
Abstract
Osteoporosis is a skeletal disorder with enhanced bone fragility, usually affecting the elderly. It is very rare in children and young adults and the definition is not only based on a low BMD (a Z-score < − 2.0 in growing children and a Z-score ≤ − 2.0 or a T-score ≤ − 2.5 in young adults) but also on the occurrence of fragility fractures and/or the existence of underlying chronic diseases or secondary factors such as use of glucocorticoids. In the absence of a known chronic disease, fragility fractures and low BMD should prompt extensive screening for secondary causes, which can be found in up to 90% of cases. When fragility fractures occur in childhood or young adulthood without an evident secondary cause, investigations should explore the possibility of an underlying monogenetic bone disease, where bone fragility is caused by a single variant in a gene that has a major role in the skeleton. Several monogenic forms relate to type I collagen, but other forms also exist. Loss-of-function variants in LRP5 and WNT1 may lead to early-onset osteoporosis. The X-chromosomal osteoporosis caused by PLS3 gene mutations affects especially males. Another recently discovered form relates to disturbed sphingolipid metabolism due to SGMS2 mutations, underscoring the complexity of molecular pathology in monogenic early-onset osteoporosis. Management of young patients consists of treatment of secondary factors, optimizing lifestyle factors including calcium and vitamin D and physical exercise. Treatment with bone-active medication should be discussed on a personalized basis, considering the severity of osteoporosis and underlying disease versus the absence of evidence on anti-fracture efficacy and potential harmful effects in pregnancy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is a skeletal disorder mainly affecting elderly people and characterized by low bone mass and abnormal bone microarchitecture, resulting in enhanced skeletal fragility and increased risk of fractures [1]. The fractures with ensuing morbidity and mortality have significant personal and economic implications worldwide. Osteoporosis was previously considered an illness affecting mainly post-menopausal women, but primary and secondary osteoporosis have more recently emerged also as important pediatric disorders [2, 3]. The prevalence of osteoporosis in young persons is considered to be low but the true prevalence is unknown and dependent on the applied definition.
A low areal BMD on a DXA scan without fractures or without underlying diseases may not necessarily imply increased bone fragility and is usually associated with a low risk of fractures in the short term [14]. This situation is different in patients with a chronic disease that impacts bone health, such as inflammatory diseases [rheumatoid arthritis (RA), inflammatory bowel disease (IBD), chronic obstructive pulmonary disease (COPD)] and diseases related to poor nutrition or nutritional deficiencies [celiac disease, cystic fibrosis (CF), anorexia nervosa (AN)] and endocrine disorders (Cushing’s syndrome, hyperparathyroidism, hyperthyroidism, type 1 diabetes, hypogonadism) [3,4,5]. These can lead to bone fragility due to the underlying disease, disease-associated co-morbidities, like malnutrition, or due to the applied therapies. Such secondary disturbances in modeling and remodeling of the skeleton during growth will have persisting long-term consequences later in life with reduced peak bone mass and structural deterioration of bone [6,7,8]. The prevalence of osteoporosis, defined as a BMD Z-score below − 2.0, has recently been reported to be as high as 45% in adults with Cushing’s disease [9] and in young adults with cystic fibrosis [10].
In young adults presenting with fragility fractures and low BMD without known chronic diseases, an underlying secondary factor can often be identified, depending on the depth of investigations and the type of hospital setting. However, fragility fractures may also be a presenting symptom of a monogenic form of osteoporosis. Fractures presenting in childhood or early adulthood without an evident secondary cause should prompt investigations for an underlying monogenetic bone disease such as osteogenesis imperfecta (OI) and several recently discovered other genetic entities. The diagnosis of idiopathic osteoporosis should thus be reserved only to cases where secondary and known monogenic causes have been appropriately excluded.
In this narrative review we discuss the definition of early-onset osteoporosis and its most common non-genetic and genetic causes and present a plan of action for evaluation and treatment.
Definition of Early-Onset Osteoporosis
Skeletal mass increases rapidly during childhood and especially adolescence; 90% of peak bone mass is acquired by age 18 [8, 11, 12]. Peak bone mass has been regarded an important determinant of osteoporosis and fracture risk later in adulthood [11]. In adults, osteoporosis is defined as a BMD measured by dual energy X-ray absorptiometry (DXA) with a T-score below or equal to − 2.5. For persons younger than 50 years both T-scores and Z-scores are used. The International Society for Clinical Densitometry (ICSD) proposed a BMD Z-score below or equal to − 2.0 to define low BMD in those below 40 years [13]. The IOF also defines low BMD in persons below age 20 years as Z-scores below − 2.0 and as T-score of below − 2.5 in those 20 years and older in association with a chronic disease known to affect bone metabolism [14].
In children the diagnosis of osteoporosis is more complex and BMD measurement by DXA is significantly impacted by the patient’s height and timing of pubertal development. Fractures are also common in childhood, especially around the time of rapid growth prior and during puberty, and one needs to take into consideration the normal fracture pattern and prevalence before regarding fractures as a sign of osteoporosis. Therefore, the diagnosis is based not only on low BMD that has been appropriately adjusted for height and/or skeletal maturity but also requires a fracture history indicative of higher-than-normal bone fragility and knowledge on underlying diseases and secondary factors.
Table 1 shows the criteria for a DXA scan assessment and for the diagnosis of early-onset osteoporosis in children and adults. In children, recurrent fractures are often the result of deficient calcium intake or vitamin D deficiency [15] but also other acquired or genetic disorders with disturbed mineral homeostasis (e.g., hypophosphatemia, hyperparathyroidism, osteomalacia, hypophosphatasia) need to be considered [16, 17]. Importantly, several monogenic forms of early-onset osteoporosis exist, as will be discussed more in detail in this review. Pathological fractures may occur due to localized bone abnormalities such as Paget’s disease of bone, fibrous dysplasia or malignancy. These can be excluded by appropriate laboratory work-up and radiological examinations. Secondary causes of osteoporosis should be searched for, especially in young individuals with low BMD and fragility fractures and these investigations should contain a minimum set of laboratory investigations and be guided by the patient’s symptoms and previous medical history.
Early-Onset Osteoporosis as a Sequelae of Other Illness
Many chronic diseases of childhood and young adulthood can lead to low BMD. These include inflammatory diseases such as RA, IBD, COPD and diseases related to poor nutrition or nutritional deficiencies (anorexia nervosa, celiac disease, cystic fibrosis, vitamin D deficiency), endocrine disorders (Cushing’s syndrome, hyperthyroidism, hyperparathyroidism, type 1 diabetes mellitus, hypogonadism) and chronic infectious (HIV), renal, liver or neurological diseases. Table 2 lists major causes of secondary osteoporosis in the young. In the following paragraphs we will briefly discuss some of these conditions potentially leading to early-onset osteoporosis, including inflammatory diseases (RA, IBD and COPD), cancer, and anorexia nervosa, as well as osteoporosis related to glucocorticoid therapy. In addition, we summarize some recent findings in pregnancy and lactation-associated osteoporosis (PLAO).
Osteoporosis in Chronic Inflammatory Diseases
The etiology of fragility fractures and low BMD in chronic inflammatory diseases rheumatic disorders, chronic lung diseases, inflammatory bowel disease (Crohn’s disease, Ulcerative Colitis) is also multifactorial, including effects of the underlying disease, systemic inflammation, use of glucocorticoids, low body weight, malabsorption, low physical activity and delayed puberty and/or secondary amenorrhea. At a young age, the disease will lead to a decrease in peak bone mass while at older age there may be increase bone loss [18, 19]. The most important treatment is that of the underlying disease and supplementation of calcium and vitamin D. There are very few RCTs with osteoporosis medication on fracture outcome. A meta-analysis in inflammatory bowel disease including 13 RCTs en 925 men and women, of which only 10% was pre-menopausal, showed an increase in BMD and a decrease in fractures with bisphosphonates [20].
Osteoporosis in (Breast) Cancer
Breast cancer is the most frequent cancer in women and early diagnosis and improved treatment has resulted in recent years in increased survival with more side effects from cancer treatments including bone loss and fractures. Mechanisms for bone loss include hypogonadism through chemotherapy, the direct toxic effects of chemotherapy itself, endocrine therapy (gonadotropin releasing hormone analogs, aromatase inhibitors and tamoxifen) as well as the general effects of being ill, loss of body weight and decreased physical activity [21]. Treatment with tamoxifen prevents bone loss in postmenopausal women but is deleterious for bone health in premenopausal women, resulting in a 75% increased fracture risk [22]. In all pre- and postmenopausal women initiating aromatase inhibitor treatment and in premenopausal women initiating tamoxifen, fracture risk should be assessed and recommendations regarding exercise and calcium and vitamin D supplementation should be given. Guidelines for indications of starting bone-active medication have been published, mostly based on expert opinion and there are no clinical trials on fracture prevention in premenopausal women [21, 23].
Osteoporosis in Anorexia Nervosa (AN) and the Female Athlete Triad
Anorexia Nervosa (AN) is serious eating disorder affecting 0.3–3% of girls and young women, but also boys and men can be affected. Patients with AN have a reduced BMD and an increased risk of osteoporosis (up to 40%) and about 30% has a history of prevalent fractures [24, 25]. Bone loss results from low body fat mass and a decreased energy intake resulting in hypothalamic amenorrhea and hypogonadism with complex neuroendocrine hormone dysregulation (such as increases in ghrelin, cortisol, PYY and growth hormone (GH) with GH resistance, and decreases in levels of insulin, IGF1 and oxytocin) and changes in levels of adipokines (low leptin and high adiponectin levels) [26]. There may also be a potential negative influence on bone of increased bone marrow adiposity and preferential development of mesenchymal stem cells towards adipocytes instead of osteoblasts. AN has the highest impact on BMD at a young age when the insult to the bone happens during formation of peak bone mass. The prevalence of low BMD and increased risk of fractures is determined by age at diagnosis and menarche, duration of amenorrhea and BMI. Weight gain with regain of menses is the most important treatment goal for BMD gain but deficits often persist [26]. Other potential treatment options include (transdermal) estrogens, bisphosphonates and teriparatide, that have shown to increase BMD in small clinical trials, but no data on fracture prevention are available [27].
