0.65; p < 0.05), but negatively with C (r = − 0.66; p < 0.05). Spirochaetes were negatively correlated with Ca, Fe, Mg, Ti, Li, V, Cr, Zn, Cu, and quartz (with r values ranging from − 0.61 to − 0.67, and p < 0.05), and positively correlated with C and calcite (with correlation coefficients equal to 0.66 and 0.61, respectively and p values < 0.05). Furthermore, N showed a positive correlation with Deferribacteres phylum (r = 0.70; p < 0.05) and a negative relationship with Elusimicrobia phylum (r = − 0.67; p < 0.05), whereas Verrucomicrobia revealed a negative correlation with Co, K, Mn, and N (− 0.63 < r < − 0.65; p < 0.05). Among the Archaea phyla, Woesearchaeota was positively correlated with organic C, showing a correlation coefficient of 0.64 (p value < 0.05).
The correlation analysis results between microbial groups identified in the 11 studied vermiculations are reported in Table 3b. Positive correlations (p < 0.001) among several groups were observed: unclassified Archaea phylum with Woesearchaeota (r = 0.92), Euryarchaeota with Actinobacteria (r = 0.85), Lentisphaerae with Ignavibacteriae (r = 0.86), and Cyanobacteria/Chloroplast with Parcubacteria (r = 0.96). Spirochaetes is the only one displayed highly negative correlation with Firmicutes (r = − 0.90; p < 0.001). Moreover, Chloroflexi were positively correlated with Gemmatimonadetes (r = 0.79; p < 0.01), Firmicutes (r = 0.83; p < 0.01), and unclassified Bacteria phylum (r = 0.65; p < 0.05), but negatively with Spirochaetes (r = − 0.80; p < 0.01) and Cyanobacteria/Chloroplast (r = − 0.65; p < 0.05), whereas Woesearchaeota showed a positive correlation with Verrucomicrobia (r = 0.83; p < 0.01) and Planctomycetes (r = 0.70; p < 0.05). Proteobacteria displayed a positive correlation with Candidatus Saccharibacteria (r = 0.77; p < 0.01) and Cyanobacteria/Chloroplast (r = 0.62; p < 0.05); candidate division WPS-1 was positively related with Armatimonadetes (r = 0.79; p < 0.01), Cyanobacteria/Chloroplast (r = 0.74; p < 0.01), Parcubacteria (r = 0.81; p < 0.01), and Elusimicrobia (r = 0.68; p < 0.05), but negatively correlated with Firmicutes (r = − 0.62; p < 0.05). Unclassified Bacteria phylum showed a negative correlation with Actinobacteria, Spirochaetes, and Euryarchaeota (− 0.60 < r < − 0.70; p < 0.05), but it was positively correlated with Firmicutes (r = 0.69; p < 0.05). Latescibacteria showed a positive correlation with Gemmatimonadetes (r = 0.61; p < 0.05) and Firmicutes (r = 0.68; p < 0.05), whereas Armatimonadetes with Elusimicrobia, Hydrogenedentes and candidate division WPS-2, with correlation coefficients ranging from 0.62 to 0.71 and p value < 0.05. Omnitrophica were positively correlated (p < 0.05) with unclassified Archaea phylum (r = 0.67) and Thaumarchaeota (r = 0.62). Finally, Verrucomicrobia highlighted a positive correlation with unclassified Archaea phylum (r = 0.73; p < 0.05), Gemmatimonadetes with Firmicutes (r = 0.62; p < 0.05), and Elusimicrobia with Parcubacteria (r = 0.68; p < 0.05).
