
Abstract
Purpose
There is a need for alternative analgosedatives such as dexmedetomidine in neonates. Given the ethical and practical difficulties, protocol design for clinical trials in neonates should be carefully considered before implementation. Our objective was to identify a protocol design suitable for subsequent evaluation of the dosing requirements for dexmedetomidine in mechanically ventilated neonates.
Methods
A published paediatric pharmacokinetic model was used to derive the dosing regimen for dexmedetomidine in a first-in-neonate study. Optimality criteria were applied to optimise the blood sampling schedule. The impact of sampling schedule optimisation on model parameter estimation was assessed by simulation and re-estimation procedures for different simulation scenarios. The optimised schedule was then implemented in a neonatal pilot study.
Results
Parameter estimates were more precise and similarly accurate in the optimised scenarios, as compared to empirical sampling (normalised root mean square error: 1673.1% vs. 13,229.4% and relative error: 46.4% vs. 9.1%). Most importantly, protocol deviations from the optimal design still allowed reasonable parameter estimation. Data analysis from the pilot group (n = 6) confirmed the adequacy of the optimised trial protocol. Dexmedetomidine pharmacokinetics in term neonates was scaled using allometry and maturation, but results showed a 20% higher clearance in this population compared to initial estimates obtained by extrapolation from a slightly older paediatric population. Clearance for a typical neonate, with a post-menstrual age (PMA) of 40 weeks and weight 3.4 kg, was 2.92 L/h. Extension of the study with 11 additional subjects showed a further increased clearance in pre-term subjects with lower PMA.
Conclusions
The use of optimal design in conjunction with simulation scenarios improved the accuracy and precision of the estimates of the parameters of interest, taking into account protocol deviations, which are often unavoidable in this event-prone population.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Neonates receiving mechanical ventilation often require analgosedation because of pain and discomfort. Opiates (morphine, fentanyl) and benzodiazepines (midazolam) are regularly prescribed to meet the analgosedation needs of the ventilated neonate. However, the use of these drugs is still a matter of debate given the potential risk for serious adverse events [1]. The search for newer agents with an improved safety profile is an ongoing endeavour.
Dexmedetomidine is a centrally acting α2-agonist with sedative, anxiolytic, sympatholytic and analgosedative properties. After intravenous administration, it quickly distributes beyond total body water. It is metabolised by glucuronidation and CYP2A6 hydroxylation and subsequently excreted in urine almost exclusively as metabolite [2]. Its pharmacokinetics has been described by a two-compartmental model both in adults [3,4,5,6,7] and children [8,9,10,11,12]. Still, other investigators have also reported the disposition of dexmedetomidine according to one-compartment [13] and three-compartment [14,15,16] models. The changes in pharmacokinetics in children are often allometrically scaled, and an increase in clearance during the first few years of life has previously been associated with covariates describing developmental growth (body weight) and organ maturation, as assessed by post-menstrual age (PMA) [12, 13]. As of yet, it is unclear to what degree gestational age (GA) or post-natal age (PNA) correlate with changes in drug clearance in neonates and infants.
Currently, dexmedetomidine is licensed in Europe and the USA for analgosedation in ventilated adult ICU patients and procedural sedation in non-ventilated adults [17]. Given its predictable (adverse) effects and potential advantages when compared to benzodiazepines [18,19,20] and opiates [21], dexmedetomidine should be considered for investigation in neonates. However, a clear dose rationale is lacking prior to establishing its efficacy. Due to the limited understanding of the effect of prematurity and ontogeny of enzymes on drug disposition in pre-term and near-term neonates, it is essential to characterise the pharmacokinetics of dexmedetomidine before defining dosing recommendations for this population.
Despite the scientific and clinical imperative, the evaluation of pharmacokinetics, pharmacodynamics, efficacy and safety of drugs in vulnerable new-borns remains challenging due to ethical and practical constraints. To minimise adverse events and burden, clinical trials are commonly restricted in number of subjects, dose range, duration, frequency and blood volume to be sampled, thereby limiting the interpretation and generalisability of results. These practical issues are further compounded by rapidly changing physiological processes, which evolve significantly within the first few weeks to months of life. Overall, these processes contribute to large inter and intra-individual variability. Typically, paediatric clinical trials are designed on an empirical basis, i.e. dose levels, total patient numbers to be recruited, number of samples to be collected and sampling times are determined according to feasibility and so-called common practice, including the use of statistical power calculations and knowledge about the compound in adults or other indications. Clearly, such an approach does not guarantee the optimal use of individual patient data, especially if such data are to be utilised as a basis for dosing recommendations. By contrast, the integration of optimal design concepts with pharmacokinetic modelling and simulation offers an opportunity to maximise the information obtained in a clinical trial with a limited number of patients while minimising burden and efforts [22,23,24,25].
Optimal design allows the maximisation of precision and accuracy of the parameters of interest (e.g. clearance, area under the concentration-time curve) through more informative trial design characteristics, such as sampling frequency, sampling schedule and number of patients [26]. Application of these concepts often results in fewer samples and/or subjects needed while maintaining comparable statistical power or precision, which consequently reduces the overall burden on patients who participate in a clinical study. Furthermore, the integration of existing models with new data allows for incremental improvements in the understanding of clinical pharmacology through the so-called learn-confirm paradigm [23, 24, 27, 28]. Despite these advantages, the use of optimality principles in paediatric clinical trial design has remained limited.
