
Abstract
Anaerobic ammonium-oxidizing (anammox) process plays an important role in the nitrogen cycle of the worldwide anoxic and mesophilic habitats. Recently, the existence and activity of anammox bacteria have been detected in some thermophilic environments, but their existence in the geothermal subterranean oil reservoirs is still not reported. This study investigated the abundance, distribution and functional diversity of anammox bacteria in nine out of 17 high-temperature oil reservoirs by molecular ecology analysis. High concentration (5.31–39.2 mg l−1) of ammonium was detected in the production water from these oilfields with temperatures between 55°C and 75°C. Both 16S rRNA and hzo molecular biomarkers indicated the occurrence of anammox bacteria in nine out of 17 samples. Most of 16S rRNA gene phylotypes are closely related to the known anammox bacterial genera Candidatus Brocadia, Candidatus Kuenenia, Candidatus Scalindua, and Candidatus Jettenia, while hzo gene phylotypes are closely related to the genera Candidatus Anammoxoglobus, Candidatus Kuenenia, Candidatus Scalindua, and Candidatus Jettenia. The total bacterial and anammox bacterial densities were 6.4 ± 0.5 × 103 to 2.0 ± 0.18 × 106 cells ml−1 and 6.6 ± 0.51 × 102 to 4.9 ± 0.36 × 104 cell ml−1, respectively. The cluster I of 16S rRNA gene sequences showed distant identity (<92%) to the known Candidatus Scalindua species, inferring this cluster of anammox bacteria to be a new species, and a tentative name Candidatus “Scalindua sinooilfield” was proposed. The results extended the existence of anammox bacteria to the high-temperature oil reservoirs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
A petroleum reservoir is a typically extreme environment characterized by high temperature, high pressure, high salinity, and strictly anoxic condition. From the first description of the microorganisms in oil deposits [4], the microbiology of petroleum reservoirs subsequently developed along with petroleum exploration. A wide range of microorganisms have been cultured from oilfields with sulfate reduction, fermentation, acetogenesis, and methanogenesis as major metabolic types [40, 63]. The culture-independent researches indicated more diverse microbes than previously perceived in oilfields worldwide [1, 5, 7, 12, 15, 28, 33, 41, 42, 46]. Recently, much more attention has been paid to the microbial flora associated with the nitrogen cycle in the subterranean petroleum-rich strata because the source of nitrogen is a limiting factor to the microbial community activity under the oilfield condition. In the petroleum industry, substantial amounts of nitrate were injected into the oil reservoirs in an attempt to control the souring [21] through changing the microbial community structure in the subsurface strata. Several nitrogen-cycle-related microorganisms were activated by the injected nitrate, including the nitrate-reducing interdependent with sulfite oxidation bacteria and heterotrophic nitrate-reducing bacteria [20]. Along with these processes, nitrite and ammonium were produced through the nitrate-reducing pathway [16]. Because of the well-known stable chemical property under the anoxic condition, ammonium will be accumulated in the formation water. Thus, the high concentration of ammonia was detected frequently in the production water from the oilfield, which is a problem in production water treatment for recycled water flooding and potentially imposed an environmental hazard. However, the detected concentration of ammonium was lower than the estimated value of stoichiometric calculation. To explain the elimination of ammonium in in situ oil reservoirs, we hypothesize that the anaerobic ammonium-oxidation (anammox) process might be responsible for consuming the ammonia in the anoxic oil strata. Because the anammox process was implicated to be most importantly responsible for the nitrogen cycle in all the anoxic habitats [11, 24, 31] and consistent with the growth condition of anammox bacteria, the subsurface oil reservoir formed a suitable niche for them.
The anammox bacteria are among the latest additions to the biogeochemical nitrogen cycle with ability to convert ammonium and nitrite into dinitrogen gas in a strictly anoxic environment. So far, five anammox genera have been identified with similar physiological property belonging to the Brocadiales order, while the phylogenetic distance is quite large with 16S rRNA gene sequence identities ranging from 87% to 99% [25]. Up to now, the presence of anammox bacteria has been demonstrated in worldwide oxygen-limited marine and freshwater systems and in the activated sludge [26, 27, 35, 47, 53, 54, 59], marine sediment and oxygen minimum zones [6, 9, 31, 32, 45, 55, 64], freshwater [22, 65], and river estuary ecosystem [8, 10]. Moreover, the phylogenetic biomarkers of the functional gene hzo and nirS encoding the key enzymes involving the anammox process were developed to investigated the functional diversity of anammox bacteria in wastewater treatment plants and natural environment [30, 32, 35, 47, 54, 57, 59], which significantly improved the understanding of the anammox bacterial community.
However, the existence of anammox process in oil reservoirs is still unknown, and the knowledge of anammox in thermophilic habitats is also very limited. Given that the anammox bacteria were cultured in the experimental trail with optimum temperature below 43°C [58], the distribution of anammox bacteria in thermophilic habitats looks unfeasible. More recently, the discovery of anammox bacteria in the deep-sea hydrothermal vent and terrestrial hot springs substantially extends the temperature limit of anammox bacterial distribution [6, 22] by proving the presence and activity of anammox bacteria in the natural ecosystem with temperatures up to 100°C. The geothermal environment of an underground oil reservoir is a special hot habitat, while very little is known about the anammox bacterial distribution. Therefore, we initiated this survey of high-temperature oil reservoir ecosystems in order to find indications of other anammox bacteria that might thrive in the thermophilic environment and provide further information on bacterial nitrogen cycle in oil reservoirs. In this study, various oil reservoir samples were collected in Shengli Oilfield and observed by chemical and molecular ecology methods (i.e., 16S rRNA gene and hzo phylogeny). The existence of anammox bacteria was detected in the hot habitat to develop a general view of ecology and biogeography characteristics of the anammox bacterial population in the geothermal and anoxic subsurface petroleum reservoir.