The NMDS biplot (Fig. 3), based on the microbiological and geochemical-mineralogical [17] characteristics of the analyzed vermicular deposits, showed a clear separation of the confidence ellipses grouping the Tourist and Fossil branches. The vermicular deposits from the active trail revealed intermediate characteristics, as highlighted by the partial overlapping of its confidence ellipse with the other two. Between the two most abundant minerals (calcite and quartz), calcite characterized the vermiculations from the four trails, whereas quartz mainly those of the Tourist and Active trails. Among the 24 elements (total Al, Ba, C, Ca, Co, Cr, Cu, Fe, K, Li, Mg, Mn, Mo, N, Na, Ni, P, S, Si, Sr, Ti, V, and Zn and organic C) analyzed, N, S, and organic C, mostly abundant in the vermiculations from the lightened trails (Paradise and Tourist), together with C, and to a lesser extent Mo, P, and Sr, showed a strong relationship with bacterial communities.

NMDS analysis, with confidence ellipses (α = 0.05) for the four branches [Active (A, blue), Fossil (F, yellow), Paradise (P, violet), and Tourist (T, green)], based on the total microbial community (red labels) and the geochemical characteristics (black labels) of the same vermiculations, as reported in Addesso et al. [17]
Confocal microscopic observations performed on samples A4 (Fig. 4a, b), F1 (Fig. 4c), and T1 (Fig. 4d) provided interesting information about the distribution and density of microbial communities (green-colored zones) on the mineral surface. As revealed by FESEM images (Online Resource 3), microbial structures were found mainly associated with clay minerals.

CLSM images of A4 (a, b), F1 (c), and T1 (d) vermiculation samples, showing the presence of microbial clusters (green-colored zones) dyed with SYBR Green staining
Discussion
Although vermiculations represent a perfect substratum suitable for microbes, probably participating also to their formation as mediators of geochemical processes [10, 20], very little is known about the microbiota of such enigmatic deposits. In this context, our study provides, for the first time, an overview on the microbial life associated with vermiculations from non-sulfidic karst systems.
The NGS approach revealed a biodiversity comparable to those observed in several matrices from different caves [10, 19, 20, 33,34,35]. Proteobacteria (41.3–54.8%), represented (in decreasing order) by Gamma-, Beta-, Alpha-, and Deltaproteobacteria classes, was the dominant phylum, likely in relation to the wide ranges in metabolism and phenotype, offering the capability to degrade a broad spectrum of organic substrates and to adapt to and thrive in the hostile cave environment [2]. The presence of Proteobacteria is often associated with Fe-Mn deposits [36, 37], both chemical elements were observed in vermiculations from Pertosa-Auletta Cave [17], and mainly related to the geochemical characteristics of the substratum, as highlighted by the NMDS. The carotenoid-producing gammaproteobacterial Xanthomonadales order was also detected, typical of yellow-colored colonies found in caves [38, 39]. Among Alphaproteobacteria, the Rhizobiales order, represented by members able to fix nitrogen and to oxidize iron and manganese, and the Rhodospirillales order, equally participating to the nitrogen cycle, were observed. They are typical surficial microorganisms [2], but, as suggested by Lavoie et al. [40], their presence in caves can be related to the migration of microorganisms from above lying soils, and once in the cave they start an adaptation process to the new surrounding environmental conditions. Similar to vermiculations from the Pertosa-Auletta Cave, those in the sulfuric acid Fetida Cave (Apulia, Italy) showed a great abundance of Proteobacteria (44-46%), but with copious microbial communities belonging to Deltaproteobacteria (25%) and Epsilonproteobacteria (16%), respectively, dominated by Desulfobacterales and Campylobacterales, involved in the sulfur cycle [19, 20].
Acidobacteria represented the second most abundant phylum, whose genetic and metabolic diversity is comparable to the highly diverse Proteobacteria [41,42,43]. Acidobacteria often occur together with chemolithoautotrophic Gammaproteobacteria, suggesting a mutualistic association between them: Acidobacteria gain energy oxidizing the reduced organic compounds (chemoorganotrophy) obtained from Proteobacteria autotrophic metabolism, an ecological advantage in cave oligotrophic environments [44]. Only in the green P1 vermiculation, in the lightened Paradise branch, the most represented phylum after Proteobacteria was Actinobacteria (33.8%), with Actinomycetales order, clearly different from the other vermiculations (1.9–10.3%), confirmed also by PCAs. The abundance of Actinobacteria in this vermiculation is justified by their association with Cyanobacteria, a well-known relationship in lightened subterranean environments [45].