We aimed to improve the dose rationale for dexmedetomidine in pre-term and near-term patients. Thus, the objective of this study was to perform a pilot dose-finding study of dexmedetomidine in mechanically ventilated neonates, optimised for blood sampling requirements. Results of the pilot study were used to refine existing models and improve the design of a follow-up trial.
Methods
A clinical trial protocol to assess the pharmacokinetics of dexmedetomidine in a pilot group of ventilated neonates was approved by the central ethics committee. Informed consent was provided by each participating infant’s parent or legal guardian. The research was conducted in accordance with the guidelines of the Declaration of Helsinki.
The overall workflow in this study is presented in 'Fig. 1. In brief, clinical trial requirements and procedures were based upon practical limitations set out by the ethics committee. Full details on the inclusion/exclusion criteria, drug administration, venous sampling procedures and bioanalytical method can be found in the supplemental file. The initial dose was determined by extrapolation of a previously published non-linear mixed effects model describing the pharmacokinetics of dexmedetomidine in older children [12]. Thereafter, this model was used to find optimal sampling times using ED-optimality (i.e. the maximisation of the expectation (E) of the determinant (D) of the information matrix), as implemented in PopED® v1.20 [29]. For comparison, empirical sampling times were also used based on general pharmacokinetic principles. Accuracy and precision of parameter estimation based on the optimised and empirical sampling times were determined by simulating individual concentration-time profiles and subsequent re-estimating the pharmacokinetic model parameters using the simulated data. Simulation scenarios included the evaluation of the impact of protocol deviations such as missed samples. The optimised sampling schedule was implemented and executed in a pilot trial of 6 patients with PMA between 34 and 44 weeks and a minimum weight of 2 kg. Data from this pilot was used for model evaluation and subsequent model refinement. The refined model was then validated against data from an extended population of 10 neonates. Simulations and estimations were all performed in NONMEM® v7.3 [30]. Pre- and post-processing of data and statistical and graphical analysis were performed in R® v2.10 [31].

Steps undertaken during the trial design optimisation, execution and evaluation. The dashed arrow represents the approach of applying optimal design without prospective trial design evaluation by clinical trial simulations. PK, pharmacokinetics
Initial dose selection
The rationale for the first dose in neonates with PMA between 34 and 44 weeks was based on an extrapolation and simulation analysis using a previously published population pharmacokinetic model by Potts et al. (n = 95 critically ill infants and children) [12]. In this model, all structural parameters were allometrically scaled using a fixed exponent of 0.75 on clearance parameters (CL, Q) and 1 on volumes of distribution (V1, V2) (Eqs. 1–4). A sigmoidal function was used to describe maturation of clearance, including a maturation half-life (TM50) which represents the postmenstrual age (PMA) at which maturation has reached 50% of the adult value, and an exponent (Hill factor, N) describing the steepness of the maturation curve (Eq. 1). The model also includes a factor that reflects the change in clearance in postoperative cardiac patients, Finf, which is not applicable to this work.
where CLstd, Vstd1, Qstd and Vstd2 are the standardised or typical population parameter values, CL = clearance, V1 = central volume of distribution, V2 = peripheral volume of distribution, Q = intercompartmental clearance, PMA = postmenstrual age, TM50 = maturation half-life, WT is body weight and Finf was set to 1 as no cardiac surgery patients were to be included in our study.
Interindividual variability (η) was included on all structural parameters (CL, V1, V2, Q) as a random, normally distributed variable (η) with variance Ω2 (Eq. 5). Residual variability was described by a random, normally distributed variable (ε) with variance σ2 (Eq. 6).
Potts et al. used their model to simulate dexmedetomidine concentrations based on doses previously reported in literature, leading them to conclude the target concentration range of 0.4–0.8 μg/L is safe and efficacious. In the current study, we have assumed that the relationship between exposure and response to dexmedetomidine was similar in the neonatal population as in the older paediatric population [32]. This target therapeutic optimum of 0.6 μg/L, with a safety measure of maintaining plasma concentrations below 1 μg/L at all times in at least 95% of patients, was set for our simulations. The goal was to derive a single or simplified dosing regimen for all subjects, if possible, to minimise the chance of dosing errors, even when considering under- or overprediction of clearance of about 20% (Supplement Table S1). Simulated patients (n = 200) were expected to fall within typical distributions of weight (WT) as defined by the correlation between WT and PMA in the Fenton growth curves [33]. In addition, a 20% larger standard deviation (SD) of WT per PMA, as compared to those from the Fenton growth curves, was assumed to account for possible deviations of sick patients from the typical healthy population.
Optimised and empirical sampling times
PopED® was configured to find optimal sampling schedules to optimise for information with regard to the parameter of interest, i.e. clearance. A minimum of seven samples (excluding pre-dose sample) per patient was considered based on the required blood volume per sample, maximum sampling blood volume and number of patients to be included. An extra sample was allowed if weight-based volume limits for blood sampling were not exceeded, totalling 8 samples per individual. For patients weighing > 2.3 kg, this extra sample was taken around 24 h after stopping of the infusion, as it was assumed that most information regarding drug clearance could be obtained around that time point. Due to numerical difficulties in performing optimisation in PopED for the full range of expected weight and PMA values and the difficulties to restrict the demographic inclusion criteria of the patients for the pilot phase of the clinical trial, a set of demographic characteristics of PMA and WT were randomly selected from reasonable values based on the Fenton growth curves and used in PopED for sampling time optimisation, i.e. optimisation was performed for a hypothetical but pre-defined sample of 6 neonates (see supplemental file).