Materials and Methods
Sample Collection and Physicochemical Analyses
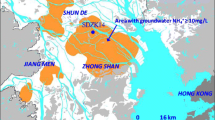
Shengli Oilfield is the second largest in China, located at Yellow River Delta in Shandong province in Eastern China. In total, 17 sampling wells distributed in an area of at least 2,000 km2 were selected to represent contrasting geographical and biogeochemical attributes and oil production operation. Seven wells (S3–S9) were in the middle of the Yellow River Delta and two wells (S1, S2) in the vicinity of Bohai Sea (Fig. 1). The formation-water samples were collected directly from the well head in sterilized steel screw-cap bottles after flushing the lines for 30 min. The bottles were filled with oil/water mixture, sealed, and immediately stored in a large plastic box with ice and transported to the laboratory. The residual oil was removed by heating the sample to 50°C for 15 min and by phase separation in 2 l sterilized separatory funnel. Microbial biomass was collected from 2 l water phase by centrifugation at 15,000×g, 4°C. The chemical properties of the formation water were measured using Hach kits by following the manufacturer’s instructions (Hach, USA).

Location map for sampling sites in Shengli Oilfield
Generation of 16S rRNA Gene and hzo Clone Library
Total community DNA was extracted from cell pellets using the lysozyme/proteinase K/sodium dodecyl sulfate treatment followed by standard phenol/chloroform extractions. Nucleic acids were purified by the DNA purification kit (Promega, USA). Sample DNA was extracted several times, pooled, and concentrated. Polymerase chain reaction (PCR) amplification of 16S rRNA gene was performed by two protocols. Firstly, the initial PCR amplification was performed with forward primer Pla46F [43] in combination with the reverse primer 1037R [39], and the nested PCR was performed with the anammox-specific primers Amx 368F [3, 53] and Amx 820R [3, 52] according to the previous study [8]. Secondly, PCR was performed with the primer sets Brod541F [45] in combination with Amx820R [52] according to the previous study [23, 56]. The hzo gene fragments were amplified according to the previous study with two sets of primers hzocl1F1 and hzocl1R2 [54], and Ana-hzo1F and Ana-hzo2F [47]. PCR products from different primer sets were purified by gel cut procedure, and a combined library for the same sample with pMD-18T-vector (Takara, Japan) was constructed.
Sequencing and Phylogenetic Analysis
The insertion of 16S rRNA gene and hzo fragments was screened and retrieved by PCR amplification with the primer set of M13F and M13R. The different numbers of positive clones in each library were randomly selected for sequencing on an automated ABI Prism 3730 DNA analyzer by using M13 universal sequencing primers and the Big Dye Terminate kit (Applied Sciences, Foster City, CA). The sequences without chimeras were initially submitted to the FASTA program (version 3) [44] to determine their closest phylogenetic relatives. For 16S rRNA gene, DNA sequences were manually compiled and aligned using CLUSTAL X [62]. For hzo gene, nucleic acid sequences were first translated into amino acids, and the resulting protein sequences were aligned to their nearest neighbor using CLUSTAL X. Phylogenetic trees were constructed based on the Kimura two-parameter model [29] and the neighbor-joining algorithm [49] using the MEGA 4.0 software [60]. Bootstrap analysis with 1,000 replicates was applied to assign confidence levels to the nodes of the consensus trees.
Statistical Analyses
Operational taxonomic units (OTUs) for 16S rRNA and hzo gene diversity analysis were defined by 3% differences in nucleotide and amino acid sequences, as determined by using the furthest neighbor algorithm in DOTUR program [50]. DOTUR was also used to generate Chao, Shannon, and Simpson index for each clone library. The coverage of clone libraries was calculated from the equation \( C{ } = { }\left[ {\;{1} - \left( {{{{{n_1}}} \left/ {N} \right.}} \right)\;} \right]\;\; \times { 100} \) [13]. Community classification of the anammox assemblages was determined with principal coordinates analyses (PCoA) [37]. Correlations between the anammox bacterial communities and environmental factors were determined by the canonical correspondence analysis (CCA) using the software CANOCO (ver. 4.5, Microcomputer Power, Ithaca, NY, USA) [61].
Real-Time PCR Assay
Gene copy numbers of total bacterial and anammox bacterial 16S rRNA gene in each sample were quantified with the quantitative PCR (qPCR) analysis. Real-time PCR was performed by using a FastStart Universal SYBR Green Master PCR mix (Roche Applied Science, Mannheim, Germany) in 96-well optical plates placed in an ABI 7000 Sequence detection system (Applied Biosystems, Foster City, CA). qPCR of total bacteria was performed with the primers 8F [2] and 338R [36] under the condition previously described [12, 35], and the qPCR assay of anammox bacteria was performed with the primers AMX-808-F and AMX-1040-R [17] under the adjusted condition previously described [35]. Each reaction was performed in a 25-μl volume containing 1 μl of DNA template, 2.5 μl bovine serum albumin (0.1%), 0.5 μl of each primer (20 μM), and 12.5 μl of the ready reaction mix. The PCR cycle was started with 2 min at 50°C, 3 min at 95°C, followed by 45 cycles of 95°C for 30 s, annealing for 30 s at 55°C, and extension for 30 s at 72°C [35]. Cycle thresholds were determined by comparison with standard curves constructed using several concentrations of positive clone. The plasmid DNA concentration was determined on a Biophotometer (Eppendorf, Germany), and the copy number of target gene was calculated directly from the concentration of the extracted plasmid DNA. Tenfold serial dilutions of a known copy number of the plasmid DNA were prepared to generate an external standard curve. The R 2 values were greater than 0.98 for the curves. Samples and standards were prepared in triplicate. The specificity of the PCR amplification was determined by the melting curve and gel electrophoresis.
Nucleotide Sequence Accession Numbers
The GenBank accession numbers of 16S rRNA and hzo gene sequences are HM208755 to HM208788 and HM208789 to HM208820, respectively.
Results
Characteristics of Petroleum Reservoirs
The investigated oil reservoirs are between 55°C and 75°C and on average 1,200–1,900 m below the ground surface. The sampling wells experienced three different stages of crude oil recovery processes, namely primary production (S6, S7), secondary production (S1–S4, S8, and S9), and tertiary production (S5) (Table 1). Sampling wells S6 and S7 were of primary production containing water mainly from an aquifer in the strata. Wells S1–S5, S8, and S9 were developed for 5–10 years with water flooding to maintain the strata pressure and allow the extraction of residual oil. In particular, well S5 has been flooded for 2 years with injection of aqueous polyacrylamide solution. Overall, the formation water was carbonate type with slightly alkaline pH values between 7.3 and 8.1 under surface atmospheric pressure. The major physicochemical parameters for the geological formation water are presented in Table 1, showing low to moderate concentrations of sulfate, phosphate, and nitrate in the production water. The ammonium measured was high in the formation water (Table 1). Nitrite was near detection limit. Sulfide and ionic iron in S6 and S7 were higher than the other water samples.