Young athletes who participate in intense athletic activities like running and ballet may have reduced energy intake, amenorrhea, and low BMD, collectively called the ‘Female Athlete Triad’. When these young females are not considered to have AN, this triad is often not recognized as a cause for fractures and low BMD. Female athletes often present with one or more of these triad components, and early intervention is essential. In a consensus statement from 2014 a set of recommendations was presented to provide clinical guidelines for screening, diagnosis, and treatment of the Female Athlete Triad [28].
Glucocorticoid-Induced Osteoporosis
Glucocorticoid-induced osteoporosis in children and young adults is usually seen in patients with immune-mediated diseases, such as rheumatic disorders, chronic lung diseases, inflammatory bowel disease (Crohn’s disease, ulcerative colitis), chronic liver and kidney diseases, skin diseases, and in organ transplantation, diseases that are in themselves also a cause of osteoporosis. The negative effects of the glucocorticoids on bone are multifactorial, including increased apoptosis of osteoblasts and osteocytes with decreased apoptosis of osteoclasts, negative effects on muscle function, sex-steroids and a decreased calcium absorption in the gut and decreased calcium re-absorption in the kidney. Despite the negative effects on bone health, they may also have some favorable effects on bone by controlling the activity of the underlying disease [29] although this has not been adequately studied in young adults and children. A clinically significant number of children with rheumatic disorders developed incident vertebral fractures in the 3 years after starting glucocorticoids (incidence rate 4.4 per 100 person-years) [30]. Almost half of the fractures were asymptomatic and thus would not have come to clinical attention in the absence of radiographic screening. Guidelines from the American College of Rheumatology in 2017 advise to perform clinical fracture risk assessment in all children and young adults within six months of starting glucocorticoid therapy and to perform a DXA scan in young adults below 40 years of age (but not in children) when there is a history of osteoporotic fractures or other significant risk factors for fracture [29]. In adults of 40 years or above they advise to use FRAX with glucocorticoid dose correction and BMD testing within six months of starting glucocorticoids [31].
Pregnancy and Lactation
During a normal pregnancy there is a temporary decrease in BMD with a stronger loss during breastfeeding. In the spine the loss of BMD is about 5–10% with a spontaneous recovery within 6–12 months. Several case reports have been published on the occurrence of “transient osteoporosis of the hip” (TOH) and of vertebral fractures during pregnancy and lactation (pregnancy and lactation-associated osteoporosis, PLAO). PLAO is a severe type of premenopausal osteoporosis which predominantly occurs in the last trimester of pregnancy or immediately postpartum. Almost 25% of patients with PLAO will sustain a subsequent fracture, and this fracture risk correlates with the number of fractures at the time of diagnosis [32]. Sometimes underlying secondary factors can be found. There is often a spontaneous improvement in BMD after delivery and cessation of breast feeding. Recent studies using bone biopsies suggest a possible defect in the functioning osteoblasts [33]. Pre-existing secondary causes of osteoporosis should always be ruled out and while some patients will improve spontaneously, others will need treatment with either antiresorptives or with anabolic treatment [19]. In some patients an underlying genetic predisposition may be identified, e.g., with pathogenic variants in LRP5, suggesting a pre-existing monogenetic form of osteoporosis with an exacerbation due to pregnancy, resulting in vertebral fractures [34]. PLAO may thus present as a rare presentation of early-onset osteoporosis that becomes apparent during the times of skeletal stress of pregnancy and lactation. After exclusion of secondary and genetic causes a persisting low BMD more than 6 months after cessation of pregnancy and lactation may indicate idiopathic osteoporosis.
Idiopathic Osteoporosis
This is a diagnosis per exclusionem when no underlying chronic disease or secondary factors can be found for fragility fractures associating with a low BMD. This condition is most likely multifactorial and should be differentiated from situations in which low BMD is present without fractures e.g., in constitutionally lean persons. Bone biopsies may show decreased bone formation in some patients [35]. Using high-resolution pQCT some similarities were found consistent with mild forms of OI with a reduction in volumetric BMD and changes in microstructure, however without changes in bone geometry [36]. Vertebral fractures are common in idiopathic male osteoporosis and have been associated with increased cortical porosity in iliac crest bone biopsies [35]. It is likely that a proportion of patients with idiopathic osteoporosis may have an underlying genetic cause.
It is important to bear in mind that in several instances so-called idiopathic osteoporosis has in fact a monogenic cause that can escape detection when only limited genetic testing is performed. Using a NGS panel in 123 young adults, rare or novel variants were found in 11 patients in the included candidate genes (COL1A1, WNT1, PLS3 and DKK1) as well as a high prevalence of known pathogenic variants in LRP5 in 22 patients [37]. Variants in LRP5 have previously been identified in children [38] and in adult males with idiopathic osteoporosis [18]. The diagnosis of idiopathic osteoporosis should thus be reserved only to cases where secondary and known monogenic causes have been appropriately excluded.
Genetic Determinants of Osteoporosis
During childhood, the skeleton undergoes rapid changes in both longitudinal growth and in bone modeling. Renewal of the bone tissue (remodeling) continues even after growth plates have fused and adult height has been reached. The remodeling process requires coordinated activity of osteoblasts, osteoclasts and osteocytes and integrity of the various signaling pathways that control the differentiation and function of these bone cells. In addition, adequate supply of minerals and normal hormonal control of mineral homeostasis are needed for appropriate bone mineralization [39].
Because of the complexity of the cellular networks in the skeleton, the genetic defects leading to skeletal fragility are numerous and variable in presentation. Monogenic low bone mass disorders can result e.g., from defects in osteoblastic bone formation, from increased osteoclastic bone resorption, or from abnormalities in the mineralization process. Osteogenesis Imperfecta (OI), although being a rare disease, is the most common inherited bone disease with low bone mass and increased fractures, often associated with some extra-skeletal features such as blue sclerae [40]. OI is caused by defects in type I collagen itself, or its posttranslational modification. In more than 90% of OI cases the gene defect involves one of the two genes (COL1A1 or COL1A2) encoding the two α-chains of type I collagen while the remaining cases show a very heterogeneous genetic background and various inheritance patterns [41, 42]. In the present article we have chosen to focus on other types of monogenic bone fragility that are not directly caused by defects in type I collagen and should be considered in differential diagnosis when evaluating a child or a young adult with early-onset osteoporosis.
Monogenic Bone Fragility Due to Impaired WNT-Signaling Activity
Genetic entities with defective WNT signaling have emerged as an important subgroup of monogenic bone fragility disorders. The spectrum of monogenic skeletal disorders directly related to the WNT signaling pathway is still increasing and includes disorders with both high and low bone mass [43]. Some genetic forms lead to severe skeletal fragility, impaired growth and deformities already in childhood and genetic evaluations are usually initiated in infancy or early childhood. However, others present only in adolescence or early adulthood with increased susceptibility to fractures without any significant extra-skeletal manifestations, height deficit, deformities, or laboratory abnormalities. For example, patients with biallelic LRP5 and WNT1 mutations present with severe skeletal fragility, growth impairment and deformities in early childhood [44, 45], while subjects with heterozygous mutations in these genes often have normal growth, lack deformities but sustain fractures and have low BMD during later childhood or in early adulthood [45,46,47].
LRP5
Biallelic rare variants in LRP5 cause the autosomal recessive osteoporosis–pseudoglioma syndrome (OPPG, MIM 259770), characterized by generalized childhood-onset osteoporosis and blindness [44]. Already in early studies on LRP5 and OPPG, and in many studies thereafter, it has been noticed that carriers of heterozygous rare LRP5 variants also have reduced bone mass but usually lack eye manifestations or may have a milder eye phenotype in the form of vitreoretinopathy [34, 44, 48]. Several studies have identified individuals with childhood or early adulthood onset symptomatic osteoporosis caused by rare heterozygous LRP5 variants [38, 47]. In addition, even common LRP5 single nucleotide variants have been linked to childhood bone mass accrual, childhood fractures, and peak bone mass in cohort studies and in genome-wide association studies on BMD and fractures [38, 49, 50].
Studies evaluating characteristics of autosomal dominant osteoporosis caused by rare heterozygous loss-of-function LRP5 variants usually include only a small number of affected individuals. A recent study on a large cohort (372 individuals) of subjects with early-onset osteoporosis identified rare LRP5 or LRP6 variants in 8.3% [47]. Detailed assessment of skeletal characteristics and treatment responses in those harboring a rare variant showed significant heterogeneity both in bone parameters and in efficacy of therapies. Analysis of bone metabolism revealed low bone formation markers in individuals carrying rare LRP5 or LRP6 variants, in line with decreased WNT signaling [47]. Another study evaluating the impact of two common LRP5 single nucleotide polymorphisms (rs4988300 and rs634008) on bone turnover markers in a cohort of 328 unrelated osteoporosis patients with or without fractures, found that the bone formation marker PINP levels and BMD were lower in patients with the GG genotype of rs4988300 and the TT genotype of rs634008 than in patients with the other genotypes [51]. However, no significant difference in b-CTX levels was observed between different genotypes.
In patients with osteoporosis-pseudoglioma syndrome due to biallelic LRP5 variants the response to bisphosphonate treatment is usually good [52, 53], but little is known about the treatment responses in those harboring heterozygous LRP5 variants. Studies have also explored whether common gene variants in LRP5 could affect response to bisphosphonate treatment, but although some variants associated with baseline BMD, no effect on treatment response was observed [54, 55]. Analysis of osteoanabolic treatment with teriparatide in two individuals with an LRP5 or LRP6 variant indicated acceleration of bone turnover during treatment [47].
Studies have suggested that LRP5 variants may also lead to altered insulin sensitivity, impaired glucose tolerance and hyperlipidemia [56,57,58] but the clinical relevance of this connection remains uncertain.
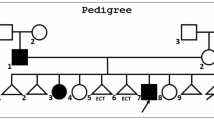
Functional studies have in some instances confirmed reduced WNT signaling by these LRP5 variants [59]. However, as rare variants in several genes, including LRP5, can be found in the general population that are predicted to be (likely) pathogenic or of undetermined significance it is not always easy to link these variants to the patient’s osteoporosis, especially when segregation analysis in the family is not possible due to lack of large pedigrees or when family members have low BMD due to other (non-genetic) causes [47]. Rare pathogenic LRP5 variants have also been described in patients with pregnancy- and lactation-associated osteoporosis (PLAO) [34].