Uncertainty in the parameter distribution for clearance was set to 20% in order to account for potential differences between patient populations, i.e. extrapolation of the model parameters beyond its original population range. Introducing uncertainty on one or more parameters requires the use of ED-optimality, defined as the maximisation of the expected value of the determinant of the Fisher Information Matrix (FIM) [29]. PopED was set to use different search types, including random, stochastic gradient and line search with ED-optimisation for sampling schedule only.
Empirical sampling times were set to time points at 0.5, 1, 4, 8, 12, 24 and 36 h post start of the infusion along with a pre-dose sample at time point 0, resulting in a total of 8 samples. This sampling schedule was based on the accepted practice that most information on pharmacokinetic parameters can be obtained during the start of the infusion and during the elimination phase.
Simulation scenarios
To determine the impact of sampling schedule optimisation on the accuracy and precision of parameter estimates, multiple trial scenarios (1a-d, 2) were considered (Supplement Table S2). Trial scenario 1 was based on the sampling scheme derived from PopED® and compared to trial scenario 2, based on the empirical sampling schedule described earlier. During trial scenario evaluation (1a-d), attention was given to the impact of potential protocol deviations during the clinical study, such as missing samples or failure in patient recruitment. First, one or two optimised blood samples per subject were removed at random to investigate the impact of total number of samples per patient on the final parameter estimates. Subsequently, one out of six subjects was excluded at random from the optimised sampling scenario.
For each scenario, dexmedetomidine plasma concentrations were simulated at the optimised or empirical sampling times using the population pharmacokinetic model. The simulated sparse sampling profiles were subsequently used to re-estimate model parameters. Results were compared to the original parameter values used for simulations. The performance of each scenario in terms of accuracy and precision of parameter estimates was determined by calculating the normalised root mean square error (NRMSE, Eq. 7) and relative error (RE, Eq. 8), respectively. An error larger than 30% was deemed unacceptable for either metrics.
where x = obtained parameter estimates from a given scenario, tv = true parameter value from Potts et al., n = number of replicates for x.
Pharmacokinetic modelling of pilot data, dose refinement and external validation
To assess the adequacy of the model-based approach for this paediatric subpopulation, the pilot trial data were initially analysed using individual concentrations predicted by post hoc estimates of individual parameters within the distributions set by Potts et al. [12]. If bias was identified in model predictions, model parameters were then re-estimated to determine which parameters were affected. In that case, parameters assumed to be stable across populations were to be fixed with the remaining parameter(s) estimated from the pilot data.
Model performance was assessed by goodness-of-fit plots of population predictions (PRED) and individual predictions (IPRED) vs. observed concentrations (DV) and conditional weighted residuals (CWRES) vs. time and PRED. A visual predictive check (VPC) was performed by simulating 1000 concentration vs. time profiles for each individual and plotting the resulting 95% prediction interval of concentrations over time, along with the observed concentrations. No separate covariate analysis was attempted as the data were considered too sparse to identify additional covariate effects.
External validation was performed by evaluating the adjusted model based on goodness of fit plots and visual and numerical predictive checks (N = 1000) against a cohort of 10 neonates included in an extension of our pilot trial. In this extension, a loading dose was used, and the continuous infusion was allowed to be titrated according to apparent clinical efficacy. This step involved no minimisation or re-estimation of the model parameters except for the calculation of the post hoc individual estimates. Both loading dose and initial continuous infusion rate were based on the pharmacokinetic parameters obtained in the initial pilot trial of 6 neonates, under the assumption that target steady-state concentrations were efficacious (see the “Initial dose selection” section).
Results
Initial dosing regimen
Simulations revealed that a continuous infusion of 0.3 μg/kg/h over 24 h should result in adequate exposure in most of the population within 4 h, while ensuring a low probability of overexposure in the pilot group (Fig. 2). Using this dosing regimen, concentrations should reach steady-state conditions after approximately 10 h, with the median concentrations just below 0.6 μg/L, 95% of the population achieving concentrations below 1.0 μg/L and 83% of the population reaching concentrations above 0.4 μg/L. More extensive coverage on our dosing simulations is found in the online supplement (Fig. S1).

Predicted concentrations over time after continuous infusion of 0.3 μg kg−1 h−1 dexmedetomidine over 24 h in neonates with post-menstrual age (PMA) in the range between 34 and 44 weeks. Red line depicts the median predicted concentration, whereas the dashed blue line represents the 95% prediction interval. Blue shaded area and dashed red line represent the therapeutic window and therapeutic target concentration, respectively
Optimisation of trial design
The optimisation of the sampling schedule using ED-optimality was computer intensive, with nearly a 3-h computational time per scenario. Increases in optimisation complexity such as optimising across all weight and PMA ranges resulted in extensive run-times (longer than a week) and had to be abandoned. Sampling times (Table 1) at 15 min after start of infusion was found to be informative for all subjects irrespective of age or weight, whereas samples at 0.5 and 48 h after start of the infusion, equal to 24 h after stopping infusion, were identified as highly informative for most subjects. No practical limitations were identified which precluded the implementation of the optimised sampling times, e.g. consecutive sample collection within less than a 10-min interval.