Diversity of Anammox Bacteria in Petroleum Reservoirs
Seventeen samples with different production history from Shengli Oilfield were analyzed by PCR with anammox bacterial specific primer sets, but only nine samples were positive for anammox bacteria with the PCR primer sets used in this study. Subsequently, the nine samples were used to construct the 16S rRNA gene and hzo sequence libraries. Fifty to 80 clones of each gene library per sample were screened by PCR and then sequenced. The positive clones were obtained from 22 to 35 of 16S rRNA gene libraries and 23 to 34 of hzo gene libraries, with three to five and three to four OTUs, respectively (Table 2). The coverage value of all libraries ranged between 0.95 and 1.00, indicating that the majority of the phylotypes in the clone libraries was detected (Table 2). Based on both Shannon and Chaol estimator indices, the anammox bacterial community diversity of samples S3 and S4 was the highest, and that of samples S1 and S2 was the lowest, which was consistent with the property of S3 and S4 with the lowest temperature but S1 and S2 with the highest temperature.
Phylogenetic Affiliation of Anammox Bacteria in Oilfields
Anammox bacterial diversity was constructed with the community DNA obtained and then 16S rRNA gene clone library from the formation water or production water of the oil strata. Screening of 241 sequences without chimera against the FASTA database indicated that the clones matched sequences related to anammox bacterial genera: Candidatus Scalindua [53], Candidatus Kuenenia [59], Candidatus Brocadia [51], and Candidatus Jettenia [47]. Most of the sequence types had relatively high levels of similarity with their closest counterparts in public databases, whereas six phylotypes showed less than 92% sequence similarity to their nearest database entries and may belong to unknown phylotypes [48]. The phylogenetic relationships of the anammox bacteria-like 16S rDNA sequences from this study and their closest relatives are represented in a consensus tree (Fig. 2). Most of the OTUs fell into the Candidatus Scalindua cluster containing the species: Candidatus Scalindua brodae [53], Candidatus Scalindua sorokinii [31], and Candidatus Scalindua wagneri [53] with sequence identities of 90.1–99.6%, and this cluster was supported by 99% bootstrap value (Fig. 2). The sequences related to Candidatus sorokinii/brodae were similar to the sequence type Menez Gwen (AM941036), recovered from the hydrothermal deep-sea vent recently [6]. In addition, six phylotypes in this subcluster were further divided into two groups that are distantly affiliated with each other (Fig. 2). The remaining sequences were more diverse and distributed within the Candidatus Scalindua wagneri subcluster, such as the Colne Estuary (EU394279) in the UK [10] and the Cape Fear River Estuary (FJ490132) in North Carolina [8], which were isolated from freshwater systems. The second large fraction of sequences were grouped within Candidatus Kuenenia genus (Fig. 2); the most abundant phylotypes, S4E6 and S2B10, in this cluster were affiliated with the sequence obtained from California Leonard hot spring clone HRH850 from a 52°C spring [22] and Rainbow hydrothermal chimney clone (AM941023) which was the first finding of anammox bacteria in the hot habitat of an active deep-sea vent with environmental fluid temperatures up to 100°C [6]. In addition, the Brodcadia cluster included the clones from high-temperature wells S1, S2, and S8, which were closely related to the sequences of California Leonard hot spring clone HRH852 [22] and Geothermal stream clone HAuD-MB/2-35 from hot water above 50°C in a Japanese gold mine [19]. The other sequences belonging to Jettenia cluster were isolated from lower temperature oilfields, closely related to the sequences from freshwater environments (Fig. 2) of the Cape Fear River Estuary in the UK [8]. Interestingly, cluster A containing clones S6G9, S7G9, S1B7, and S2E8 were distantly (90.1–92.6%) related to the known anammox bacteria, while this cluster was adjacent to the Scalindua cluster in the topology structure of the phylogenetic tree (Fig. 2), which were retrieved from the no-water-flooding and highest temperature oilfields, respectively. Samples S3, S4, S6, and S7 yielded four sequence types forming cluster B (Fig. 2), affiliated with the Rainbow hydrothermal chimney clone CH10MOM07.5 (AM941027) from the high-temperature deep-sea vent and the Nevada Patua-Hazen hot spring clone HRH860 from a 51°C spring [22], inferring a potentially new anammox bacterial cluster [6].

Phylogenetic tree of the anammox bacterial 16S rRNA genes retrieved from Shengli Oilfield (shown in boldface) and the referred sequences in the EMBL database with the divisions listed to the right. The clones were designated by the sample name and the randomly picked clone series number. The topology shown was calculated with the neighbor-joining method. Bootstrap values (n = 1,000 replicates) of ≥50% were reported near the corresponding nodes. The scale bar represented 0.05 nucleic acid substitutions per nucleotide position. Planctomyces maris (AJ231184), Planctomyces limnophilus (X62911), and Nitrosomonas europaea (AF353160) were used as out-groups
Functional Diversity of Anammox Bacteria by hzo Gene Analysis
The HZO enzyme (hydrazine oxidoreductase) catalyzes the initial hydrazine-forming reaction during the anammox biochemical process. The encoding gene hzo cluster 1 [54] was proven to be an appropriate and feasible biomarker for phylogenetic analysis of anammox bacteria in the enrichment cultures and environmental samples [34, 54]. To detect the functional diversity of anammox bacteria in the petroleum reservoirs, clone libraries of hzo gene were constructed by using the community DNA from the formation-water samples based on the cluster 1 hzo specific primers sets. The deduced amino acid sequences of 243 nucleic acid sequences could be grouped into 12 OTUs based on 3% substitutions/difference of amino acid sequences. The comparative analysis indicated that the closest matched HZO sequences belong to the anammox bacteria Candidatus Anammoxoglobus, Candidatus Kuenenia, Candidatus Jettenia, and Candidatus Scalindua (Fig. 3), but no sequence type fell into the anammox bacteria Candidatus Brocadia. The phylogenetic relationships of the HZO-like sequences in this study and their closest relatives were represented in a consensus tree, and the amino acid sequences of HZO cluster 2a [54] were used as out-groups to root the tree (Fig. 3). The most abundant clones were grouped in the Scalindua cluster, which was 96.5–98.9% similar to HZO sequences of Candidatus Scalindua sp. [54]. Adjacent to this cluster, cluster D was distantly related to other HZO phylotypes (Fig. 3). Consistent with cluster C of 16S rRNA gene tree, this cluster had the same phylogenetic topology status in the HZO tree, so it was likely that sequences of these two clusters represented the unknown anammox bacteria. Cluster D sequences also formed a slightly distant group affiliated with other detected hzo genes. However, no corresponding cluster sequences to cluster C were found in the 16S rRNA gene libraries (Figs. 2 and 3). Most sequences from water-flooding and lower temperature oilfields were grouped within Jettenia cluster, showing 97.6–99.5% sequence identities to the HZO sequence of Candidatus Jettenia asiatica [47]. But some of no-water-flooding oilfield sequences fell into Anammoxoglobus cluster with 98.2–99.6% identities to the HZO sequence of Candidatus Anammoxoglobus sp. [54]. Consistent with the 16S rRNA gene phylogeny, most of the highest temperature oilfield sequence types were related to the HZO sequence of Candidatus Kuenenia stuttgartiensis with 97.3–99.8% sequence identities [59]. Interestingly, no sequences related to Candidatus Brocadia were detected in any of the samples used in this study.