WNT1
Several WNT ligands are expressed in bone tissue and regulate bone homeostasis and are hence relevant for osteoporosis pathogenesis [60]. Based on genome-wide association studies on BMD and fractures, WNT16 was discovered as an important ligand for WNT signaling in bone [61]. WNT16 variants associate with cortical bone thickness, BMD, and osteoporotic fracture risk and may also impact peak bone mass [62,63,64]. However, WNT16 variants have not been linked to monogenic osteoporosis, possibly implying that other WNT ligands have overlapping functions.
In 2013, we and several other groups identified WNT1 as a key ligand to the WNT pathway in the regulation of bone formation and bone homeostasis. While biallelic loss-of-function mutations led to severe autosomal recessive OI-like phenotype with severe short stature, fractures, deformities and in some instances, developmental defects in the central nervous system, heterozygous WNT1 mutations were reported to cause autosomal dominant osteoporosis [45, 65, 66]. Since then, several cases with recessive OI caused by WNT1 mutations have been reported [67,68,69,70], confirming the severe OI type III -like phenotype in these patients. Ptosis has been suggested as a specific hallmark of this disease [67].
In contrast, WNT1-associated autosomal dominant early-onset osteoporosis, caused by heterozygous variants, has been reported less frequently. In our analyses involving a large Finnish cohort of 25 WNT1 mutation-positive children and adults, all with the same heterozygous missense WNT1 variant p.Cys218Gly, we have obtained in-depth information regarding the presenting features and progression of bone fragility, tissue-level bone characteristics, biomarkers and extra-skeletal manifestations. Affected children present in childhood usually with normal growth, only mildly reduced BMD but often with frequent long bone fractures and mild radiographic changes in long bone morphology, the fibulae being particularly thin [46]. Vertebral compression fractures are rare in childhood and early adulthood but practically all individuals have increased kyphosis and spinal compression fractures after the age of 50 years [71].
Histomorphometric analyses of transiliac bone biopsies demonstrated low-turnover osteoporosis [45]. Immunohistochemistry of bone biopsies showed altered expression of FGF23, sclerostin and phosphor-β-catenin and histology showed abnormal osteocyte morphology [72]. Quantitative back-scattering electron imaging (qBEI) showed heterogeneous matrix mineralization in children but homogeneous and increasing mineralization in adults [73]. Teriparatide treatment had only a minor effect on mineralization and seemed to increase bone marrow adiposity [73, 74]. Another study reported myelofibrosis in one young adult with WNT1 osteoporosis and increased bone marrow fibrosis in other individuals with the same heterozygous WNT1 variant [75]. It is unclear whether this is indicative of the importance of intact WNT1 signaling for the bone marrow niche or a sign of imbalanced maturation of the hematopoietic stem cell—osteoblast lineage.
While traditional biomarkers for bone turnover tend to be normal in WNT1 mutation-positive subjects with osteoporosis, the patients have a unique miRNA profile in serum [76]. In search for other potential biomarkers for this type of osteoporosis we showed that both intact and C-terminal FGF23 were significantly elevated in WNT1 mutation-positive subjects, while concentrations of the two WNT pathway-associated markers Sclerostin and DKK1 did not differ from age-matched controls [76].
A two-year teriparatide treatment in three adults showed increased bone formation but, as mentioned earlier, also a tendency to increased bone marrow adiposity [74]. Overall, the treatment results with conventional osteoporosis medications have not been optimal as several of the WNT1 mutation-positive adults have developed significant skeletal pathology with extensive spinal compression fractures despite several years of treatment [71]. Novel anabolic treatments, targeting specifically the WNT pathway may provide improved treatment results. Evidence for this in humans is still lacking but experiments in the WNT1 murine model, “the Swaying mouse” [77], suggest that sclerostin antibody is effective in increasing bone mass and decreasing peripheral fractures [78].
Although no major extra-skeletal features are seen in patients with heterozygous WNT1 variants, there are features suggesting cartilaginous alterations, such as vertebral endplate deterioration with frequent Schmorl nodes and changes at the knee articular cartilage [71, 79]. In addition, bone marrow biopsies indicated increased reticulin and altered granulopoiesis as signs of abnormal bone marrow function [75].
X-Chromosomal Osteoporosis Due to PLS3 Mutations
In 2013 Dijk et al. described a novel monogenic form of osteoporosis that involved predominantly boys and men in five families [80]. The causative gene defect involved the PLS3 gene, encoding Plastin 3. The gene’s X-chromosomal location explained why PLS3 mutations affected mainly hemizygous males while the heterozygous females did not present significant bone fragility. Since the original description, several other families and single patients have been reported [81,82,83,84,85,86,87,88,89]. Based on these, it is evident, that PLS3 mutations cause in affected males severe, early-onset and progressive osteoporosis predominated by multiple spinal compression fractures. Peripheral fractures are also common and may also present as atypical femur fractures after use of bisphosphonates [90, 91]. Despite being an X-chromosomal disorder, even females with a heterozygous PLS3 variant can present with significant peripheral fractures and vertebral compressions especially later in adulthood [92]. However, Kämpe et al. [82] also described a young girl who presented with recurrent peripheral fractures, extremely low BMD (lumbar spine BMD Z-score − 6.6 at 6 years) and a heterozygous de novo PLS3 variant. This indicates that PLS3 variants should be considered especially in males but even in females with early-onset osteoporosis. Regarding the nature of the reported variants, the studies have identified both missense and nonsense variants but also partial or total deletions of the gene [83, 89] as well as a partial duplication of the gene [85] in individuals with early-onset osteoporosis.
The mechanisms leading from pathogenic PLS3 variants to the clinical phenotype of the disorder have not yet been fully uncovered. PLS3 plays an important role in the maintenance of the intracellular actin cytoskeleton. PLS3 has been suggested to be important for the osteocytes’ mechanosensing properties. Osteocyte shape is dependent on actin filaments and osteocyte processes are rich in actin [93], suggesting that this actin-bundling protein could indeed be especially important for osteocyte function. PLS3 has also been implicated in bone matrix mineralization. Matrix vesicles, crucial in the mineralization process, are formed by budding from the tip of mineralizing cell microvilli. These microvilli contain a dense bundle of cross-linked actin microfilaments as a structural core. Thouverey et al. showed that in a mineralizing osteocyte-like cell line PLS3 was expressed both in the budding matrix vesicles and in the apical microvilli from which the vesicles were formed [94].
Detailed evaluation of transiliac bone biopsies obtained from individuals with PLS3-related osteoporosis show low bone turnover often with increased unmineralized osteoid [81, 82] and in quantitative backscattering electron imaging, a very variable mineralization pattern in childhood and more uniform increase in mineralization with age in adults [73, 83]. Evaluations of circulating biomarker profiles in patients with PLS3-related osteoporosis showed surprisingly normal conventional bone marker concentrations, increased DKK1 concentration and a specific miRNA profile with alterations also in some miRNAs linked to the WNT signaling pathway and TGF-beta signaling pathway [76, 95].
Recently some data emerging from studies on a PLS3 knock-out mouse model were described [96]. Based on μCT scanning, the PLS3-deficient mice exhibited moderate osteopenia at 12 weeks. More detailed skeletal evaluation at various ages revealed that PLS3-deficiency in mice only recapitulated the cortical bone phenotype of the human PLS3-related osteoporosis by negatively affecting the early stage of cortical bone acquisition, the cortical thickness in both tibia and femur being significantly reduced in PLS3-deficient mice in all age groups. In contrast, no significant differences between wildtype and PLS3-deficient littermates were detected in trabecular bone mass or in histomorphometric parameters at 12 weeks [96]. It therefore remains uncertain whether this model is suitable for e.g., preclinical trials testing various treatment options.
Osteoporosis Caused by SGMS2 Variants
The combination of early-onset osteoporosis and doughnut-shaped sclerotic skull lesions was described in 1974 [97]. This and subsequent reports confirmed the existence of a specific autosomal dominant disorder which was later termed as “osteoporosis with calvarial doughnut lesions” (CDL) (OMIM #126550) and was characterized by osteopenia, multiple pathologic fractures, elevated serum alkaline phosphatase, doughnut-shape calvarial lesions and dental caries. Mutations in the gene SGMS2 were recently identified as the cause of this rare autosomal dominant disorder [98]. SGMS2 encodes sphingomyelin synthase 2, an enzyme involved in sphingolipid metabolism. Sphingomyelin is a major lipid of the plasma membrane and enriched in microdomains of the plasma membrane that are critical for signal transduction. Mutations in SGMS2 lead to changes in the sphingomyelin synthase enzyme function and, through mostly unknown mechanisms, to a significant disturbance in bone metabolism and mineralization.
Individuals with a heterozygous SGMS2 mutation had sustained since childhood peripheral and spinal fractures [98]. Histomorphometric evaluation of patients’ bone biopsies showed a decrease in bone volume, reduced mineral content, heterogeneity of matrix mineralization, and importantly, a very disturbed matrix lamellarity with woven-bone appearance. Several subjects displayed neurological symptoms, transient facial nerve palsy being particularly common, suggesting that these extra-skeletal disease manifestations may be a distinctive feature of SGMS2-related osteoporosis [98].
Importantly, the phenotype varied significantly depending on the nature and location of the SGMS2 mutation. The recurrent Arg50* stop-gain variant was reported in four unrelated families in the original publication [98]. It has thereafter been reported in at least two additional families with CDL [99] confirming this to be the “hot-spot” for osteoporosis-causing variants. In contrast, in two families a missense mutation in the same gene led to a much more severe disorder with spondylometaphyseal skeletal dysplasia, significant calvarial hyperostosis, severe short stature and skeletal fragility since early infancy [98]. Since only a few reports on mutation-positive subjects have been published, the full spectrum of SGMS2 mutation-associated skeletal pathology remains to be elucidated.
Early-Onset Osteoporosis as a Polygenic Disorder
Polygenic risk scores are used to take into consideration the sum effect of several gene variants that may contribute to the overall risk for a certain phenotype. A recent study on individuals with a significant childhood fracture history but no identifiable monogenic cause had an increased burden of common fracture risk alleles compared to the general population [100]. This suggests that some patients with presumed monogenic osteoporosis do in fact have a polygenic etiology for bone fragility, making diagnostics and genetic counseling much more demanding. Further research is required to develop clinically usable tools for estimating polygenic contribution to early-onset osteoporosis in an individual patient and family and to separate monogenic forms from polygenic forms. Interestingly, some attempts to identify the underlying genetic cause for osteoporosis in patients with presumed monogenic form of osteoporosis have in fact identified two or several rare and potentially pathogenic variants in the sequenced candidate genes, e.g., heterozygous variants in both WNT1 and PLS3 in the same individual [101]. These studies are limited by lack of functional data exploring the significance of the identified variants and the links between gene variants and the phenotype thus remain uncertain. With increasing genetic testing in various patient cohorts with mild to moderate early-onset osteoporosis it has become evident that more tools are needed also in clinical settings to determine the true significance of the detected rare variants.