Commonly found in soil systems, Actinobacteria may have an important ecological role in biogeochemical cycles of cave ecosystems, mediating mineralization processes [34] and producing bioactive compounds, such as antimicrobials that allow the biotic control on other populations [46]. Cuezva et al. [7] demonstrated they are able to capture CO2 from the atmosphere and precipitate CaCO3 polymorphs, as shown in FESEM images of the same samples reported in Addesso et al. [17]. In particular, Actinomycetales are able to degrade recalcitrant organic compounds [47]. The relative humidity and availability of endo- and exogenous organic matter in the Paradise branch can explain their colonization success. In fact, here, the moisture reaches approximately 100%, due to the presence of an underground river nearby, promoting the proliferation of Actinomycetes [48]. Moreover, the Paradise trail is lit and frequented by tourists who, together with photoautotrophic communities growing close to artificial light systems, bring an important input in terms of organic compounds, facilitating heterotrophic populations, including Actinomycetales [49].
The aerobic chemolithoautotrophic nitrite-oxidizing Nitrospirae and the anaerobic ammonium-oxidizing Planctomycetes, together with Firmicutes, able to reduce/oxidize sulfur, as well as chemo- or phototrophic Chloroflexi, were also found elsewhere in small amounts. Moreover, numerous less-represented taxonomic groups (with relative abundance < 1%) were observed in the 11 vermiculations investigated and their ecological role in this kind of ecosystem is still debated [2]. Among them, Archaea were also present, with the Thaumarchaeota, Euryarchaeota, and Woesearchaeota phyla, despite the archaeal DGGE profile highlighted a major number of bands in terms of core species richness. The same were found in considerable amount (< 4.3%) in Fetida Cave [20], where the relative abundances change (Proteobacteria > Planctomycetes > Acidobacteria > Chloroflexi > Bacteroidetes > Actinobacteria > Nitrospirae), likely due to the more extreme acidophilic environment, promoting the development of some bacterial groups rather than others. Despite the scarcity of knowledge about the archaeal group in cave ecosystems, it is well known that they give a relevant contribution to the global carbon nitrogen and sulfur cycles [22, 50, 51]. This may explain both the strong association between Euryarchaeota and N, and the relation of unclassified Archaea phylum groups, Thaumarchaeota, and Woesearchaeota with C and organic C highlighted by NMDS.
The Simpson index displays values close to 1 for all the samples, considering the dominant groups in the community and excluding the rare ones, indicating a low biodiversity and a high dominance. From the NGS results, it emerges that the dominant groups are unclassified already at the phylum level and this increases with the taxonomic level specificity. Values close to 7 were, instead, obtained for Shannon index, sensible also to the rare species, abundantly present in all the samples and certainly important from an ecological point of view.
Overall, geochemical and microbiological characteristics of the studied vermiculations differed among branches of the Pertosa-Auletta Cave, with the greatest differences observed between those from tourist and unvisited branches. Anyway, macroelements (C, N, S, and P), as well as the organic matter, were mostly abundant in the vermiculations from the Paradise and Tourist branches, highlighting the presence of more abundant biomass in lightened trails, where the photoautotrophs proliferate. In these samples, also Mo and Sr were more abundant, indicating that a specialized microbial community could have resulted from some microbial lineages able to oxidize minerals containing such elements [52]. However, F4 sample showed a high abundance of Nitrospirae phylum compared to the other vermicular deposits that displayed also a high correlation with molybdenum, probably due to its content in the membrane-associated enzyme of the nitrite-oxidizing system [53]. Furthermore, the higher content of organic C in vermiculations from Fossil and Tourist trails [17] may explain the major abundance of Nitrospirae in such locations, where the availability of ammonia by ammonificators can increase the presence of nitrites, in turn usable by nitrite-oxidizing Nitrospirae group bacteria [33]. From Pearson correlation analysis, several associations emerge between biological and geochemical properties, as well as among the taxonomic groups, especially the rarest, but they are not at all easy to explain, due to the lack of information about their biogeochemical role in the cave ecosystem [33].