Phylogenetic tree of the deduced anammox bacterial HZO protein sequences from Shengli Oilfield (shown in boldface) and the referred sequences in the EMBL database with the putative divisions listed to the right. The clones were designated by the sample name and the randomly picked clone series number. The topology shown was calculated with the neighbor-joining method. Bootstrap values (n = 1,000 replicates) of ≥50% were reported near the corresponding nodes. The scale bar represented 0.10 amino acid substitutions per amino acid position. The HZO sequences of Planctomycete KSU-1 (BAF98478) and Candidatus Kuenenia stuttgartiensis (CAJ71086) were used as out-groups
Quantification of Anammox Bacteria
The abundance of total bacteria and anammox bacteria in the oilfield production water was estimated based on the quantification of bacterial 16S rRNA genes using the qPCR method [12, 17, 35]. The amounts of total bacterial and anammox bacterial 16S rRNA gene copy numbers ranged from 2.3 ± 0.18 × 104 to 7.3 ± 0.65 × 106 ml−1 and from 6.6 ± 0.51 × 102 to 4.9 ± 0.36 × 104 (Fig. 4), respectively. Based on the estimation that 3.6 copy numbers of 16S rRNA gene per bacterial cell genome [18], the total bacterial density was calculated from 6.4 ± 0.5 × 103 to 2.0 ± 0.18 × 106 cells ml−1. Assuming that the anammox bacteria contained one copy of the 16S rRNA operon based on Candidatus Kuenenia stuttgartiensisis genome [59], the anammox bacterial density was calculated to be 6.6 ± 0.51 × 102 to 4.9 ± 0.36 × 104 cells ml−1. The anammox bacteria was a small fraction of the bacterial community in Shengli Oilfield production waters, as the anammox bacteria accounted for 2.5% to 10.3% of all bacteria detected. The highest concentration of total bacteria and anammox bacteria was found in the production water from S4 (Fig. 4), where the lowest temperature and moderate time scale of water-flooding operation were observed (Table 1). Correlation analysis showed that the total bacteria and anammox bacteria populations were highly related to the temperature gradient. The lowest bacterial abundance was found in sample S2, where the highest temperature and longest term of flooding were observed.

Abundance of total bacterial and anammox bacterial 16S rRNA gene copy numbers in production water from Shengli Oilfield. Mean values and standard deviations were calculated from triplicate assay within a single qPCR setup (more detailed information available in “Materials and Methods”)
Anammox Bacterial Communities Classification
Significant difference of the anammox bacterial assemblages in the oilfields was revealed by the PCoA test (p < 0.005) [38] based on the phylogenetic tree of 16S rRNA gene and HZO protein sequences. The results indicated that anammox bacterial assemblage fell into four groups, S5 for chemical flooding, S3 and S4 for lower temperature and water-flooding oilfield, S6 and S7 for no-water-flooding oilfield, and S1, S2, S8, and S9 for high-temperature water-flooding oilfield (p < 0.032) (Fig. 5a). Both S6 and S7 represented no-water-flooding oil reservoirs, and S5 represented chemical-flooding oilfield, showing obvious difference from the other samples from the water-flooding oilfield along the first principal coordinate (P1) which explained 32.9% of the total anammox bacterial community variability among all the sampling oilfields (Fig. 5a). The PCoA analysis of the 16S rRNA gene and HZO protein sequences of anammox bacterial assemblages showed a slightly different classification (Fig. 5b), with S9 closer to S8 in the former analysis, but S8 was closer to S2 in the latter one.

PCoA ordination diagrams of the anammox bacterial assemblages calculated with 16S rRNA gene sequences (a) and HZO protein sequences (b) from production water of Shengli Oilfield. Shown are the plots of the first two principal coordinate axes (P1 and P2) for PCoA and the distributions of the anammox bacterial assemblages (designated with the sampling wells) in response to these axes
Spatial Distribution of Anammox Bacterial Communities
To find the correlations between the distribution of anammox bacterial community and the environmental variables of the oilfields, canonical correspondence analysis was conducted based on the 16S rRNA gene and HZO protein sequences and the physiochemical parameters recovered in Shengli Oilfield. The results showed that the CCA axe in 16S rRNA gene deduced scheme explained 40.1% but the CCA axe in HZO sequence deduced scheme explained 54.6% of the cumulative variance of the correlation between the environmental factors and the anammox bacterial community distribution (Fig. 6). The CCA analysis indicated that all of the anammox bacterial assemblages fell into four groups, with the CCA1 distinguishing the anammox bacterial assemblages of S1, S2, S8 and S9, S6, and S7 together and S5 distant to others, while the CCA2 axe found S3 and S4 as one assemblage (Fig. 6). The oil recovery process contributed another important effect (p = 0.005, 1,000 Monte Carlo permutations) to the distinction of the anammox bacterial community and environment relationship, which revealed that the distribution of the anammox bacterial assemblages of S1, S2, S8, and S9 were positively correlated with the water flooding but in the opposite direction to the distribution of S6 and S7, and the distribution of S5 was positively correlated with chemical flooding (Fig. 6). The temperature and the concentration of ammonium cation appeared to be significant (p = 0.048, 1,000 Monte Carlo permutations) to the anammox bacterial distribution, and the environmental parameter relationship with the S1, S2, S8, and S9 were positively correlated with the two factors. Other environmental factors did not contribute significantly (p > 0.500) to the anammox bacterial community, while the combination of these environmental factors provided additionally 55.0% of the total CCA explanatory power.