Evaluation of Osteoporosis
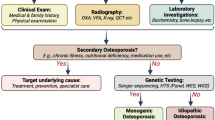
Once a diagnosis of osteoporosis has been established, based on the criteria suggested in Table 1, a thorough evaluation needs to take place. This involves a complete medical history and physical and laboratory examination to search for underlying chronic diseases and secondary factors. As discussed previously, many chronic diseases of childhood and young adulthood can lead to low BMD and fractures. These conditions often influence bone health and BMD through multiple mechanisms, including systemic inflammation, malnutrition, sex hormone deficiency, delayed puberty, and low mobility. Furthermore, several medications that are directly or indirectly harmful to bone, may be used, including glucocorticoids, anti-epileptic drugs or cancer treatment.
When no chronic condition is known, a thorough laboratory evaluation should be aimed at identifying secondary causes. The suggested laboratory tests for basic and extended screening are shown in Table 3. The main goal of this evaluation is to identify potential treatable secondary conditions. When no evidence of secondary factors can be found, a monogenetic cause should be considered and evaluated with appropriate genetic tools, as discussed in a separate paragraph. Bone turnover markers may help in diagnostic evaluation but are more helpful especially in adults in monitoring disease course and treatment response. Careful histological and histomorphometric evaluation of a transiliac bone biopsy is a standardized method to obtain detailed information regarding the bone metabolic activity and is often a very useful tool in patient evaluation. Its use is, however, limited because of the invasive nature and the required expertise in sample evaluation.
Approaches to Genetic Testing
A thorough clinical examination of a patient with early-onset osteoporosis may give some indications regarding the underlying genetic cause, for example blue sclerae, joint laxity and dentinogenesis imperfecta in OI or bony protrusions in the skull or a history of transient facial nerve palsy in SGMS2-related osteoporosis. However, clinical presentations vary and in most of the monogenic forms, no specific clinical signs have been reported. Family history, both regarding the patient’s parents and siblings, but also the patient’s own children, may give helpful clues and aide in establishing the genetic nature of the disease. In most situations, however, a genetic evaluation is in any case necessary to confidently exclude or confirm a heritable disease.
As mentioned, approximately 90% of all patients with OI have mutations in COL1A1 or COL1A2, the two genes encoding type I collagen. These should be included in genetic screening since the presentation of OI can be variable and in many instances the patients lack the typical clinical appearance of OI. Several other monogenic forms of bone fragility have been recognized and it is therefore advisable to use one of the several commercially available, or in-house, gene panels for OI and monogenic osteoporosis. Another possibility is to perform exome sequencing or whole genome sequencing and filter the sequencing data to detect variants of known clinical importance in the selected genes. This approach would allow the re-analysis of the sequencing data as novel disease-causing genes are identified.
At least patients with a positive family history for early-onset osteoporosis would benefit from a thorough genetic investigation, as identification of the defective gene can help to establish long-term prognosis, enable genetic counseling, and may influence treatment decisions. Since possibilities for genetic studies vary in different centers, guidelines for approaches and prioritizing need to be established locally. Recent developments in testing capacity and prices are promising and with decreasing costs, genetic testing can be more widely implemented in our clinical practices.
Treatment Considerations in Early-Onset Osteoporosis
Because osteoporosis is rare in the young, only few large-scale studies with pharmacological treatment have been performed and most of these studies had BMD and not fractures as a primary outcome. Concerning supplementation with calcium and vitamin D, studies with fracture outcome are lacking but an increase in BMD has been observed in some smaller scale studies [102, 103]. In case of insufficiency, also of other vitamins and minerals, supplementation is a pragmatic approach as well as other lifestyle advises related to use of alcohol, smoking and exercise. In fact, in children and adolescents with recurrent fractures, studies have indicated low calcium intake, vitamin D deficiency and inadequate physical activity to be a major contributing factor [15] and these should be addressed before other medications are considered.
Treatment of underlying diseases or secondary factors appears to be beneficial for bone and BMD increases have been observed e.g., with diet in celiac disease, (9% increase of radius BMD after one year), anti-TNF in IBD, estrogens for amenorrhea, surgery for primary hyperparathyroidism and Cushing’s disease, treatment of hyperthyroidism and malnutrition [19]. When treatment of the underlying cause is not possible or effective and fracture risk appears high, antiresorptive and anabolic drugs can be considered, taking into account potential adverse effects in pregnancies in women of childbearing age. The use of zoledronic acid in premenopausal breast cancer patients has clearly shown in the Austrian Breast and Colorectal cancer study group 12 trial (ABCSG-12) to prevent adjuvant endocrine therapy related bone loss [104] but evidence for fracture prevention is limited. A management algorithm for early breast cancer patients including premenopausal women on adjuvant endocrine therapy was recently published [23]. Treatment with bisphosphonates has also shown to improve BMD in several other underlying conditions of osteoporosis in young people such as anorexia nervosa (mainly at the lumbar spine), IBD, cystic fibrosis, thalassemia major and glucocorticoid-induced bone loss but fracture data are mostly lacking [14]. For guidance on treatment of glucocorticoid-induced osteoporosis in the young we refer to guidelines of the IOF-ECTS in 2012 and de American College of Rheumatology in 2017 [31, 105]. In PLAO, a retrospective, multicenter study in 52 women showed that BMD increased without pharmacological treatment but more so during treatment with bisphosphonates and with even higher increase in BMD with teriparatide although in all three groups about 19% developed a new fracture during follow-up of 36 months [106]. A similar larger increase in LS BMD was reported in a retrospective study of 32 PLAO women with multiple fractures treated with teriparatide for one year (15.5% ± 6.6) compared to controls (7.5% ± 7.1) [107].
Awaiting further RCTs with fracture reduction as a primary outcome in young persons with osteoporosis, a personalized approach is needed depending on the patient and the condition, where an absence of evidence should not be equal to evidence of absence of effects. The management of children and young adults with osteoporosis and fragility fractures requires a patient-centered multidisciplinary approach with a team of health professionals, optimally with expertise in both pediatric and young adult bone disease.
Apart from OI, even less data from human treatment trials is available for monogenic forms of osteoporosis. These treatment-related aspects have been briefly discussed in the separate paragraphs dealing with each genetic form. In young women with known osteoporosis and fragility fractures that desire future pregnancy it is important to discuss timing of pregnancy regarding the effect of pregnancy and especially of lactation on their bone health and the timing of bone-active medication. In a recent small-scale case–control study no major teratogenic effects of bisphosphonates were observed but potential negative effect on rates of neonatal complications and live birth rate could not be excluded [108]. Because of retention of bisphosphonates in bone it is generally advised not to start bisphosphonate treatment if when there are plans for future pregnancy within 1 year [109]. There is no data in humans on the safety of teriparatide, denosumab or romosozumab in pregnant women but since these drugs are not retained in bone it can be assumed that after stopping them before a pregnancy, they will not have teratogenic effects. What is not known is whether their effects will remain if no after-treatment with bisphosphonates is given, nor is it known if there is the same risk of stopping denosumab as in postmenopausal women on a rebound of bone turnover and occurrence of multiple vertebral fractures [110].
Concluding Remarks
Early-onset osteoporosis continues to be a diagnostic challenge. Careful clinical, radiological and biochemical evaluation is needed to detect underlying secondary causes. These should always precede a suspicion of a monogenic form of osteoporosis. Further, when establishing a diagnosis of idiopathic osteoporosis, a careful genetic evaluation is also needed to exclude the known monogenic forms of osteoporosis. Recent developments in advanced radiological imaging techniques that can measure volumetric BMD and bone microstructure in cancellous and cortical bone and estimate bone strength, such as high-resolution pQCT, may in the future give more insight into underlying bone defects and may limit the need for invasive bone biopsies. Because there is very limited evidence of anti-fracture efficacy of bone-active drugs it is important to consider their use in a personalized approach, after implementing optimal lifestyle and calcium and vitamin D supplementation and treatment of the underlying disorder while considering plans for future pregnancy in females of child-bearing age.
In the genetic forms of early-onset osteoporosis, careful characterization of the associated phenotypes, tissue-level pathology and the involved cellular mechanisms are of great value. Such studies can lead to discoveries that will benefit not only patients with these particular rare disorders but may prove efficacious even in the treatment of other patients with early-onset osteoporosis or patients with postmenopausal osteoporosis. Genetic diagnosis provides the affected individuals and their families information about the cause of osteoporosis and the mode of inheritance. The results will also affect the patients' medical care and follow-up. A specific genetic diagnosis enables early detection and timely preventive measures also in other family members who are affected by the same genetic defect. A multidisciplinary approach with a team of experts including e.g., pediatric and adult internists and endocrinologists, orthopedic surgeons, obstetricians and geneticists is usually needed for optimal care of these young patients with osteoporosis and fragility fractures. In order to expand the knowledge on the rare forms of osteoporosis in children and young adults, international collaboration is important, as is increasingly being implemented for example in the European Networks for Rare Bone Conditions (ERN BOND) and rare endocrine disorders (ERN ENDO) and within scientific consortia like GEFOS and GENOMOS and the GEMSTONE COST action (http://www.gefos.org; http://www.genomos.eu; https://cost-gemstone.eu).