Based on NRMSE and RE plots, scenario 1 was deemed to perform equally well in the case of a loss of 1 subject (i.e. 5 instead of 6 subjects recruited) or 1 sample for each subject (i.e. 5 instead of 6 samples collected for each subject). Further details are presented in the online supplement Figs. S2 and S3. Figure 3 shows the median and 95% confidence interval of NRMSE and RE, revealing that optimised scenario 1 was superior to empirical scenario 2 in precision (median NRMSE of CLstd 229.1% vs. 8610.2%, N 9269.4% vs. 7970.9%, TM50 109.0% vs. 62,029.9%, V1std 49.6% vs. 99.7%, Qstd 267.8% vs. 553.4%, V2std 113.5% vs. 112.4%) and showed similar accuracy (median RE of CLstd 42.8% vs. 10.0%, N 346.2% vs. 38.8%, TM50 24.5% vs. 20.0%, V1std 3.1% vs. 10.5%, Qstd 8.6% vs. 14.1%, V2std 4.9% vs. 2.2%) for most population parameters. The parameter of interest (CLstd) was re-estimated with acceptable accuracy and precision in scenario 1, while precision in scenario 2 was much lower (Table 2). The exponent of the maturation function (Hill factor, N) proved to be consistently imprecise in both scenarios, but more accurate in scenario 2.

Accuracy as normalised root mean square errors (NRMSEs) and precision as relative errors (REs) of parameters for scenarios 1 (left panels a and c) and 2 (right panels b and d). Red circle: median; black lines: 95% confidence interval. A NRMSE and RE of 0 signifies perfect accuracy and precision. Red dashed lines demarcate the 30% acceptable level for both NRMSE and RE. Parameters: maturation half-life (TM50), clearance (CL), Hill coefficient of the maturation function (N), central volume (V1), inter-compartmental clearance (Q), peripheral volume (V2)
Pilot clinical trial
A total of six patients were included, three males and three females, all Caucasian, with weights ranging 2.25–4.1 kg (birth weight 1.9–4.3 kg), PMA 34–44 weeks, GA 34–40 weeks and PNA 0–23 days. Three patients were admitted for respiratory distress syndrome and one for hernia diaphragmatica, oesophageal atresia repair and respiratory syncytial virus infection each. One adverse event of mild bradycardia (< 96 beats per minute) was reported, 17 h after the start of the continuous infusion. According to the protocol, the dexmedetomidine infusion was halved and the bradycardia episode was resolved. All but one patient needed fentanyl (range, 0.5–3 μg/kg/h) as rescue analgesic (83%). In total, 45 samples were collected from these patients at times as reported in Table 1. One patient was not sampled pre-dose due to a catheter flushing error, leading to a shift in subsequent sampling times. During PK analysis, the actual sampling times relative to the dose were used instead of the planned sampling times. Another patient missed one sample because the arterial catheter was removed. All other samples were taken within a 10-min window of the optimal sampling schedule.
Model evaluation and external validation
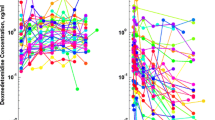
When evaluating the predictive performance of the population PK model without estimating parameter values, predicted concentrations based on post-hoc individual parameter estimates did not match up in three out of six patients (data not shown). No explanatory covariate or population characteristic could be identified for this discrepancy. Some trends could be observed in CWRES vs. time and PRED, although with such sparse data, these trends could be an artefact or random noise. Nevertheless, variability in drug disposition was captured adequately by the model as shown by the visual predictive check. However, the model over-predicted dexmedetomidine concentrations consistently in this group of mechanically ventilated neonates between 34 and 44 weeks (Fig. S4), who showed higher clearance values of roughly 20% on average, as compared to the predicted values obtained by extrapolation [12, 34].
Due to the bias in population predictions, it was decided that the model required a refinement for this younger population. Subsequent fitting procedures showed that the data did not allow full parameter estimation and thus parameters had to be fixed. CLstd was estimated in the original model taking into account a large range of weights, but their maturation function was estimated on a population all with PMA above the TM50. Thus, it seemed plausible to re-estimate the maturation to account for the difference in clearance in this age group. Parameters were therefore re-estimated using CLstd fixed at the previously reported values [12]. This approach improved the goodness-of-fit, with predictions much closer to the observed data (Fig. 4). Trends in CWRES over time and PRED were also improved (Fig. 4). The visual predictive check (Fig. 5) shows that the observations were mostly within the expected 95% confidence interval. Apart from the change in maturation parameters, the peripheral volume seemed to be significantly increased in these patients, possibly due to their disease status, although with such small sample size the interpretation of the source for such differences is limited (Table 3).

Goodness of fit plots for the refined model. PRED, population predictions. IPRED, individual predictions. CWRES, conditional weighted residuals. Concentrations are shown in μg/L. Time after dose is in hours. Blue dots depict the observed data points. Red line is a trend line

Individual visual predictive check for the adjusted model. Blue dots and blue line depict the observed concentrations. Blue shaded area represents the 95% prediction interval of simulations using the adjusted model. Red dashed lines show the 95% prediction interval from simulations using the model by Greenberg et al. [13]
For external validation of the refined model, an additional 11 neonates were included, with mean PMA 36.2 weeks (range 34–40, SD 2.1) and mean weight 2.8 kg (range 2.2–3.8, SD 0.6). Model population predictions versus observations showed a bias towards overpredictions, pointing to an underpredicted clearance (see Fig. S5 in supplement). The mean clearance in this patient population was estimated to be 26.6% higher than that observed in older neonates.