CCA ordination plots for the first two dimensions to show the relationship between the anammox bacterial diversity with environmental parameters analyzed using a 16S rRNA gene sequence and b HZO protein sequence in the production water of Shengli Oilfield. Correlations between environmental variables and CCA axes are represented by the length and angle of arrows (environmental factor vectors). Temp temperature, WF water flooding, CF chemical flooding
Discussion
The combined data of clone libraries of both 16S rRNA and hzo genes confirmed the occurrence of anammox bacteria in the anoxic geothermal oilfield environment at Shengli Oilfield in China. Our results confirmed that Candidatus Brocadia sp. and Candidatus Kuenenia sp. could be detected in oil reservoirs with temperature up to 75°C and were the first to detect the anammox bacterial species Candidatus “Scalindua sinooilfield” in such habitat. It is consistent with other findings observed in the deep-sea hydrothermal vent at Mid-Atlantic Ridge [6] and in Great Basin hot springs of Northern California and Western Nevada [22]. By applying more specific primer sets for anammox bacterial 16S rRNA gene and the functional biomarker hzo gene, anammox bacteria were successfully detected in the oil reservoir ecosystem. This is the first report for the existence of anammox bacteria in the high-temperature oil reservoir. Therefore, anammox bacteria are present not only in the marine environments at generally lower ambient temperatures [55, 64] but also in the high-temperature environment, which expands our knowledge of the biogeography of anammox bacteria.
The diversity of anammox bacteria depends on the oil production processes and physicochemical conditions in situ. Most phylotypes detected in relatively higher temperature wells belong to Kuenenia and Brocadia clusters. Although the highest optimum temperature was at 43°C in enrichment culture [58], Candidatus Kuenenia species were also obtained from the hydrothermal vent with fluid temperatures more than 100°C and the hot springs with higher than 52°C [6, 22]. In this study, the Brocadia-like organisms were detected in the water samples from wells S1, S2, and S8 with temperature up to 75°C, which was the first report of the Brocadia-like organisms in such habitat. Interestingly, the sequence detected in the no-water-flooding wells belonged to the Anammoxoglobus cluster closely related to Candidatus “Anammoxoglobus propionicus” which had capability to oxidize simple organic acids (acetate and propionate) at a higher rate and without a transient nitrite accumulation [27]. This property may be critical for them to thrive in an oil reservoir environment because the crude oil consists of abundant hydrocarbons which can be converted to short-chain fatty acids by microbial fermentation process in the oil reservoirs under primary operation without anthropogenic influences, such as water flooding or chemical injection [14, 15].
For a more comprehensive analysis of the functional diversity of anammox bacteria in oil reservoirs, the hzo sequences were retrieved to compare the phylogenetic diversity of anammox bacterial 16S rRNA gene. Consistent with previous results [34, 35, 54], the topologies of the phylogenetic tree of the obtained hzo was congruent with retrieved 16S rRNA gene phylogeny, suggesting that the hzo gene is a good alternative biomarker for detecting the anammox bacteria in oil reservoirs. More importantly, our results indicated that the phylogenetic diversities based on 16S rRNA and hzo genes are complementary to each other for Brocadia-like organisms in the 16S rRNA gene library and Anammoxoglobus-like ones in hzo library. Besides the sequences closely related to the known anammox bacteria, two clusters of sequences retrieved were apparently more distantly related to the sequences from cultured and uncultured anammox bacteria. Firstly, cluster A 16S rRNA gene sequences were distantly related to other Candidatus Scalindua-like phylotypes. Based on the sequence identities of up to 97% of the 16S rRNA gene commonly used to define different species [48], it was proposed that the sequences of this cluster comprise a new species within the genus Candidatus Scalindua, for which the name Candidatus “Scalindua sinooilfield” is proposed provisionally. Given the cluster D hzo sequences have similar topology structure compared to the cluster A 16S rRNA gene phylogeny, we suggest that this cluster belongs to the new anammox bacterial genus Candidatus “Scalindua sinooilfield”. Assuming this hypothesis is correct, the distinctness of the Candidatus “Scalindua sinooilfield” cluster could also explain that cluster D hzo sequences have distinctly different lengths compared to other hzo genes of Candidatus Scalindua species. In addition, the cluster C hzo sequences show further separation from other hzo-like sequences, whereas the corresponding sequence cluster could not be found in the 16S rRNA gene phylogenetic tree, which might be due to the differences of specificity and efficiency of the PCR primer sets of these two biomarkers [34]. Thus, to further investigate the functional diversity of anammox bacteria in various ecosystems, more effective primers need to be designed for the hzo or other important anammox genes, such as the putative cytochrome cd1-containing nitrite reductase gene (nirS) proven to be an alternative biomarker for Scalindua-like sequences recently [32].
The abundance of anammox bacteria decreased in the oil reservoirs as the temperature increased. The lowest concentration was detected in samples with moderate water flooding, which was comparable with the anammox bacterial numbers in high-temperature deep-sea vents [6]. However, the significant variation of anammox bacterial abundance was observed in different oilfields with temperatures ranging from 55°C to 75°C. The mean concentration of anammox bacteria was obviously lower than that in marine ecosystem [8, 17, 55]. This discrepancy might be related to the inhibition of high temperature to the growth of anammox bacteria. Meanwhile, the water-flooding and chemical-flooding operations seem to affect their number. The anammox bacterial density in the moderate water-flooding oilfield was higher than that in the no-water-flooding and the longest water flooding. The chemical-flooding oilfield S5 was lower by 90 times than that in the no-chemical-flooding oilfields. The abundance of anammox bacteria in primary production well samples was between those of the chemical-flooding sample and the water-flooding samples.