References
Kanis JA, Gluer CC (2000) An update on the diagnosis and assessment of osteoporosis with densitometry. Committee of Scientific Advisors International Osteoporosis Foundation. Osteoporos Int 11:192–202
Mäkitie RE, Costantini A, Kämpe A, Alm JJ, Mäkitie O (2019) New insights into monogenic causes of osteoporosis. Front Endocrinol (Lausanne) 25(10):70. https://doi.org/10.3389/fendo.2019.00070
Mäkitie O (2013) Causes, mechanisms and management of paediatric osteoporosis. Nat Rev Rheumatol 9(8):465–475. https://doi.org/10.1038/nrrheum.2013.45
Chotiyarnwong P, McCloskey EV (2020) Pathogenesis of glucocorticoid-induced osteoporosis and options for treatment. Nat Rev Endocrinol 16(8):437–447. https://doi.org/10.1038/s41574-020-0341-0
Galindo-Zavala R, Bou-Torrent R, Magallares-López B, Mir-Perelló C, Palmou-Fontana N, Sevilla-Pérez B, Medrano-San Ildefonso M, González-Fernández MI, Román-Pascual A, Alcañiz-Rodríguez P, Nieto-Gonzalez JC, López-Corbeto M, Graña-Gil J (2020) Expert panel consensus recommendations for diagnosis and treatment of secondary osteoporosis in children. Pediatr Rheumatol Online J 18(1):20. https://doi.org/10.1186/s12969-020-0411-9
Laakso S, Valta H, Verkasalo M, Toiviainen-Salo S, Mäkitie O (2014) Compromised peak bone mass in patients with inflammatory bowel disease—a prospective study. J Pediatr 164(6):1436–43.e1. https://doi.org/10.1016/j.jpeds.2014.01.073
Golden NH, Abrams SA, Committee on Nutrition (2014) Optimizing bone health in children and adolescents. Pediatrics 134(4):e1229-43. https://doi.org/10.1542/peds.2014-2173
Zhu X, Zheng H (2021) Factors influencing peak bone mass gain. Front Med 15(1):53–69. https://doi.org/10.1007/s11684-020-0748-y
Rahaman SH, Jyotsna VP, Kandasamy D, Shreenivas V, Gupta N, Tandon N (2018) Bone health in patients with Cushing’s syndrome. Indian J Endocrinol Metab 22(6):766–769
Cairoli E, Eller-Vainicher C, Morlacchi LC, Tarsia P, Rossetti V, Pappalettera M, Arosio M, Chiodini I, Blasi F (2019) Bone involvement in young adults with cystic fibrosis awaiting lung transplantation for end-stage respiratory failure. Osteoporos Int 30(6):1255–1263
Weaver CM, Gordon CM, Janz KF, Kalkwarf HJ, Lappe JM, Lewis R, O’Karma M, Wallace TC, Zemel BS (2016) The National Osteoporosis Foundation’s position statement on peak bone mass development and lifestyle factors: a systematic review and implementation recommendations. Osteoporos Int 27(4):1281–1386. https://doi.org/10.1007/s00198-015-3440-3 (Erratum in: Osteoporos Int. 2016 Apr;27(4):1387)
Berger C, Goltzman D, Langsetmo L, Joseph L, Jackson S, Kreiger N, Tenenhouse A, Davison KS, Josse RG, Prior JC, Hanley DA (2010) Peak bone mass from longitudinal data: implications for the prevalence, pathophysiology, and diagnosis of osteoporosis. J Bone Miner Res 25:1948–1957
Lewiecki EM, Gordon CM, Baim S, Leonard MB, Bishop NJ, Bianchi ML, Kalkwarf HJ, Langman CB, Plotkin H, Rauch F, Zemel BS, Binkley N, Bilezikian JP, Kendler DL, Hans DB, Silverman S (2008) International society for clinical densitometry 2007 adult and pediatric official positions. Bone 43(6):1115–1121. https://doi.org/10.1016/j.bone.2008.08.106
Ferrari S, Bianchi ML, Eisman JA, Foldes AJ, Adami S, Wahl DA, Stepan JJ, de Vernejoul MC, Kaufman JM, IOF Committee of Scientific Advisors Working Group on Osteoporosis Pathophysiology (2012) Osteoporosis in young adults: pathophysiology, diagnosis, and management. Osteoporos Int 23(12):2735–48. https://doi.org/10.1007/s00198-012-2030-x
Mäyränpää MK, Viljakainen HT, Toiviainen-Salo S, Kallio PE, Mäkitie O (2012) Impaired bone health and asymptomatic vertebral compressions in fracture-prone children: a case-control study. J Bone Miner Res 27(6):1413–1424. https://doi.org/10.1002/jbmr.1579
Whyte MP (2016) Hypophosphatasia—aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 12(4):233–246. https://doi.org/10.1038/nrendo.2016.14
Hannan FM, Newey PJ, Whyte MP, Thakker RV (2019) Genetic approaches to metabolic bone diseases. Br J Clin Pharmacol 85(6):1147–1160. https://doi.org/10.1111/bcp.13803
Ferrari SL, Deutsch S, Baudoin C, Cohen-Solal M, Ostertag A, Antonarakis SE, Rizzoli R, de Vernejoul MC (2005) LRP5 gene polymorphisms and idiopathic osteoporosis in men. Bone 37(6):770–775
Pepe J, Body JJ, Hadji P, McCloskey E, Meier C, Obermayer-Pietsch B, Palermo A, Tsourdi E, Zillikens MC, Langdahl B, Ferrari S (2020) Osteoporosis in Premenopausal Women: A Clinical Narrative Review by the ECTS and the IOF. J Clin Endocrinol Metab 105(8):dgaa306. https://doi.org/10.1210/clinem/dgaa306
Yao L, Wang H, Dong W, Liu Z, Mao H (2017) Efficacy and safety of bisphosphonates in management of low bone density in inflammatory bowel disease: a meta-analysis. Medicine (Baltimore) 96(3):e5861. https://doi.org/10.1097/MD.0000000000005861
Hadji P, Aapro MS, Body JJ, Gnant M, Brandi ML, Reginster JY, Zillikens MC, Glüer CC, de Villiers T, Baber R, Roodman GD, Cooper C, Langdahl B, Palacios S, Kanis J, Al-Daghri N, Nogues X, Eriksen EF, Kurth A, Rizzoli R, Coleman RE (2017) Management of aromatase inhibitor-associated bone loss (AIBL) in postmenopausal women with hormone sensitive breast cancer: joint position statement of the IOF, CABS, ECTS, IEG, ESCEO IMS, and SIOG. J Bone Oncol 7:1–12. https://doi.org/10.1016/j.jbo.2017.03.001
Kyvernitakis I, Kostev K, Hadji P (2018) The tamoxifen paradoxinfluence of adjuvant tamoxifen on fracture risk in pre- and postmenopausal women with breast cancer. Osteoporos Int 29(11):2557–2564
Waqas K, Ferreira JL, Tsourdi E, Body J, Hadji P, Zillikens M.C. Updated guidance on the management of cancer treatment-induced bone loss (CTIBL) in pre- and postmenopausal women with early-stage breast cancer. J Bone Oncol 2021, in press
Solmi M, Veronese N, Correll CU, Favaro A, Santonastaso P, Caregaro L, Vancampfort D, Luchini C, De Hert M, Stubbs B (2016) Bone mineral density, osteoporosis, and fractures among people with eating disorders: a systematic review and meta-analysis. Acta Psychiatr Scand 133(5):341–351
Miller KK, Grinspoon SK, Ciampa J et al (2005) Medical findings in outpatients with anorexia nervosa. Arch Intern Med 165(5):561–566
Miller KK (2013) Endocrine effects of anorexia nervosa. Endocrinol Metab Clin North Am 42(3):515–528. https://doi.org/10.1016/j.ecl.2013.05.007
Robinson L, Aldridge V, Clark EM, Misra M, Micali N (2017) Pharmacological treatment options for low bone mineral density and secondary osteoporosis in anorexia nervosa: a systematic review of the literature. J Psychosom Res 98:87–97. https://doi.org/10.1016/j.jpsychores.2017.05.011
De Souza MJ, Nattiv A, Joy E (2014) Expert panel, 2014 Female athlete triad coalition consensus statement on treatment and return to play of the female athlete triad: 1st international conference held in San Francisco, California, May 2012 and 2nd international conference held in Indianapolis, Indiana, May 2013. Br J Sports Med 48:289. https://doi.org/10.1136/bjsports-2013-093218
Buckley L, Humphrey MB (2018) Glucocorticoid-induced osteoporosis. N Engl J Med 379(26):2547–2556
LeBlanc CM, Ma J, Taljaard M, Roth J, Scuccimarri R, Miettunen P, Lang B, Huber AM, Houghton K, Jaremko JL, Ho J, Shenouda N, Matzinger MA, Lentle B, Stein R, Sbrocchi AM, Oen K, Rodd C, Jurencak R, Cummings EA, Couch R, Cabral DA, Atkinson S, Alos N, Rauch F, Siminoski K, Ward LM, Canadian STeroid-Associated Osteoporosis in Pediatric Population (STOPP) Consortium (2015) Incident vertebral fractures and risk factors in the first three years following glucocorticoid initiation among pediatric patients with rheumatic disorders. J Bone Miner Res 30(9):1667–75. https://doi.org/10.1002/jbmr.2511
Buckley L, Guyatt G, Fink HA, Cannon M, Grossman J, Hansen KE, Humphrey MB, Lane NE, Magrey M, Miller M, Morrison L, Rao M, Robinson AB, Saha S, Wolver S, Bannuru RR, Vaysbrot E, Osani M, Turgunbaev M, Miller AS, McAlindon T (2017) 2017 American College of rheumatology guideline for the prevention and treatment of glucocorticoid-induced osteoporosis. Arthritis Rheumatol 69(8):1521–1537. https://doi.org/10.1002/art.40137
Kyvernitakis I, Reuter TC, Hellmeyer L, Hars O, Hadji P (2018) Subsequent fracture risk of women with pregnancy and lactation-associated osteoporosis after a median of 6 years of follow-up. Osteoporos Int 29(1):135–142. https://doi.org/10.1007/s00198-017-4239-1