Confocal microscopy images showed a localization of DNA only in specific sites, recognizable in the green zones. This was confirmed also by FESEM images, as reported in Addesso et al. [17], showing the clayey deposits always associated with biogenic filamentous material, not ruling out the possibility that the microbes can interact or influence their behavior and evolution in the environment [54, 55].
The findings of the present study support the theory formulated by Jones et al. [10], suggesting that microorganisms play an active role in vermiculation genesis, producing organic matter and secondary minerals, enriching the calcite matrix, trapping and binding sediment particles and dissolving, through etching or pitting, the rock. This may happen in different environments, from sulfuric acid to normal karst caves. However, beyond the biological evidences, the possibility of coexistence of several processes remains. For example, decalcification of rock walls, due to the dissolution processes caused by the acidity of seeping or condensation waters, can contribute to create the primordial calcite matrix [56,57,58,59]; thereafter, neutralization of electrical charges in the small particles, associated to wet-dry phenomena, can determine the different morphologies [16, 60]. Nevertheless, further studies are required to clarify to what extent some processes prevail over others, determining the variety of vermiculations described.
The present study, describing the microbiota present in the vermicular deposits of the Pertosa-Auletta Cave and its relationships with geochemistry of vermiculations, fills the gap characterizing these topics in karst caves. The analyses carried out indicate a certain diversity of biological communities living in vermicular deposits, with a considerable percentage of unclassified lineages, already at the phylum level, demonstrating once more that the underground ecosystem hosts still a high number of unknown taxa. Proteobacteria and Acidobacteria were the predominant phyla, as generally observed in such environments, whereas Actinobacteria showed an increased growth due to the high humidity conditions and the input of organic matter from the considerable presence of tourists in the show cave. The involvement of such communities in the biogeochemical cycles is indisputable and the highlighted biological evidences confirm a tight interaction between biotic and abiotic factors in the formation of vermiculations. The obtained findings represent a crucial step for the protection and conservation of such unique ecological niches, making still more intriguing the knowledge and comprehension pathway of vermiculations.
References
Lee NM, Meisinger DB, Aubrecht R, Kovacik L, Saiz-Jimenez C, Baskar S, Baskar R, Liebl W, Porter ML, Engel AS (2012) Caves and karst environments. In: Bell EM (ed) Life at extremes: environments, organisms and strategies for survival. CABI, Wallingford, pp 320–344
Tomczyk-Żak K, Zielenkiewicz U (2016) Microbial diversity in caves. Geomicrobiol J 33:20–38. https://doi.org/10.1080/01490451.2014.1003341
Kováč Ľ (2018) Caves as oligotrophic ecosystems. In: Moldovan OT, Kováč Ľ, Halse S (eds) Cave Ecology. Springer International Publishing, Cham, pp 297–307
Northup DE, Lavoie KH (2001) Geomicrobiology of caves: a review. Geomicrobiol J 18:199–222. https://doi.org/10.1080/01490450152467750
Galdenzi S, Maruoka T (2003) Gypsum deposits in the Frasassi caves, central Italy. J Cave Karst Stud 65(2):111–125