Discussion
ED-optimality in neonatal and paediatric research
Despite the increase in the uptake of optimal design in recent years, its use in paediatric clinical research for is rather limited [35, 36]. Here we aimed to establish the dose rationale for dexmedetomidine in mechanically ventilated neonates using optimality principles in conjunction with a population pharmacokinetic modelling approach.
In contrast to empirical dose selection and sampling schemes, a practice which often leads to inaccurate conclusions about the magnitude and relevance of pharmacokinetic covariate factors, modelling and simulation tools have been advocated to mitigate many of the issues imposed by empirical protocol designs [37, 38]. In fact, some examples are available on the successful use of adult pharmacokinetic data as a basis for modelling and extrapolation to neonates [34, 39]. Nevertheless, it has been shown by Wang et al. that the use of prior pharmacokinetic data from children younger than 2 years of age to support model parameter estimation resulted in lower prediction bias, but paediatric data are not always available during early clinical development.
Optimisation procedures were limited to sampling schedule based on a minimal number of seven samples per subject taking into account the clinical context and practical limitations. Most informative samples were found during the accumulation phase to steady-state and after the end of infusion, corresponding to time points at which information regarding parameters for distribution and clearance, respectively, is more prevalent. It should be noted, however, that focus was given primarily to the optimisation of the estimates of clearance, as it is the parameter that determines the overall systemic exposure.
The implementation of a comprehensive set of simulation scenarios allowed the identification of weak spots in trial design, providing the investigators with the opportunity to mitigate eventual risks and explore alternative approaches. Our analysis also revealed that the loss of 1 or 2 samples per subject and failure to recruit one subject would still lead to similar parameter estimates based on the optimised sampling schedule. On the other hand, it became evident that incorrect dosing information could have been derived from an empirical protocol design with restricted number of patients and sparse sampling. In this sense, the approach proposed here supplants previous investigations in which optimised designs are executed without an additional evaluation step for protocol deviations. It also highlights the potential limitations of data extrapolation when patient population characteristics differ from those used in the development of a pharmacokinetic model [40, 41].
We also had to cope with some limitations in computational complexity, which may have led to less-than-optimal sampling time points. Given the possibility to estimate individual clearances in the population accurately, it was anticipated that such conditions would have a significant impact on the proposed dosing recommendations. These limitations may be fully overcome by evolving technologies, which provide increased computer processing power.
Pharmacokinetics and dosing recommendations
Dexmedetomidine exposure as witnessed in the pilot study was largely in line with that expected from the model extrapolations. However, evidence arising from the collected data showed a bias in model predictions for the initial pilot and external validation cohorts, which prompted us to refine the model to improve accuracy and ensure suitable dosing recommendations for prospective patients.
As the cohort size in neonatal clinical pharmacology studies is usually small, identifying covariate effects due to differences in baseline demographic or clinical characteristics is challenging. Hence, the characterisation of potential sources of variability or bias may not always be feasible. In this pilot study, baseline characteristics, such as GA or PNA, did not seem to explain the bias. Given the heterogeneity in this patient population, it cannot be excluded that interindividual differences in disease status are likely to have contributed more to the observed pharmacokinetic differences than baseline demographic factors. It is possible that future investigations in larger study populations will elucidate the relevance of age-related covariates or other baseline characteristics that determine differences in drug disposition in pre-term neonates.
This example highlights therefore the importance of the learning-confirming paradigm when applying extrapolation concepts, including flexible protocol procedures to ensure appropriate data are generated.
From a modelling perspective, the bias in the observed concentrations indicated a clear discrepancy in the estimates associated with the parameters describing the maturation processes, possibly related to subjects being pre-term or because of the underlying clinical condition. Consequently, we have re-estimated the maturation parameters, while fixing the portion of clearance predicted by weight to its value, as described by Potts et al. The original publication was based on data from 95 children between the age of 1 week and 14 years but included limited data from children below or around TM50. The inclusion of patients with low PMA extended the information regarding the maturation process. The re-estimation of the parameters of the maturation function allowed accurate prediction of dexmedetomidine exposure in this group of patients and was deemed a necessary step to ensure accurate dose selection in even younger neonates, planned to be enrolled at a later stage of the trial (minimum PMA of 28 weeks).
In order to further investigate whether prematurity and maturation processes were associated with the observed differences in pre-term neonates, we have attempted to analyse the data using the pharmacokinetic model proposed by Greenberg et al. [13]. We performed simulations using their model along with the protocol design (i.e. dosing regimen, covariates and time points) in the pilot study. The 95% prediction intervals from these simulations were included in the individual VPC plots (Fig. 5). According to Greenberg et al., the pharmacokinetics of dexmedetomidine in pre-term and term neonates can be described by a one-compartment model, including a standard allometric function to correlate clearance and volume to changes in body weight and a slightly different maturation function based on PMA. These simulations show that their model predicts concentrations during the upswing phase as well as our two-compartment model, whereas concentrations during the later elimination phase are under-predicted. Underprediction of drug levels during the elimination phase may have consequences for the assessment of prolonged dexmedetomidine (side-)effects when lowering the infusion rate or stopping the infusion.