Cohen A, Kamanda-Kosseh M, Dempster DW, Zhou H, Müller R, Goff E, Colon I, Bucovsky M, Stubby J, Nickolas TL, Stein EM, Recker RR, Lappe JM, Shane E (2019) Women with pregnancy and lactation-associated osteoporosis (PLO) have low bone remodeling rates at the tissue level. J Bone Miner Res 34(9):1552–1561
Campos-Obando N, Oei L, Hoefsloot LH, Kiewiet RM, Klaver CC, Simon ME, Zillikens MC (2014) Osteoporotic vertebral fractures during pregnancy: be aware of a potential underlying genetic cause. J Clin Endocrinol Metab 99(4):1107–1111. https://doi.org/10.1210/jc.2013-3238
Ostertag A, Cohen-Solal M, Audran M, Legrand E, Marty C, Chappard D, de Vernejoul MC (2009) Vertebral fractures are associated with increased cortical porosity in iliac crest bone biopsy of men with idiopathic osteoporosis. Bone 44(3):413–417
Rolvien T, Stürznickel J, Schmidt FN, Butscheidt S, Schmidt T, Busse B, Mundlos S, Schinke T, Kornak U, Amling M, Oheim R (2018) Comparison of bone microarchitecture between adult osteogenesis imperfecta and early-onset osteoporosis. Calcif Tissue Int 103(5):512–521
Collet C, Ostertag A, Ricquebourg M, Delecourt M, Tueur G, Isidor B, Guillot P, Schaefer E, Javier RM, Funck-Brentano T, Orcel P, Laplanche JL, Cohen-Solal M (2017) Primary osteoporosis in young adults: genetic basis and identification of novel variants in causal genes. JBMR Plus 2(1):12–21
Hartikka H, Mäkitie O, Männikkö M, Doria AS, Daneman A, Cole WG, Ala-Kokko L, Sochett EB (2005) Heterozygous mutations in the LDL receptor-related protein 5 (LRP5) gene are associated with primary osteoporosis in children. J Bone Miner Res 20(5):783–789. https://doi.org/10.1359/JBMR.050101
Arnold A, Dennison E, Kovacs CS, Mannstadt M, Rizzoli R, Brandi ML, Clarke B, Thakker RV (2021) Hormonal regulation of biomineralization. Nat Rev Endocrinol. https://doi.org/10.1038/s41574-021-00477-2
Marini JC, Forlino A, Bächinger HP, Bishop NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM, Krakow D, Montpetit K, Semler O (2017) Osteogenesis imperfecta. Nat Rev Dis Primers 18(3):17052. https://doi.org/10.1038/nrdp.2017.52
Cundy T (2012) Recent advances in osteogenesis imperfecta. Calcif Tissue Int 90(6):439–449. https://doi.org/10.1007/s00223-012-9588-3
Besio R, Chow CW, Tonelli F, Marini JC, Forlino A (2019) Bone biology: insights from osteogenesis imperfecta and related rare fragility syndromes. FEBS J 286(15):3033–3056. https://doi.org/10.1111/febs.14963
Huybrechts Y, Mortier G, Boudin E, Van Hul W (2020) WNT signaling and bone: lessons from skeletal dysplasias and disorders. Front Endocrinol (Lausanne) 9(11):165. https://doi.org/10.3389/fendo.2020.00165
Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Jüppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML, Osteoporosis-Pseudoglioma Syndrome Collaborative Group (2001) LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 107(4):513–23. https://doi.org/10.1016/s0092-8674(01)00571-2
Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, Lu JT, Pekkinen M, Wessman M, Heino TJ, Nieminen-Pihala V, Aronen M, Laine T, Kröger H, Cole WG, Lehesjoki AE, Nevarez L, Krakow D, Curry CJ, Cohn DH, Gibbs RA, Lee BH, Mäkitie O (2013) WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med 368(19):1809–1816. https://doi.org/10.1056/NEJMoa1215458
Mäkitie RE, Haanpää M, Valta H, Pekkinen M, Laine CM, Lehesjoki AE, Schalin-Jäntti C, Mäkitie O (2016) Skeletal characteristics of WNT1 osteoporosis in children and young adults. J Bone Miner Res 31(9):1734–1742. https://doi.org/10.1002/jbmr.2841
Stürznickel J, Rolvien T, Delsmann A, Butscheidt S, Barvencik F, Mundlos S, Schinke T, Kornak U, Amling M, Oheim R (2021) Clinical phenotype and relevance of LRP5 and LRP6 variants in patients with early-onset osteoporosis (EOOP). J Bone Miner Res 36(2):271–282. https://doi.org/10.1002/jbmr.4197
Somer H, Palotie A, Somer M, Hoikka V, Peltonen L (1988) Osteoporosis-pseudoglioma syndrome: clinical, morphological, and biochemical studies. J Med Genet 25(8):543–549. https://doi.org/10.1136/jmg.25.8.543
Saarinen A, Välimäki VV, Välimäki MJ, Löyttyniemi E, Auro K, Uusen P, Kuris M, Lehesjoki AE, Mäkitie O (2007) The A1330V polymorphism of the low-density lipoprotein receptor-related protein 5 gene (LRP5) associates with low peak bone mass in young healthy men. Bone 40(4):1006–1012. https://doi.org/10.1016/j.bone.2006.11.010
Estrada K, Styrkarsdottir U, Evangelou E, Hsu YH, Duncan EL, Ntzani EE, Oei L, Albagha OM, Amin N, Kemp JP, Koller DL, Li G, Liu CT, Minster RL, Moayyeri A, Vandenput L, Willner D, Xiao SM, Yerges-Armstrong LM, Zheng HF, Alonso N, Eriksson J, Kammerer CM, Kaptoge SK, Leo PJ, Thorleifsson G, Wilson SG, Wilson JF, Aalto V, Alen M, Aragaki AK, Aspelund T, Center JR, Dailiana Z, Duggan DJ, Garcia M, Garcia-Giralt N, Giroux S, Hallmans G, Hocking LJ, Husted LB, Jameson KA, Khusainova R, Kim GS, Kooperberg C, Koromila T, Kruk M, Laaksonen M, Lacroix AZ, Lee SH, Leung PC, Lewis JR, Masi L, Mencej-Bedrac S, Nguyen TV, Nogues X, Patel MS, Prezelj J, Rose LM, Scollen S, Siggeirsdottir K, Smith AV, Svensson O, Trompet S, Trummer O, van Schoor NM, Woo J, Zhu K, Balcells S, Brandi ML, Buckley BM, Cheng S, Christiansen C, Cooper C, Dedoussis G, Ford I, Frost M, Goltzman D, González-Macías J, Kähönen M, Karlsson M, Khusnutdinova E, Koh JM, Kollia P, Langdahl BL, Leslie WD, Lips P, Ljunggren Ö, Lorenc RS, Marc J, Mellström D, Obermayer-Pietsch B, Olmos JM, Pettersson-Kymmer U, Reid DM, Riancho JA, Ridker PM, Rousseau F, Slagboom PE, Tang NL, Urreizti R, Van Hul W, Viikari J, Zarrabeitia MT, Aulchenko YS, Castano-Betancourt M, Grundberg E, Herrera L, Ingvarsson T, Johannsdottir H, Kwan T, Li R, Luben R, Medina-Gómez C, Palsson ST, Reppe S, Rotter JI, Sigurdsson G, van Meurs JB, Verlaan D, Williams FM, Wood AR, Zhou Y, Gautvik KM, Pastinen T, Raychaudhuri S, Cauley JA, Chasman DI, Clark GR, Cummings SR, Danoy P, Dennison EM, Eastell R, Eisman JA, Gudnason V, Hofman A, Jackson RD, Jones G, Jukema JW, Khaw KT, Lehtimäki T, Liu Y, Lorentzon M, McCloskey E, Mitchell BD, Nandakumar K, Nicholson GC, Oostra BA, Peacock M, Pols HA, Prince RL, Raitakari O, Reid IR, Robbins J, Sambrook PN, Sham PC, Shuldiner AR, Tylavsky FA, van Duijn CM, Wareham NJ, Cupples LA, Econs MJ, Evans DM, Harris TB, Kung AW, Psaty BM, Reeve J, Spector TD, Streeten EA, Zillikens MC, Thorsteinsdottir U, Ohlsson C, Karasik D, Richards JB, Brown MA, Stefansson K, Uitterlinden AG, Ralston SH, Ioannidis JP, Kiel DP, Rivadeneira F (2012) Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet 44(5):491–501. https://doi.org/10.1038/ng.2249
Wang QF, Bi HS, Qin ZL, Wang P, Nie FF, Zhang GW (2020) Associations of LRP5 gene with bone mineral density, bone turnover markers, and fractures in the elderly with osteoporosis. Front Endocrinol (Lausanne) 25(11):571549. https://doi.org/10.3389/fendo.2020.571549
Tüysüz B, Bursalı A, Alp Z, Suyugül N, Laine CM, Mäkitie O (2012) Osteoporosis-pseudoglioma syndrome: three novel mutations in the LRP5 gene and response to bisphosphonate treatment. Horm Res Paediatr 77(2):115–120. https://doi.org/10.1159/000336193
Papadopoulos I, Bountouvi E, Attilakos A, Gole E, Dinopoulos A, Peppa M, Nikolaidou P, Papadopoulou A (2019) Osteoporosis-pseudoglioma syndrome: clinical, genetic, and treatment-response study of 10 new cases in Greece. Eur J Pediatr 178(3):323–329. https://doi.org/10.1007/s00431-018-3299-3
Marozik P, Alekna V, Rudenko E, Tamulaitiene M, Rudenka A, Mastaviciute A, Samokhovec V, Cernovas A, Kobets K, Mosse I (2019) Bone metabolism genes variation and response to bisphosphonate treatment in women with postmenopausal osteoporosis. PLoS ONE 14(8):e0221511. https://doi.org/10.1371/journal.pone.0221511
Kruk M, Ralston SH, Albagha OM (2009) LRP5 Polymorphisms and response to risedronate treatment in osteoporotic men. Calcif Tissue Int 84(3):171–179. https://doi.org/10.1007/s00223-008-9207-5
Saarinen A, Saukkonen T, Kivelä T, Lahtinen U, Laine C, Somer M, Toiviainen-Salo S, Cole WG, Lehesjoki AE, Mäkitie O (2010) Low density lipoprotein receptor-related protein 5 (LRP5) mutations and osteoporosis, impaired glucose metabolism and hypercholesterolaemia. Clin Endocrinol (Oxf) 72(4):481–488. https://doi.org/10.1111/j.1365-2265.2009.03680.x
Kim SP, Frey JL, Li Z, Goh BC, Riddle RC (2017) Lack of Lrp5 signaling in osteoblasts sensitizes male mice to diet-induced disturbances in glucose metabolism. Endocrinology 158(11):3805–3816. https://doi.org/10.1210/en.2017-00657