Engel AS, Stern LA, Bennett PC (2004) Microbial contributions to cave formation: new insights into sulfuric acid speleogenesis. Geol 32:369. https://doi.org/10.1130/G20288.1
Cuezva S, Fernandez-Cortes A, Porca E, Pašić L, Jurado V, Hernandez-Marine M, Serrano-Ortiz P, Hermosin B, Cañaveras JC, Sanchez-Moral S, Saiz-Jimenez C (2012) The biogeochemical role of Actinobacteria in Altamira Cave, Spain. FEMS Microbiol Ecol 81:281–290. https://doi.org/10.1111/j.1574-6941.2012.01391.x
Tisato N, Torriani SFF, Monteux S, Sauro F, de Waele J, Tavagna ML, D’Angeli IM, Chailloux D, Renda M, Eglinton TI, Bontognali TRR (2015) Microbial mediation of complex subterranean mineral structures. Sci Rep 5:15525. https://doi.org/10.1038/srep15525
Bontognali TRR, D’Angeli IM, Tisato N, et al (2016) Mushroom speleothems: stromatolites that formed in the absence of phototrophs. Front Earth Sci 4. https://doi.org/10.3389/feart.2016.00049
Jones DS, Lyon EH, Macalady JL (2008) Geomicrobiology of biovermiculations from the Frasassi cave system, Italy. J Cave Karst Stud 70(2):78–93
Miller AZ, Garcia-Sanchez AM, Martin-Sanchez PM, Costa Pereira MF, Spangenberg JE, Jurado V, Dionísio A, Afonso MJ, Iglé sias Chaminé HI, Hermosin B, Saiz-Jimenez C (2018) Origin of abundant moonmilk deposits in a subsurface granitic environment. Sedimentology 65:1482–1503. https://doi.org/10.1111/sed.12431
Jameson RA (1991) Management considerations for clay vermiculations. Proc of the Nat Cave Manag Symp Am Cave Conserv Assoc, Bowling Green, pp 39–144
Hedges J (1993) A review on vermiculations. Bol Soc Venez Espeleol 27:2–6
Hill CA, Forti P (1997) Cave minerals of the world. National Speleological Society, Huntsville
Parenzan P (1961) Sulle formazioni argillo-limose dette vermicolari. Atti Symp Int di Spel Varenna Mem 5:120–125
Bini A, Gori MC, Gori S (1978) A critical review of hypotheses on the origin of vermiculations. IJS 10:11–33. https://doi.org/10.5038/1827-806X.10.1.2
Addesso R, Bellino A, D’Angeli IM et al (2019) Vermiculations from karst caves: the case of Pertosa-Auletta system (Italy). CATENA 182:104178. https://doi.org/10.1016/j.catena.2019.104178
Camassa MM, Febbroriello P (2003) Le foval della Grotta Zinzulusa in Puglia (SE-Italia). Thalass Salentina 26(suppl):207–218
D’Angeli IM, Waele JD, Ieva MG, et al (2017) Next-generation sequencing for microbial characterization of biovermiculations from a sulfuric acid cave in Apulia (Italy). Proc of the 17th Int Congr of Speleol 1:377–380
D’Angeli IM, Ghezzi D, Leuko S, Firrincieli A, Parise M, Fiorucci A, Vigna B, Addesso R, Baldantoni D, Carbone C, Miller AZ, Jurado V, Saiz-Jimenez C, de Waele J, Cappelletti M (2019) Geomicrobiology of a seawater-influenced active sulfuric acid cave. PLoS One 14:e0220706. https://doi.org/10.1371/journal.pone.0220706
D’Angeli IM, Parise M, Vattano M et al (2019) Sulfuric acid caves of Italy: a review. Geomorphology 333:105–122. https://doi.org/10.1016/j.geomorph.2019.02.025
Parenzan P (1965) Le formazioni vermicolari della grotta di Sant’Angelo di Statte (Taranto). Atti IX Congr. Naz. di Speleol., Trieste. Memoria 7(2):101–104
Snaidr J, Amann R, Huber I, Ludwig W, Schleifer KH (1997) Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl Environ Microbiol 63:2884–2896
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. John Wiley & Sons, Inc., Chichester, pp 115–175
Großkopf R, Janssen PH, Liesack W (1998) Diversity and structure of the methanogenic community in anoxic rice paddy soil microcosms as examined by cultivation and direct 16S rRNA gene sequence retrieval. Appl Environ Microbiol 64:960–969. https://doi.org/10.1128/AEM.64.3.960-969.1998