Even though the current investigation focused on the pharmacokinetics and dose rationale for dexmedetomidine, an initial assessment can be made of the efficacy and safety findings in our trial as compared to those reported by Chrysostomou et al. [42]. In their study, additional sedation was required only in 10% of the population, as compared to 83% in ours. This difference in efficacy may be due to the fact that Chrysostomou et al. used a loading dose, which may have yielded efficacious concentrations much sooner after the start of the infusion. However, one cannot exclude potential differences in the sensitivity of clinical scales, as they have measured N-PASS, instead of Comfort-neo score and Numeric Rating Scale. With regard to the safety, one patient (17%) in our trial showed an episode of bradycardia, which was promptly resolved by reducing the infusion rate. A similar event was reported by Chrysostomou et al. (12%), which reflects the known secondary pharmacological effects of dexmedetomidine.
This research relied on a set of assumptions and as such has some limitations. First, we have focused on the pharmacokinetic characteristics of dexmedetomidine in neonates as the basis for dose selection. Ideally, one might consider exploring the dose-exposure and exposure-response relationships to define an appropriate neonatal dose or dose range. On the other hand, evaluation of these relationships in neonates without the confounding of co-medication, rescue medication and other relevant clinical procedures is not feasible or ethical. We have therefore assumed comparable PKPD relationships across the different age groups [43, 44]. Second, we only compared the utility of different optimal and empirical sampling scenarios. Optimality criteria can be applied to other aspects of a protocol, such as patient numbers and dose level. However, a comprehensive evaluation of all protocol variables was beyond the scope of our analysis, as most of these variables were predefined by ethics committee restrictions. Due to time constraints and limited computational power at the time of performing this study, the number of simulations was limited to 100 replicates per sampling scenario. With ever-increasing computing power, these limitations can be overcome and more elaborate optimisation scenarios can be evaluated within the time allotted for trial design. Lastly, we recognise that despite the number of samples and patients available from the pilot phase, the data was not sufficient to re-estimate all model parameters. Inclusion of patients from the follow-up phase of the trial will allow further improvement.
In conclusion, our study provides the dose rationale for pre-term and term neonates neonatal patients, taking into consideration ethics committee restrictions. The use of simulation scenarios showed the relevance of optimal sampling for the characterisation of the pharmacokinetics of dexmedetomidine and enabled the selection of the best candidate trial design, which was then implemented, executed and analysed successfully. The large differences between optimal and non-optimal sampling schedules highlight the urgency for the use of flexible protocol designs in conjunction with model-based approaches in mainstream neonatal trials. Due to the higher clearance of dexmedetomidine, the dose was increased to 0.4 μg/kg/h in the follow-up phase of this trial. As shown, results from the follow-up phase revealed an even further increase in clearance in the enrolled subjects. While the perceived benefit-risk balance should guide protocol procedures and restrictions, our work shows that such an empirical approach may lead to inappropriate dose rationale. Without the intermediate modelling step, these vulnerable patients would have been under-dosed by about 45%.
References
Thewissen L, Allegaert K (2011) Analgosedation in neonates: do we still need additional tools after 30 years of clinical research? Arch Dis Child Educ Pract 96(3):112–118
Gertler R, Brown HC, Mitchell DH, Silvius EN (2001) Dexmedetomidine: a novel sedative-analgesic agent. Proc Baylor Univ Med Cent 14(1):13–21
Rolle A, Paredes S, Cortı LI, Anderson BJ, Quezada N, Solari S et al (2018) Dexmedetomidine metabolic clearance is not affected by fat mass in obese patients. Br J Anaesth 120(5):969–977
Li A, Yuen VM, Sheng Y, Standing JF, Kwok PCL, Leung MKM et al (2018) Pharmacokinetic and pharmacodynamic study of intranasal and intravenous dexmedetomidine. Br J Anaesth 120(5):960–968
Smuszkiewicz P, Wiczling P, Ber J, Warzybok J, Małkiewicz T, Matysiak J, Klupczyńska A, Trojanowska I, Kokot Z, Grześkowiak E, Krzyzanski W, Bienert A (2018) Pharmacokinetics of dexmedetomidine during analgosedation in ICU patients. J Pharmacokinet Pharmacodyn 45(2):277–284