Foer D, Zhu M, Cardone RL, Simpson C, Sullivan R, Nemiroff S, Lee G, Kibbey RG, Petersen KF, Insogna KL (2017) Impact of gain-of-function mutations in the low-density lipoprotein receptor-related protein 5 (LRP5) on glucose and lipid homeostasis. Osteoporos Int 28(6):2011–2017. https://doi.org/10.1007/s00198-017-3977-4
Korvala J, Jüppner H, Mäkitie O, Sochett E, Schnabel D, Mora S, Bartels CF, Warman ML, Deraska D, Cole WG, Hartikka H, Ala-Kokko L, Männikkö M (2012) Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Med Genet 10(13):26. https://doi.org/10.1186/1471-2350-13-26
Witte F, Dokas J, Neuendorf F, Mundlos S, Stricker S (2009) Comprehensive expression analysis of all Wnt genes and their major secreted antagonists during mouse limb development and cartilage differentiation. Gene Expr Patterns 9(4):215–223. https://doi.org/10.1016/j.gep.2008.12.009
Movérare-Skrtic S, Henning P, Liu X, Nagano K, Saito H, Börjesson AE, Sjögren K, Windahl SH, Farman H, Kindlund B, Engdahl C, Koskela A, Zhang FP, Eriksson EE, Zaman F, Hammarstedt A, Isaksson H, Bally M, Kassem A, Lindholm C, Sandberg O, Aspenberg P, Sävendahl L, Feng JQ, Tuckermann J, Tuukkanen J, Poutanen M, Baron R, Lerner UH, Gori F, Ohlsson C (2014) Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat Med 20(11):1279–88. https://doi.org/10.1038/nm.3654
Zheng HF, Tobias JH, Duncan E, Evans DM, Eriksson J, Paternoster L, Yerges-Armstrong LM, Lehtimäki T, Bergström U, Kähönen M, Leo PJ, Raitakari O, Laaksonen M, Nicholson GC, Viikari J, Ladouceur M, Lyytikäinen LP, Medina-Gomez C, Rivadeneira F, Prince RL, Sievanen H, Leslie WD, Mellström D, Eisman JA, Movérare-Skrtic S, Goltzman D, Hanley DA, Jones G, St Pourcain B, Xiao Y, Timpson NJ, Smith GD, Reid IR, Ring SM, Sambrook PN, Karlsson M, Dennison EM, Kemp JP, Danoy P, Sayers A, Wilson SG, Nethander M, McCloskey E, Vandenput L, Eastell R, Liu J, Spector T, Mitchell BD, Streeten EA, Brommage R, Pettersson-Kymmer U, Brown MA, Ohlsson C, Richards JB, Lorentzon M (2012) WNT16 influences bone mineral density, cortical bone thickness, bone strength, and osteoporotic fracture risk. PLoS Genet 8(7):e1002745. https://doi.org/10.1371/journal.pgen.1002745
Gori F, Lerner U, Ohlsson C, Baron R (2015) A new WNT on the bone: WNT16, cortical bone thickness, porosity and fractures. Bonekey Rep 13(4):669. https://doi.org/10.1038/bonekey.2015.36
Medina-Gomez C, Kemp JP, Estrada K, Eriksson J, Liu J, Reppe S, Evans DM, Heppe DH, Vandenput L, Herrera L, Ring SM, Kruithof CJ, Timpson NJ, Zillikens MC, Olstad OK, Zheng HF, Richards JB, St Pourcain B, Hofman A, Jaddoe VW, Smith GD, Lorentzon M, Gautvik KM, Uitterlinden AG, Brommage R, Ohlsson C, Tobias JH, Rivadeneira F (2012) Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet. 8(7):e1002718. https://doi.org/10.1371/journal.pgen.1002718
Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, Fischer B, Yigit G, Janda CY, Becker J, Breer S, Altunoglu U, Grünhagen J, Krawitz P, Hecht J, Schinke T, Makareeva E, Lausch E, Cankaya T, Caparrós-Martín JA, Lapunzina P, Temtamy S, Aglan M, Zabel B, Eysel P, Koerber F, Leikin S, Garcia KC, Netzer C, Schönau E, Ruiz-Perez VL, Mundlos S, Amling M, Kornak U, Marini J, Wollnik B (2013) Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet 92(4):565–74. https://doi.org/10.1016/j.ajhg.2013.02.010
Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, Fernandez BA, Elsayed SM, Elsobky E, Verma I, Nair S, Turner EH, Smith JD, Jarvik GP, Byers PH (2013) WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. Am J Hum Genet 92(4):590–597. https://doi.org/10.1016/j.ajhg.2013.02.009
Nampoothiri S, Guillemyn B, Elcioglu N, Jagadeesh S, Yesodharan D, Suresh B, Turan S, Symoens S, Malfait F (2019) Ptosis as a unique hallmark for autosomal recessive WNT1-associated osteogenesis imperfecta. Am J Med Genet A 179(6):908–914. https://doi.org/10.1002/ajmg.a.61119
Hayat A, Hussain S, Bilal M, Kausar M, Almuzzaini B, Abbas S, Tanveer A, Khan A, Siddiqi S, Foo JN, Ahmad F, Khan F, Khan B, Anees M, Mäkitie O, Alfadhel M, Ahmad W, Umair M (2020) Biallelic variants in four genes underlying recessive osteogenesis imperfecta. Eur J Med Genet. 63(8):103954. https://doi.org/10.1016/j.ejmg.2020.103954
Kausar M, Siddiqi S, Yaqoob M, Mansoor S, Makitie O, Mir A, Khor CC, Foo JN, Anees M (2018) Novel mutation G324C in WNT1 mapped in a large Pakistani family with severe recessively inherited Osteogenesis Imperfecta. J Biomed Sci 25(1):82. https://doi.org/10.1186/s12929-018-0481-x (Erratum in: J Biomed Sci. 2019 Apr 28;26(1):31)
Umair M, Alhaddad B, Rafique A, Jan A, Haack TB, Graf E, Ullah A, Ahmad F, Strom TM, Meitinger T, Ahmad W (2017) Exome sequencing reveals a novel homozygous splice site variant in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr Res 82(5):753–758. https://doi.org/10.1038/pr.2017.149
Mäkitie RE, Niinimäki T, Nieminen MT, Schalin-Jäntti C, Niinimäki J, Mäkitie O (2017) Impaired WNT signaling and the spine-Heterozygous WNT1 mutation causes severe age-related spinal pathology. Bone 101:3–9. https://doi.org/10.1016/j.bone.2017.04.001
Wesseling-Perry K, Mäkitie RE, Välimäki VV, Laine T, Laine CM, Välimäki MJ, Pereira RC, Mäkitie O (2017) Osteocyte protein expression is altered in low-turnover osteoporosis caused by mutations in WNT1 and PLS3. J Clin Endocrinol Metab 102(7):2340–2348. https://doi.org/10.1210/jc.2017-00099
Fratzl-Zelman N, Wesseling-Perry K, Mäkitie RE, Blouin S, Hartmann MA, Zwerina J, Välimäki VV, Laine CM, Välimäki MJ, Pereira RC, Mäkitie O (2021) Bone material properties and response to teriparatide in osteoporosis due to WNT1 and PLS3 mutations. Bone 146:115900. https://doi.org/10.1016/j.bone.2021.115900
Välimäki VV, Mäkitie O, Pereira R, Laine C, Wesseling-Perry K, Määttä J, Kirjavainen M, Viljakainen H, Välimäki MJ (2017) Teriparatide treatment in patients with WNT1 or PLS3 mutation-related early-onset osteoporosis: a pilot study. J Clin Endocrinol Metab 102(2):535–544. https://doi.org/10.1210/jc.2016-2423
Mäkitie RE, Niinimäki R, Kakko S, Honkanen T, Kovanen PE, Mäkitie O (2018) Defective WNT signaling associates with bone marrow fibrosis-a cross-sectional cohort study in a family with WNT1 osteoporosis. Osteoporos Int 29(2):479–487. https://doi.org/10.1007/s00198-017-4309-4
Mäkitie RE, Kämpe A, Costantini A, Alm JJ, Magnusson P, Mäkitie O (2020) Biomarkers in WNT1 and PLS3 osteoporosis: altered concentrations of DKK1 and FGF23. J Bone Miner Res 35(5):901–912. https://doi.org/10.1002/jbmr.3959
Joeng KS, Lee YC, Jiang MM, Bertin TK, Chen Y, Abraham AM, Ding H, Bi X, Ambrose CG, Lee BH (2014) The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet 23(15):4035–42. https://doi.org/10.1093/hmg/ddu117
Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, Munivez E, Ambrose C, Lee BH (2017) Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest 127(7):2678–2688
Lehtovirta S, Mäkitie RE, Casula V, Haapea M, Niinimäki J, Niinimäki T, Peuna A, Lammentausta E, Mäkitie O, Nieminen MT (2019) Defective WNT signaling may protect from articular cartilage deterioration—a quantitative MRI study on subjects with a heterozygous WNT1 mutation. Osteoarthritis Cartilage 27(11):1636–1646. https://doi.org/10.1016/j.joca.2019.07.001
van Dijk FS, Zillikens MC, Micha D, Riessland M, Marcelis CL, de Die-Smulders CE, Milbradt J, Franken AA, Harsevoort AJ, Lichtenbelt KD, Pruijs HE, Rubio-Gozalbo ME, Zwertbroek R, Moutaouakil Y, Egthuijsen J, Hammerschmidt M, Bijman R, Semeins CM, Bakker AD, Everts V, Klein-Nulend J, Campos-Obando N, Hofman A, te Meerman GJ, Verkerk AJ, Uitterlinden AG, Maugeri A, Sistermans EA, Waisfisz Q, Meijers-Heijboer H, Wirth B, Simon ME, Pals G (2013) PLS3 mutations in X-linked osteoporosis with fractures. N Engl J Med 369(16):1529–1536. https://doi.org/10.1056/NEJMoa1308223
Laine CM, Wessman M, Toiviainen-Salo S, Kaunisto MA, Mäyränpää MK, Laine T, Pekkinen M, Kröger H, Välimäki VV, Välimäki MJ, Lehesjoki AE, Mäkitie O (2015) A novel splice mutation in PLS3 causes X-linked early onset low-turnover osteoporosis. J Bone Miner Res 30(3):510–518. https://doi.org/10.1002/jbmr.2355
Kämpe AJ, Costantini A, Mäkitie RE, Jäntti N, Valta H, Mäyränpää M, Kröger H, Pekkinen M, Taylan F, Jiao H, Mäkitie O (2017) PLS3 sequencing in childhood-onset primary osteoporosis identifies two novel disease-causing variants. Osteoporos Int 28(10):3023–3032. https://doi.org/10.1007/s00198-017-4150-9