Medlin L, Elwood HJ, Stickel S, Sogin ML (1988) The characterization of enzymatically amplified eukaryotic 16S-like rRNA-coding regions. Gene 71:491–499. https://doi.org/10.1016/0378-1119(88)90066-2
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: PCR Protocols. Elsevier, pp 315–322
Muyzer G, de Waal EC, Uitterlinden AG (1993) Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. https://doi.org/10.1093/bioinformatics/btq461
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963. https://doi.org/10.1093/bioinformatics/btr507
R Core Team (2019) R: A language and environment for statistical computing. R Foundation for Statistical Computing. R version 3.6.0
De Mandal S, Chatterjee R, Kumar NS (2017) Dominant bacterial phyla in caves and their predicted functional roles in C and N cycle. BMC Microbiol 17:90. https://doi.org/10.1186/s12866-017-1002-x
Gonzalez-Pimentel JL, Miller AZ, Jurado V, Laiz L, Pereira MFC, Saiz-Jimenez C (2018) Yellow coloured mats from lava tubes of La Palma (Canary Islands, Spain) are dominated by metabolically active Actinobacteria. Sci Rep 8:1944. https://doi.org/10.1038/s41598-018-20393-2
Hershey OS, Barton HA (2018) The microbial diversity of caves. In: Moldovan OT, Kováč Ľ, Halse S (eds) Cave Ecology. Springer International Publishing, Cham, pp 69–90
Northup DE, Barns SM, Yu LE, Spilde MN, Schelble RT, Dano KE, Crossey LJ, Connolly CA, Boston PJ, Natvig DO, Dahm CN (2003) Diverse microbial communities inhabiting ferromanganese deposits in Lechuguilla and Spider Caves. Environ Microbiol 5:1071–1086. https://doi.org/10.1046/j.1462-2920.2003.00500.x
Spilde MN, Northup DE, Boston PJ, Schelble RT, Dano KE, Crossey LJ, Dahm CN (2005) Geomicrobiology of cave ferromanganese deposits: a field and laboratory investigation. Geomicrobiol J 22:99–116. https://doi.org/10.1080/01490450590945889
Portillo MC, Gonzalez JM, Saiz-Jimenez C (2008) Metabolically active microbial communities of yellow and grey colonizations on the walls of Altamira Cave, Spain. J Appl Microbiol 104:681–691. https://doi.org/10.1111/j.1365-2672.2007.03594.x
Porca E, Jurado V, Žgur-Bertok D, Saiz-Jimenez C, Pašić L (2012) Comparative analysis of yellow microbial communities growing on the walls of geographically distinct caves indicates a common core of microorganisms involved in their formation. FEMS Microbiol Ecol 81:255–266. https://doi.org/10.1111/j.1574-6941.2012.01383.x
Lavoie KH, Winter AS, Read KJH, Hughes EM, Spilde MN, Northup DE (2017) Comparison of bacterial communities from lava cave microbial mats to overlying surface soils from Lava Beds National Monument, USA. PLoS One 12:e0169339. https://doi.org/10.1371/journal.pone.0169339
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Quaiser A, Ochsenreiter T, Lanz C, Schuster SC, Treusch AH, Eck J, Schleper C (2003) Acidobacteria form a coherent but highly diverse group within the bacterial domain: evidence from environmental genomics: Environmental genomics of Acidobacteria. Mol Microbiol 50:563–575. https://doi.org/10.1046/j.1365-2958.2003.03707.x
Barns SM, Cain EC, Sommerville L, Kuske CR (2007) Acidobacteria phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl Environ Microbiol 73:3113–3116. https://doi.org/10.1128/AEM.02012-06
Meisinger DB, Zimmermann J, Ludwig W, Schleifer KH, Wanner G, Schmid M, Bennett PC, Engel AS, Lee NM (2007) In situ detection of novel Acidobacteria in microbial mats from a chemolithoautotrophically based cave ecosystem (Lower Kane Cave, WY, USA). Environ Microbiol 9:1523–1534. https://doi.org/10.1111/j.1462-2920.2007.01271.x