Zhong W, Zhang Y, Zhang MZ, Huang XH, Li Y, Li R, Liu QW (2018) Pharmacokinetics of dexmedetomidine administered to patients with end-stage renal failure and secondary hyperparathyroidism undergoing general anaesthesia. J Clin Pharm Ther 43(3):414–421
Yoo H, Iirola T, Vilo S, Manner T, Aantaa R (2015) Mechanism-based population pharmacokinetic and pharmacodynamic modeling of intravenous and intranasal dexmedetomidine in healthy subjects. Eur J Clin Pharmacol 71(10):1197–1207
Damian MA, Hammer GB, Elkomy MH, Frymoyer A, Drover DR, Su F (2018) Pharmacokinetics of dexmedetomidine in infants and children after orthotopic liver transplantation. Anesth Analg. https://doi.org/10.1213/ANE.0000000000003761
Perez-Guille M-G, Toledo-Lopez A, Rivera-Espinosa L, Alemon-medina R, Murata C, Lares-asseff I et al (2018) Population pharmacokinetics and pharmacodynamics of dexmedetomidine in children undergoing ambulatory surgery. Pediatr Anesth 127(3):716–723
Wiczling P, Bartkowska-sniatkowska A, Szerkus O, Siluk D, Rosada-kurasin J, Warzybok J et al (2016) The pharmacokinetics of dexmedetomidine during long-term infusion in critically ill pediatric patients. A Bayesian approach with informative priors. J Pharmacokinet Pharmacodyn 43:315–324
Liu HC, Lian QQ, Wu FF, Wang CY, Sun W, Zheng LD, Schüttler J, Ihmsen H (2017) Population pharmacokinetics of dexmedetomidine after short intravenous infusion in Chinese children. Eur J Drug Metab Pharmacokinet 42(2):201–211
Potts AL, Anderson BJ, Warman GR, Lerman J, Diaz SM, Vilo S (2009) Dexmedetomidine pharmacokinetics in pediatric intensive care--a pooled analysis. Paediatr Anaesth 19(11):1119–1129
Greenberg RG, Wu H, Laughon M, Capparelli E, Rowe S, Zimmerman KO, Smith PB, Cohen-Wolkowiez M (2017) Population pharmacokinetics of dexmedetomidine in infants. J Clin Pharmacol 57(9):1174–1182
Talke P, Anderson B (2018) Pharmacokinetics and pharmacodynamics of dexmedetomidine-induced vasoconstriction in healthy volunteers. Br J Clin Pharmacol 84(6):1364–1372
Colin PJ, Hannivoort LN, Eleveld DJ, Reyntjens KMEM, Absalom AR, Vereecke HEM, Struys MMRF (2017) Dexmedetomidine pharmacokinetic–pharmacodynamic modelling in healthy volunteers: 1. Influence of arousal on bispectral index and sedation. Br J Anaesth 119:200–210
Kuang Y, Xiang Y, Guo C-X, Zhang R, Yang G-P, Hou G et al (2016) Population pharmacokinetics study of dexmedetomidine in Chinese adult patients during spinal anesthesia. Int J Clin Pharmacol Ther 54:200–207
Weerink MAS, Struys MMRF, Hannivoort LN, Barends CRM, Absalom AR, Colin P (2017) Clinical pharmacokinetics and pharmacodynamics of dexmedetomidine. Clin Pharmacokinet 56(8):893–913
Vázquez-Reta JA, Jiménez Ferrer MC, Colunga-Sánchez A, Pizarro-Chávez S, Vázquez-Guerrero AL, Vázquez-Guerrero AR (2011) Midazolam versus dexmedetomidine for sedation for upper gastrointestinal endoscopy. Rev Gastroenterol Mex 76(1):13–18
Riker RR, Shehabi Y, Bokesch PM, Ceraso D, Wisemandle W, Koura F et al (2009) Dexmedetomidine vs midazolam for sedation of critically ill patients: a randomized trial. JAMA. 301(5):489–499
Pandharipande PP, Pun BT, Herr DL, Maze M, Girard TD, Miller RR, Shintani AK, Thompson JL, Jackson JC, Deppen SA, Stiles RA, Dittus RS, Bernard GR, Ely EW (2007) Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients: the MENDS randomized controlled trial. JAMA. 298(22):2644–2653
Aziz NA, Chue MC, Yong CY, Hassan Y, Awaisu A, Hassan J et al (2011) Efficacy and safety of dexmedetomidine versus morphine in post-operative cardiac surgery patients. Int J Clin Pharm 33(2):150–154
Manolis E, Osman TE, Herold R, Koenig F, Tomasi P, Vamvakas S, Raymond AS (2011) Role of modeling and simulation in pediatric investigation plans. Paediatr Anaesth 21(3):214–221
Roberts JK, Stockmann C, Balch A, Yu T, Ward RM, Spigarelli MG, Sherwin CMT (2015) Optimal design in pediatric pharmacokinetic and pharmacodynamic clinical studies. Pediatr Anesth 25(3):222–230
Bellanti F, Di Iorio VL, Danhof M, Della Pasqua O (2016) Sampling optimization in pharmacokinetic bridging studies: example of the use of deferiprone in children with beta-thalassemia. J Clin Pharmacol 56(9):1094–1103
Barrett JS, Bishai R, Bucci-Rechtweg C, Cheung A, Corriol-Rohou S, Haertter S, James A, Kovacs SJ, Liu J, Potempa D, Strougo A, Vanevski K, for the IQ consortium - CPLG Pediatric Working Group (2018) Challenges and opportunities in the development of medical therapies for pediatric populations and the role of extrapolation. Clin Pharmacol Ther 103(3):419–433
Kimko H, Duffull S (2003) Simulation for designing clinical trials : a pharmacokinetic-pharmacodynamic modeling perspective. Marcel Dekker, New York
Cella M, Danhof M, Della Pasqua O (2012) Adaptive trials in paediatric development: dealing with heterogeneity and uncertainty in pharmacokinetic differences in children. Br J Clin Pharmacol 74(2):346–353
Sheiner LB (1997) Learning versus confirming in clinical drug development. Clin Pharmacol Ther 61(3):275–291