Kämpe AJ, Costantini A, Levy-Shraga Y, Zeitlin L, Roschger P, Taylan F, Lindstrand A, Paschalis EP, Gamsjaeger S, Raas-Rothschild A, Hövel M, Jiao H, Klaushofer K, Grasemann C, Mäkitie O (2017) PLS3 deletions lead to severe spinal osteoporosis and disturbed bone matrix mineralization. J Bone Miner Res 32(12):2394–2404. https://doi.org/10.1002/jbmr.3233
Costantini A, Krallis PΝ, Kämpe A, Karavitakis EM, Taylan F, Mäkitie O, Doulgeraki A (2018) A novel frameshift deletion in PLS3 causing severe primary osteoporosis. J Hum Genet 63(8):923–926. https://doi.org/10.1038/s10038-018-0472-5
Costantini A, Skarp S, Kämpe A, Mäkitie RE, Pettersson M, Männikkö M, Jiao H, Taylan F, Lindstrand A, Mäkitie O (2018) Rare copy number variants in array-based comparative genomic hybridization in early-onset skeletal fragility. Front Endocrinol (Lausanne) 10(9):380. https://doi.org/10.3389/fendo.2018.00380
Balasubramanian M, Fratzl-Zelman N, O’Sullivan R, Bull M, Fa Peel N, Pollitt RC, Jones R, Milne E, Smith K, Roschger P, Klaushofer K, Bishop NJ (2018) Novel PLS3 variants in X-linked osteoporosis: exploring bone material properties. Am J Med Genet A 176(7):1578–1586. https://doi.org/10.1002/ajmg.a.38830
Wang L, Zhai Q, Zhao P, Xiang X, Zhang X, Tian W, Li T (2018) Functional analysis of p.Ala253_Leu254insAsn mutation in PLS3 responsible for X-linked osteoporosis. Clin Genet. 93(1):178–181. https://doi.org/10.1111/cge.13081
Lv F, Ma M, Liu W, Xu X, Song Y, Li L, Jiang Y, Wang O, Xia W, Xing X, Qiu Z, Li M (2017) A novel large fragment deletion in PLS3 causes rare X-linked early-onset osteoporosis and response to zoledronic acid. Osteoporos Int 28(9):2691–2700. https://doi.org/10.1007/s00198-017-4094-0
Kannu P, Mahjoub A, Babul-Hirji R, Carter MT, Harrington J (2017) PLS3 mutations in X-linked osteoporosis: clinical and bone characteristics of two novel mutations. Horm Res Paediatr 88(3–4):298–304. https://doi.org/10.1159/000477242
van de Laarschot DM, Zillikens MC (2016) Atypical femur fracture in an adolescent boy treated with bisphosphonates for X-linked osteoporosis based on PLS3 mutation. Bone 91:148–151. https://doi.org/10.1016/j.bone.2016.07.022
Nguyen HH, van de Laarschot DM, Verkerk AJMH, Milat F, Zillikens MC, Ebeling PR (2018) Genetic risk factors for atypical femoral fractures (AFFs): a systematic review. JBMR Plus 2(1):1–11. https://doi.org/10.1002/jbm4.10024
Mäkitie RE, Niinimäki T, Suo-Palosaari M, Kämpe A, Costantini A, Toiviainen-Salo S, Niinimäki J, Mäkitie O (2020) PLS3 mutations cause severe age and sex-related spinal pathology. Front Endocrinol (Lausanne) 23(11):393. https://doi.org/10.3389/fendo.2020.00393
Tanaka-Kamioka K, Kamioka H, Ris H, Lim SS (1998) Osteocyte shape is dependent on actin filaments and osteocyte processes are unique actin-rich projections. J Bone Miner Res 13(10):1555–1568. https://doi.org/10.1359/jbmr.1998.13.10.1555
Thouverey C, Malinowska A, Balcerzak M, Strzelecka-Kiliszek A, Buchet R, Dadlez M, Pikula S (2011) Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J Proteomics 74(7):1123–1134. https://doi.org/10.1016/j.jprot.2011.04.005
Mäkitie RE, Hackl M, Weigl M, Frischer A, Kämpe A, Costantini A, Grillari J, Mäkitie O (2020) Unique, gender-dependent serum microrna profile in PLS3 gene-related osteoporosis. J Bone Miner Res 35(10):1962–1973. https://doi.org/10.1002/jbmr.4097
Yorgan TA, Sari H, Rolvien T, Windhorst S, Failla AV, Kornak U, Oheim R, Amling M, Schinke T (2020) Mice lacking plastin-3 display a specific defect of cortical bone acquisition. Bone 130:115062. https://doi.org/10.1016/j.bone.2019.115062
Royen PM, Ozonoff MB (1974) Multiple calvarial “doughnut lesions.” Am J Roentgenol Radium Ther Nucl Med 121(1):121–123. https://doi.org/10.2214/ajr.121.1.121
Pekkinen M, Terhal PA, Botto LD, Henning P, Mäkitie RE, Roschger P, Jain A, Kol M, Kjellberg MA, Paschalis EP, van Gassen K, Murray M, Bayrak-Toydemir P, Magnusson MK, Jans J, Kausar M, Carey JC, Somerharju P, Lerner UH, Olkkonen VM, Klaushofer K, Holthuis JC, Mäkitie O (2019) Osteoporosis and skeletal dysplasia caused by pathogenic variants in SGMS2. JCI Insight 4(7):e126180. https://doi.org/10.1172/jci.insight.126180
Robinson ME, Bardai G, Veilleux LN, Glorieux FH, Rauch F (2020) Musculoskeletal phenotype in two unrelated individuals with a recurrent nonsense variant in SGMS2. Bone 134:115261. https://doi.org/10.1016/j.bone.2020.115261
Manousaki D, Kämpe A, Forgetta V, Makitie RE, Bardai G, Belisle A, Li R, Andersson S, Makitie O, Rauch F, Richards JB (2020) Increased burden of common risk alleles in children with a significant fracture history. J Bone Miner Res 35(5):875–882. https://doi.org/10.1002/jbmr.3956
Rocha-Braz MGM, França MM, Fernandes AM, Lerario AM, Zanardo EA, de Santana LS, Kulikowski LD, Martin RM, Mendonca BB, Ferraz-de-Souza B (2020) Comprehensive genetic analysis of 128 candidate genes in a cohort with idiopathic, severe, or familial osteoporosis. J Endocr Soc 4(12):bvaa148. https://doi.org/10.1210/jendso/bvaa148
Peris P, Monegal A, Martinez MA, Moll C, Pons F, Guanabens N (2007) Bone mineral density evolution in young premenopausal women with idiopathic osteoporosis. Clin Rheumatol 26(6):958–961
Islam MZ, Shamim AA, Viljakainen HT, Akhtaruzzaman M, Jehan AH, Khan HU et al (2010) Effect of vitamin D, calcium and multiple micronutrient supplementation on vitamin D and bone status in Bangladeshi premenopausal garment factory workers with hypovitaminosis D: a double-blinded, randomised, placebo-controlled 1-year intervention. Br J Nutr 104(2):241–247
Gnant M, Mlineritsch B, Luschin-Ebengreuth G, Kainberger F, Kassmann H, Piswanger-Solkner JC et al (2008) Adjuvant endocrine therapy plus zoledronic acid in premenopausal women with early-stage breast cancer: 5-year follow-up of the ABCSG-12 bone-mineral density substudy. Lancet Oncol 9(9):840–849
Lekamwasam S, Adachi JD, Agnusdei D, Bilezikian J, Boonen S, Borgström F, Cooper C, Diez Perez A, Eastell R, Hofbauer LC, Kanis JA, Langdahl BL, Lesnyak O, Lorenc R, McCloskey E, Messina OD, Napoli N, Obermayer-Pietsch B, Ralston SH, Sambrook PN, Silverman S, Sosa M, Stepan J, Suppan G, Wahl DA, Compston JE, Joint IOF-ECTS GIO Guidelines Working Group (2012) A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporos Int 23(9):2257–2276. https://doi.org/10.1007/s00198-012-1958-1
Laroche M, Talibart M, Cormier C, Roux C, Guggenbuhl P, Degboe Y (2017) Pregnancy-related fractures: a retrospective study of a French cohort of 52 patients and review of the literature. Osteoporos Int 28(11):3135–3142
Hong N, Kim JE, Lee SJ, Kim SH, Rhee Y (2018) Changes in bone mineral density and bone turnover markers during treatment with teriparatide in pregnancy- and lactation-associated osteoporosis. Clin Endocrinol (Oxf) 88(5):652–658
Sokal A, Elefant E, Leturcq T, Beghin D, Mariette X, Seror R (2019) Pregnancy and newborn outcomes after exposure to bisphosphonates: a casecontrol study. Osteoporos Int 30(1):221–229
Langdahl BL (2017) Osteoporosis in premenopausal women. Curr Opin Rheumatol 29(4):410–415
Tsourdi E, Zillikens MC, Meier C, Body JJ, Gonzalez Rodriguez E, Anastasilakis AD, Abrahamsen B, McCloskey E, Hofbauer LC, Guañabens N, Obermayer-Pietsch B, Ralston SH, Eastell R, Pepe J, Palermo A, Langdahl B (2020) Fracture risk and management of discontinuation of denosumab therapy: a systematic review and position statement by ECTS. J Clin Endocrinol Metab. https://doi.org/10.1210/clinem/dgaa756
Funding
Open access funding provided by University of Helsinki including Helsinki University Central Hospital. The authors’ research is funded by the Novo Nordisk Foundation, the Sigrid Jusélius Foundation, the Finnish Pediatric Research Foundation, the Academy of Finland, and the Folkhälsan Research Foundation (to OM).
Author information
Authors and Affiliations
Contributions
OM initiated the manuscript and wrote the first draft. Both authors contributed to the writing of the manuscript, reviewed the final version and approved submission.
Corresponding author
Ethics declarations
Conflict of interest
Outi Mäkitie declares consultancy to Kyowa Kirin, Alexion, Merck and Sandoz. M. Carola Zillikens declares having received honoraria in the past for lectures or advice from Alexion, Amgen, Eli Lilly, Kyowa Kirin, Shire and UCB.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mäkitie, O., Zillikens, M.C. Early-Onset Osteoporosis. Calcif Tissue Int 110, 546–561 (2022). https://doi.org/10.1007/s00223-021-00885-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00223-021-00885-6