Albertano P, Urzì C (1999) Structural interactions among epilithic cyanobacteria and heterotrophic microorganisms in Roman hypogea. Microb Ecol 38:244–252. https://doi.org/10.1007/s002489900174
Rangseekaew P, Pathom-aree W (2019) Cave Actinobacteria as producers of bioactive metabolites. Front Microbiol 10:387. https://doi.org/10.3389/fmicb.2019.00387
Sonia M-T, Hafedh B, Abdennaceur H, Ali G (2011) Studies on the ecology of actinomycetes in an agricultural soil amended with organic residues: II. Assessment of enzymatic activities of Actinomycetales isolates. World J Microbiol Biotechnol 27:2251–2259. https://doi.org/10.1007/s11274-011-0688-4
Zvyagintsev DG, Zenova GM, Doroshenko EA, Gryadunova AA, Gracheva TA, Sudnitsyn II (2007) Actinomycete growth in conditions of low moisture. Biol Bull Russ Acad Sci 34:242–247. https://doi.org/10.1134/S1062359007030053
Cañaveras JC, Sanchez-Moral S, Soler V, Saiz-Jimenez C (2001) Microorganisms and microbially induced fabrics in cave walls. Geomicrobiol J 18:223–240. https://doi.org/10.1080/01490450152467769
Cavicchioli R (2011) Archaea — timeline of the third domain. Nat Rev Microbiol 9:51–61. https://doi.org/10.1038/nrmicro2482
Offre P, Spang A, Schleper C (2013) Archaea in biogeochemical cycles. Annu Rev Microbiol 67:437–457. https://doi.org/10.1146/annurev-micro-092412-155614
Mann H (1990) Biosorption of heavy metals by bacterial biomass. In: Volesky B (ed) Biosorption of Heavy Metals. CRC Press, Boca Raton, pp 93–138
Spieck E, Ehrich S, Aamand J, Bock E (1998) Isolation and immunocytochemical location of the nitrite-oxidizing system in Nitrospira moscoviensis. Arch Microbiol 169:225–230. https://doi.org/10.1007/s002030050565
Dong H, Jaisi DP, Kim J, Zhang G (2009) Microbe-clay mineral interactions. Am Mineral 94:1505–1519. https://doi.org/10.2138/am.2009.3246
Cuadros J (2017) Clay minerals interaction with microorganisms: a review. Clay Miner 52:235–261. https://doi.org/10.1180/claymin.2017.052.2.05
De Joly R (1934) Ruissellement et percolations. Actes Congr D’Erfoud (Maroc), Com d’Etude des Eaux Souterr, pp 54-61
Waldner F (1936) Contributo alla morfologia del limo argilloso delle caverne. Osservazioni fatte nelle Grotte di Postumia. Grotte d’Italia 2:56–60
Renault P (1953a) Exploration du Grand Aven de Canjeurs. Bul du Com Na Spéléol 3:72–79
Renault P (1953b) Dépôts vermiculés d’argille de decalcification. 1° Congr Int de Spéléol, Paris
Montoriol-Pous J (1958) Sobre el origen de las vermiculationes arcillosas. Actes 2° Cong Int de Spéléol. Bari 1:389–395
Acknowledgments
Great thanks go to the president, Dr. Francescantonio D'Orilia, and the scientific director, Prof. Mariana Amato, of MIdA Foundation for having believed in the project and to the skilled speleo-guide, Mr. Vincenzo Manisera, for the precious support in the field (cave).
Funding
Open access funding provided by Università degli Studi di Salerno within the CRUI-CARE Agreement. This work was supported by Musei Integrati dell’Ambiente Foundation, by the Spanish project MINECO [CGL2016-75590-P] with European Regional Development Fund and by University of Salerno [ORSA197159].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Addesso, R., Gonzalez-Pimentel, J.L., D’Angeli, I.M. et al. Microbial Community Characterizing Vermiculations from Karst Caves and Its Role in Their Formation. Microb Ecol 81, 884–896 (2021). https://doi.org/10.1007/s00248-020-01623-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-020-01623-5