Foracchia M, Hooker A, Vicini P, Ruggeri A (2004) POPED, a software for optimal experiment design in population kinetics. Comput Methods Prog Biomed 74(1):29–46
Beal SL, Sheiner LB, Boeckmann A, Bauer RJ (2011) NONMEM user’s guide. Icon Development Solutions, Ellicott City
R Core Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria (accessible at URL: http://www.r-project.org)
Perala M, Hirvonen H, Kalimo H, Ala-Uotila S, Regan JW, Akerman KEO et al (1992) Differential expression of two alpha 2-adrenergic receptor subtype mRNAs in human tissue. Mol Brain Res 16:57–63
Fenton TR (2003) A new growth chart for pre-term babies: Babson and Benda’s chart updated with recent data and a new format. BMC Pediatr 3:13
Wang J, Edginton AN, Avant D, Burckart GJ (2015) Predicting neonatal pharmacokinetics from prior data using population pharmacokinetic modeling. J Clin Pharmacol 55(10):1175–1183
Wang J, Avant D, Green D, Seo S, Fisher J, Mulberg A, McCune S, Burckart GJ (2015) A survey of neonatal pharmacokinetic and pharmacodynamic studies in pediatric drug development. Clin Pharmacol Ther 98(3):328–335
Davis JM, Connor EM, Wood AJ (2012) The need for rigorous evidence on medication use in pre-term infants: is it time for a neonatal rule? JAMA. 308(14):1435–1436
Harnisch L, Shepard T, Pons G, Della Pasqua O (2013) Modeling and simulation as a tool to bridge efficacy and safety data in special populations. CPT Pharmacometrics Syst Pharmacol 2(2):e28
Bellanti F, Della Pasqua O (2011) Modelling and simulation as research tools in paediatric drug development. Eur J Clin Pharmacol 67(Suppl 1):75–86
Zhao W, Le Guellec C, Benjamin DK, Hope WW, Bourgeois T, Watt KM et al (2014) First dose in neonates: are juvenile mice, adults and in vitro–in silico data predictive of neonatal pharmacokinetics of fluconazole. Clin Pharmacokinet 53(11):1005–1018
Cella M, Zhao W, Jacqz-Aigrain E, Burger D, Danhof M, Della PO (2011) Paediatric drug development: are population models predictive of pharmacokinetics across paediatric populations? Br J Clin Pharmacol 72(3):454–464
Cella M, Knibbe C, de Wildt SN, Van Gerven J, Danhof M, Della Pasqua O (2012) Scaling of pharmacokinetics across paediatric populations: the lack of interpolative power of allometric models. Br J Clin Pharmacol 74(3):525–535
Chrysostomou C, Schulman SR, Castellanos MH, Cofer BE, Mitra S, Garcia M et al (2014) A phase II/III, multicenter, safety, efficacy, and pharmacokinetic study of dexmedetomidine in pre-term and term neonates. J Pediatr 164(2):276–282.e3
Zhao W, Piana C, Danhof M, Burger D, Della Pasqua O, Jacqz-Aigrain E (2013) Population pharmacokinetics of abacavir in infants, toddlers and children. Br J Clin Pharmacol 75(6):1525–1535
Bellanti F, Del Vecchio GC, Putti MC, Cosmi C, Fotzi I, Bakshi SD et al (2016) Model-based optimisation of deferoxamine chelation therapy. Pharm Res 33(2):498–509
Acknowledgements
We are grateful to Vincenzo di Iorio for his support with the implementation of the optimal design scenarios in PopED® v1.20.
Funding
This work was supported by the Clinical Research Fund Ghent University Hospital, Ghent Belgium (grant number WR/1492/APO/001 to P. De Cock), the Agency for Innovation by Science and Technology, Flanders, Belgium (SAFEPEDRUG project; grant number IWT/SBO/130033 to P. De Cock, K. Allegaert, J. Vande Walle, P. De Paepe and A. Smits) and an unrestricted academic grant from Orion Pharma, Finland (to Ghent University Hospital, Belgium). Sven van Dijkman’s contribution to this research collaboration was possible thanks to the funding from the European Union Seventh Framework Programme FP7/2007-2013 under grant agreement no. 261060.
Author information
Authors and Affiliations
Contributions
Manuscript writing: SVD, PDC, ODP, PDP, JVW.
Trial design: PDC, SVD, PDP, ODP.
Performing clinical trial: PDC, WD, KS, AS, KA.
Data analysis: SVD, PDC, ODP
Corresponding author
Ethics declarations
The pilot clinical study described here was conducted in accordance with International Conference on Harmonization Guideline for Good Clinical Practice and Declaration of Helsinki and was approved in advance by the Ethics Committees of the participating hospitals (Ghent University Hospital, AZ Sint Jan Brugge-Oostende AV and University Hospital Leuven).
Parents or legal guardians, if applicable, have provided written informed consent before enrolment of the new-borns in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(PDF 311 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
van Dijkman, S.C., De Cock, P.A.J.G., Smets, K. et al. Dose rationale and pharmacokinetics of dexmedetomidine in mechanically ventilated new-borns: impact of design optimisation. Eur J Clin Pharmacol 75, 1393–1404 (2019). https://doi.org/10.1007/s00228-019-02708-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00228-019-02708-y