The distribution of anammox bacteria is affected by the oilfield history and production processes. Both temperature and recovery process are the two most important environmental variables affecting the distribution of anammox bacteria in the oil reservoirs. Most phylotypes detected in primary production wells belong to Scalindua cluster observed from the freshwater system, which might adapt to the oil reservoir environment in underground water system, while most phylotypes in the water-flooding and chemical-flooding oil reservoir samples fall into the Jettenia cluster, which might be originated from the injected water. This discrepancy might be related to the introduction of microorganisms by injected water or chemical [7, 33], resulting in the shift of microbial community composition. Given that the members of Candidatus Scalindua and Candidatus Jettenia genera were previously proved to be ubiquitously distributed in environments such as river and marine environments or granular sludge anammox reactor [47, 55, 64, 65], it is reasonable to have anammox bacteria in the injection water with sources of river water or recycled flooding water. This explains the existence of the putative Scalindua and Jettenia-related sequences in the production water from oilfield. Interestingly, there are more than one kind of anammox bacteria detected in most of the oil reservoirs, which contradict the niche differentiation of anammox bacteria, genera Candidatus Kuenenia, Candidatus Anammoxglobus, and Candidatus Brocadia in anoxic freshwater and Candidatus Scalindua in anoxic marine habitats [55], indicating that anammox bacteria have broader distribution than previously estimated [27, 55].
The existence of anaerobic ammonium-oxidizing bacteria in the high-temperature oil reservoir was confirmed in this study. Widely distributed and diverse 16S rRNA gene and hzo sequence types were detected in the production water samples. In addition, two clusters of sequences retrieved from high-temperature oilfield might represent a novel anammox bacterial phylotype. Anammox bacterial communities are influenced by temperature and production processes. Further study of anammox bacteria in oilfield ecosystems would allow a better understanding of the nitrogen cycle in the subterranean oil reservoir ecosystems.
References
Agrawal A, Lal B (2009) Rapid detection and quantification of bisulfite reductase genes in oil field samples using real-time PCR. FEMS Microbiol Ecol 69:301–312
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925
Amano T, Yoshinaga I, Okada K, Yamagishi T, Ueda S, Obuchi A, Sako Y, Suwa Y (2007) Detection of anammox activity and diversity of anammox bacteria-related 16S rRNA genes in coastal marine sediment in Japan. Microbes and Environments 22:232–242
Bastin ES (1926) Microorganisms in oilfields. Science 63:21–24
Bonch-Osmolovskaya EA, Miroshnichenko ML, Lebedinsky AV, Chernyh NA, Nazina TN, Ivoilov VS, Belyaev SS, Boulygina ES, Lysov YP, Perov AN, Mirzabekov AD, Hippe H, Stackebrandt E, L’Haridon S, Jeanthon C (2003) Radioisotopic, culture-based, and oligonucleotide microchip analyses of thermophilic microbial communities in a continental high-temperature petroleum reservoir. Appl Environ Microbiol 69:6143–6151
Byrne N, Strous M, Crepeau V, Kartal B, Birrien JL, Schmid M, Lesongeur F, Schouten S, Jaeschke A, Jetten M, Prieur D, Godfroy A (2009) Presence and activity of anaerobic ammonium-oxidizing bacteria at deep-sea hydrothermal vents. ISME J 3:117–123
Dahle H, Garshol F, Madsen M, Birkeland NK (2008) Microbial community structure analysis of produced water from a high-temperature North Sea oil-field. Antonie van Leeuwenhoek International Journal of General and Molecular Microbiology 93:37–49
Dale OR, Tobias CR, Song BK (2009) Biogeographical distribution of diverse anaerobic ammonium oxidizing (anammox) bacteria in Cape Fear River Estuary. Environ Microbiol 11:1194–1207
Dalsgaard T, Thamdrup B, Canfield DE (2005) Anaerobic ammonium oxidation (anammox) in the marine environment. Res Microbiol 156:457–464
Dong LF, Smith CJ, Papaspyrou S, Stott A, Osborn AM, Nedwell DB (2009) Changes in benthic denitrification, nitrate ammonification, and anammox process rates and nitrate and nitrite reductase gene abundances along an estuarine nutrient gradient (the Colne Estuary, United Kingdom). Appl Environ Microbiol 75:3171–3179
Francis CA, Beman JM, Kuypers MM (2007) New processes and players in the nitrogen cycle: the microbial ecology of anaerobic and archaeal ammonia oxidation. ISME Jounal 1:19–27
Gittel A, Sorensen KB, Skovhus TL, Ingvorsen K, Schramm A (2009) Prokaryotic community structure and sulfate reducer activity in water from high-temperature oil reservoirs with and without nitrate treatment. Appl Environ Microbiol 75:7086–7096
Good IJ (1953) The population frequencies of species and the estimation of population parameters. Biometrika 40:237–264
Grabowski A, Blanchet D, Jeanthon C (2005) Characterization of long-chain fatty-acid-degrading syntrophic associations from a biodegraded oil reservoir. Res Microbiol 156:814–821
Grabowski A, Nercessian O, Fayolle F, Blanchet D, Jeanthon C (2005) Microbial diversity in production waters of a low-temperature biodegraded oil reservoir. FEMS Microbiol Ecol 54:427–443
Greene EA, Hubert C, Nemati M, Jenneman GE, Voordouw G (2003) Nitrite reductase activity of sulphate-reducing bacteria prevents their inhibition by nitrate-reducing, sulphide-oxidizing bacteria. Environ Microbiol 5:607–617
Hamersley MR, Lavik G, Woebken D, Rattray JE, Lam P, Hopmans EC, Damste JSS, Kruger S, Graco M, Gutierrez D, Kuypers MMM (2007) Anaerobic ammonium oxidation in the Peruvian oxygen minimum zone. Limnol Oceanogr 52:923–933
Harms G, Layton AC, Dionisi HM, Gregory IR, Garrett VM, Hawkins SA, Robinson KG, Sayler GS (2003) Real-time PCR quantification of nitrifying bacteria in a municipal wastewater treatment plant. Environ Sci Technol 37:343–351
Hirayama H, Takai K, Inagaki F, Yamato Y, Suzuki M, Nealson KH, Horikoshi K (2005) Bacterial community shift along a subsurface geothermal water stream in a Japanese gold mine. Extremophiles 9:169–184
Hubert C, Voordouw G (2007) Oil field souring control by nitrate-reducing Sulfurospirillum spp. that outcompete sulfate-reducing bacteria for organic electron donors. Appl Environ Microbiol 73:2644–2652
Hubert C, Nemati M, Jenneman G, Voordouw G (2005) Corrosion risk associated with microbial souring control using nitrate or nitrite. Appl Microbiol Biotechnol 68:272–282
Jaeschke A, den Camp HJMO, Harhangi H, Klimiuk A, Hopmans EC, Jetten MSM, Schouten S, Damste JSS (2009) 16S rRNA gene and lipid biomarker evidence for anaerobic ammonium-oxidizing bacteria (anammox) in California and Nevada hot springs. FEMS Microbiol Ecol 67:343–350
Jetten M, Schmid M, van de Pas-Schoonen K, Damste JSS, Strous M (2005) Anammox organisms: enrichment, cultivation, and environmental analysis. Environ Microbiol 397:34–57
Jetten MSM (2008) The microbial nitrogen cycle. Environ Microbiol 10:2903–2909
Jetten MSM, van Niftrik L, Strous M, Kartal B, Keltjens JT, Op den Camp HJM (2009) Biochemistry and molecular biology of anammox bacteria. Crit Rev Biochem Mol Biol 44:65–84
Kartal B, van Niftrik L, Rattray J, de Vossenberg JLCMV, Schmid MC, Damste JSS, Jetten MSM, Strous M (2008) Candidatus ‘Brocadia fulgida’: an autofluorescent anaerobic ammonium oxidizing bacterium. FEMS Microbiol Ecol 63:46–55
Kartal B, Rattray J, van Niftrik LA, van de Vossenberg J, Schmid MC, Webb RI, Schouten S, Fuerst JA, Damste JSS, Jetten MSM, Strous M (2007) Candidatus “Anammoxoglobus propionicus” a new propionate oxidizing species of anaerobic ammonium oxidizing bacteria. Syst Appl Microbiol 30:39–49
Kaster KM, Bonaunet K, Berland H, Kjeilen-Eilertsen G, Brakstad OG (2009) Characterisation of culture-independent and -dependent microbial communities in a high-temperature offshore chalk petroleum reservoir. Antonie van Leeuwenhoek International Journal of General and Molecular Microbiology 96:423–439
Kimura M (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide-sequences. J Mol Evol 16:111–120
Klotz MG, Stein LY (2008) Nitrifier genomics and evolution of the nitrogen cycle. FEMS Microbiol Lett 278:146–156
Kuypers MMM, Sliekers AO, Lavik G, Schmid M, Jorgensen BB, Kuenen JG, Damste JSS, Strous M, Jetten MSM (2003) Anaerobic ammonium oxidation by anammox bacteria in the Black Sea. Nature 422:608–611
Lam P, Lavik G, Jensen MM, van de Vossenberg J, Schmid M, Woebken D, Dimitri G, Amann R, Jetten MSM, Kuypers MMM (2009) Revising the nitrogen cycle in the Peruvian oxygen minimum zone. Proc Natl Acad Sci USA 106:4752–4757
Li H, Yang SZ, Mu BZ, Rong ZF, Zhang J (2007) Molecular phylogenetic diversity of the microbial community associated with a high-temperature petroleum reservoir at an offshore oilfield. FEMS Microbiol Ecol 60:74–84
Li M, Hong YG, Klotz MG, Gu J-D (2010) A comparison of primer sets for detecting 16S rRNA and hydrazine oxidoreductase genes of anaerobic ammonium-oxidizing bacteria in marine sediments. Appl Microbiol Biotechnol 86:781–790
Li XR, Du B, Fu HX, Wang RF, Shi JH, Wang Y, Jetten MSM, Quan ZX (2009) The bacterial diversity in an anaerobic ammonium-oxidizing (anammox) reactor community. Syst Appl Microbiol 32:278–289
Loy A, Lehner A, Lee N, Adamczyk J, Meier H, Ernst J, Schleifer KH, Wagner M (2002) Oligonucleotide microarray for 16S rRNA gene-based detection of all recognized lineages of sulfate-reducing prokaryotes in the environment. Appl Environ Microbiol 68:5064–5081
Lozupone C, Hamady M, Knight R (2006) UniFrac-an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinform 7:371–384
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73:1576–1585
Ludwig W, Kirchhof G, Klugbauer N, Weizenegger M, Betzl D, Ehrmann M, Hertel C, Jilg S, Tatzel R, Zitzelsberger H, Liebl S, Hochberger M, Shah J, Lane D, Wallnöfer PR, Scheifer KH (1992) Complete 23S ribosomal RNA sequences of Gram-positive bacteria with a low DNA G+C content. System Appl Microbiol 15:487–501
Magot M (2005) Indigenous microbial communities in oil fields. In: Ollivier B, Magot M (eds) Petroleum microbiology. ASM Press, Washington, pp 21–34
Nazina TN, Kosareva IM, Petrunyaka VV, Savushkina MK, Kudriavtsev EG, Lebedev VA, Ahunov VD, Revenko YA, Khafizov RR, Osipov GA, Belyaev SS, Ivanov MV (2004) Microbiology of formation waters from the deep repository of liquid radioactive wastes Severnyi. FEMS Microbiol Ecol 49:97–107
Nazina TN, Grigor’ian AA, Shestakova NM, Babich TL, Ivoilov VS, Feng Q, Ni F, Wang J, She Y, Xiang T, Luo Z, Beliaev SS, Ivanov MV (2007) Microbiological investigations of high-temperature horizons of the Kongdian petroleum reservoir in connection with field trial of a biotechnology for enhancement of oil recovery. Mikrobiologiia 76:329–339
Neef A, Amann R, Schlesner H, Schleifer KH (1998) Monitoring a widespread bacterial group: In situ detection of planctomycetes with 16S rRNA-targeted probes. Microbiology 144:3257–3266
Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85:2444–2448
Penton CR, Devol AH, Tiedje JM (2006) Molecular evidence for the broad distribution of anaerobic ammonium-oxidizing bacteria in freshwater and marine sediments. Appl Environ Microbiol 72:6829–6832
Pham VD, Hnatow LL, Zhang SP, Fallon RD, Jackson SC, Tomb JF, DeLong EF, Keeler SJ (2009) Characterizing microbial diversity in production water from an Alaskan mesothermic petroleum reservoir with two independent molecular methods. Environ Microbiol 11:176–187
Quan ZX, Rhee SK, Zuo JE, Yang Y, Bae JW, Park JR, Lee ST, Park YH (2008) Diversity of ammonium-oxidizing bacteria in a granular sludge anaerobic ammonium-oxidizing (anammox) reactor. Environ Microbiol 10:3130–3139
Rossello-Mora R, Amann R (2001) The species concept for prokaryotes. FEMS Microbiol Rev 25:39–67
Saitou N, Nei M (1987) The neighbor-joining method: a method for constructing phylogenetic trees. Mol Biol Evol 4:406–424
Schloss PD, Handelsman J (2005) Introducing DOTUR, a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506
Schmid M, Schmitz-Esser S, Jetten M, Wagner M (2001) 16S-23S rDNA intergenic spacer and 23S rDNA of anaerobic ammonium-oxidizing bacteria: implications for phylogeny and in situ detection. Environ Microbiol 3:450–459
Schmid M, Twachtmann U, Klein M, Strous M, Juretschko S, Jetten M, Metzger JW, Schleifer KH, Wagner M (2000) Molecular evidence for genus level diversity of bacteria capable of catalyzing anaerobic ammonium oxidation. Syst Appl Microbiol 23:93–106
Schmid M, Walsh K, Webb R, Rijpstra WIC, van de Pas-Schoonen K, Verbruggen MJ, Hill T, Moffett B, Fuerst J, Schouten S, Damste JSS, Harris J, Shaw P, Jetten M, Strous M (2003) Candidatus “Scalindua brodae”, sp nov., Candidatus “Scalindua wagneri”, sp nov., two new species of anaerobic ammonium oxidizing bacteria. Syst Appl Microbiol 26:529–538
Schmid MC, Hooper AB, Klotz MG, Woebken D, Lam P, Kuypers MMM, Pommerening-Roeser A, op den Camp HJM, Jetten MSM (2008) Environmental detection of octahaem cytochrome c hydroxylamine/hydrazine oxidoreductase genes of aerobic and anaerobic ammonium-oxidizing bacteria. Environ Microbiol 10:3140–3149
Schmid MC, Risgaard-Petersen N, van de Vossenberg J, Kuypers MMM, Lavik G, Petersen J, Hulth S, Thamdrup B, Canfield D, Dalsgaard T, Rysgaard S, Sejr MK, Strous M, den Camp HJMO, Jetten MSM (2007) Anaerobic ammonium-oxidizing bacteria in marine environments: widespread occurrence but low diversity. Environ Microbiol 9:1476–1484
Schmid MC, Maas B, Dapena A, de Pas-Schoonen KV, de Vossenberg JV, Kartal B, van Niftrik L, Schmidt I, Cirpus I, Kuenen JG, Wagner M, Damste JSS, Kuypers M, Revsbech NP, Mendez R, Jetten MSM, Strous M (2005) Biomarkers for in situ detection of anaerobic ammonium-oxidizing (anammox) bacteria. Appl Environ Microbiol 71:1677–1684
Shimamura M, Nishiyama T, Shigetomo H, Toyomoto T, Kawahara Y, Furukawa K, Fujii T (2007) Isolation of a multiheme protein with features of a hydrazine-oxidizing enzyme from an anaerobic ammonium-oxidizing enrichment culture. Appl Environ Microbiol 73:1065–1072
Strous M, Kuenen JG, Jetten MSM (1999) Key physiology of anaerobic ammonium oxidation. Appl Environ Microbiol 65:3248–3250
Strous M, Pelletier E, Mangenot S, Rattei T, Lehner A, Taylor MW, Horn M, Daims H, Bartol-Mavel D, Wincker P, Barbe V, Fonknechten N, Vallenet D, Segurens B, Schenowitz-Truong C, Medigue C, Collingro A, Snel B, Dutilh BE, Op den Camp HJM, van der Drift C, Cirpus I, van de Pas-Schoonen KT, Harhangi HR, van Niftrik L, Schmid M, Keltjens J, van de Vossenberg J, Kartal B, Meier H, Frishman D, Huynen MA, Mewes HW, Weissenbach J, Jetten MSM, Wagner M, Le Paslier D (2006) Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440:790–794
Tamura T, Dudley J, Nei M, Kumarsis S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
ter Braak CJF, Šmilauer P (2002) CANOCO Reference Manual and CanoDraw for Windows User’s Guide: Software for Canonical Community Ordination (version 4.5) Microcomputer Power (Ithaca NY, USA)
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
van Hamme JD, Singh A, Ward OP (2003) Recent advances in petroleum microbiology. Microbiol Mol Biol Rev 67:503–549
Woebken D, Lam P, Kuypers MMM, Naqvi SWA, Kartal B, Strous M, Jetten MSM, Fuchs BM, Amann R (2008) A microdiversity study of anammox bacteria reveals a novel Candidatus Scalindua phylotype in marine oxygen minimum zones. Environ Microbiol 10:3106–3119
Zhang Y, Ruan XH, den Camp HJMO, Smits TJM, Jetten MSM, Schmid MC (2007) Diversity and abundance of aerobic and anaerobic ammonium-oxidizing bacteria in freshwater sediments of the Xinyi River (China). Environ Microbiol 9:2375–2382
Acknowledgements
This study was supported by Environmental Toxicology Education Fund (to J-D G), National Natural Science Foundation of China (50744016), National High Technology Research and Development Program of China (2009AA063503), China Postdoctoral Science Foundation funded project (20080440082, 200902223), Shanghai Municipal Science and Technology Commission (071607014), and Shanghai Postdoctoral Scientific Program (09R21411900). We thank Jessie Lai for laboratory assistance and Meng Li for useful discussion.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Li, H., Chen, S., Mu, BZ. et al. Molecular Detection of Anaerobic Ammonium-Oxidizing (Anammox) Bacteria in High-Temperature Petroleum Reservoirs. Microb Ecol 60, 771–783 (2010). https://doi.org/10.1007/s00248-010-9733-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-010-9733-3