
Abstract
Dispersive liquid–liquid microextraction (DLLME) is a rapid and easy technique that consumes minute amounts of organic solvents. In this work, we present chemometric study on optimization of DLLME parameters for the extraction of aldrin, endrin, lindane, α-endosulfan, 4,4′-DDT and its metabolites from honey matrix. Method quantification limits (MQLs) vary between 0.3 ng/g for 2,4′-DDE and 4,4′-DDE to 13.2 ng/g for α-endosulfan and enable determination at levels below EU-established Maximum Residue Limits. The developed method is linear (R 2 > 0.994) in the investigated range (MQL—100 ng/g), with preconcentration factors of 13.2–30.5 and good repeatability (CV ≤ 17%). A comparison with other available methods reported in the last decade is provided. The method has been applied to 19 real samples from Poland, and the results show that organochlorine pesticides (OCPs) are present in analysed honeys at levels not posing threat to human health (below 14 ng/g for sum of 4,4′-DDT and metabolites and below 5 ng/g for aldrin, endrin and lindane). To the best of our knowledge, this is the first reported application of DLLME for the determination of OCPs in honey.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Honey, regarded as a valuable natural food product of animal origin, should be free from contaminants, and its quality and safety have become a concern. Due to the persistence of OCPs in the environment, they can be introduced to honey by bees during its production [e.g. 1, 2]. Thus, it is important to monitor OCP levels in honey by the most environmentally friendly means possible; this, in turn, results from the “green analytical chemistry” approach.
Concerning the analysis of pesticides and their metabolites in food products, the majority of them is currently determined by liquid chromatography (LC) coupled with mass spectrometer detector (single or tandem) [3], except for OCPs that are better determined by GC–MS.
Solvent microextraction is a technique increasingly used these days. It permits the analysis of small amount of samples and, because of the small amount of solvent used, is more environmentally friendly. There are four main methods used in solvent microextraction: single-drop microextraction (SDME), headspace single-drop microextraction (HS-SDME), hollow-fibre-protected microextraction (HFME) and dispersive liquid–liquid microextraction (DLLME). It is the volatility and polarity of analytes that usually determine which technique is the most suitable one [4].
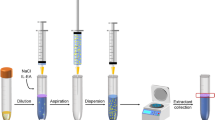
DLLME was first reported by Rezaee’s group in 2006 [5]. It involves dissolving an amount of water-insoluble extracting solvent in a water-soluble dispersive solvent such as acetone. The mixture is then injected into the water sample contained in a centrifuge tube. As the extracting solvent is insoluble in water, an emulsion is created, making greater contact area between the phases and the faster establishment of extraction equilibrium than in traditional liquid–liquid extraction. The tube is then centrifuged, and a portion of the extracting solvent is collected using a syringe and injected into a GC.
A major limitation of this technique is the choice of extracting solvent. Only solvents slightly soluble in water and denser than water (for example, tetrachloroethene, carbon tetrachloride, carbon disulphide or chlorobenzene) or high-melting liquids less dense than water (for instance, 1-undecanol or hexadecane) can be used as extractants. With regard to the second type of solvent, the sample is cooled, then the solidified drop is collected from the vial, melted and analysed—the variant called DLLME-Solidification of Floating Organic droplet, developed and reported by Leong and Huang in 2008 [6].
A major disadvantage of DLLME compared to SDME is the fact that several discrete steps must be performed, including centrifugation. This means that the method can only be semi-automated, since extraction and injection are not performed in one device.
On the other hand, this technique is very effective for the extraction of analytes such as PAHs and PCBs, which have large organic solvent–water partition coefficients. Moreover, a major factor as regards extraction efficiency and extraction time is the rate of absorption of a chemical by the extracting solvent. This is no longer a problem with DLLME. Since the surface area of the microdrops in the dispersed state is very large, equilibrium is reached within a few seconds. In the case of SDME, reaching equilibrium may take around 40 min. Hence, having weighed up these facts, DLLME was the technique of choice.
In DLLME, the factors that may affect extraction efficiency are the suitability of the extracting solvent, analyte concentrations, extracting solvent volume, extraction time, salting out effect, temperature, centrifugation time and sample pH.
The aim of this work was to develop a new, rapid, easy and reliable method for the determination of OCPs in honey, applying DLLME with GC–MS, and to optimize extraction parameters employing factorial designs. Although DLLME has been applied to honey samples for the determination of chloramphenicol and thiamphenicol [7–9] and triazines [10] by HPLC, to the best of our knowledge, this is the first reported application of DLLME for the determination of OCPs in honey.
Experimental
Chemicals and reagents
Pesticide stock standard solutions (100 ng/μL in iso-octane) of lindane, aldrin, endrin, 2,4′-DDT, 2,4′-DDD, 2,4′-DDE, 4,4′-DDT, 4,4′-DDE and α-endosulfan, as well as 2,4′-D8-DDE, 2,4′-D8-DDT, were supplied by LGC Standards (Poland). Methanol, acetone and acetonitrile (all Pestanal grade), chloroform (99.9%, Chromasolv grade) and 1-undecanol (99% pure) were purchased from Sigma-Aldrich (Poland).
Honey samples, collected from apiaries in different regions of Poland, were provided for analysis by the Bee Product Quality Testing Laboratory, Research Institute of Horticulture, Apiculture Division, Puławy, Poland.
Calibration solutions
Working standard solution of all analytes at concentrations of 1 ng/μL in acetone was prepared from stock standard solutions of individual pesticides and stored at 4 °C in the dark. A set of calibration standard solutions (in acetone) was prepared by dilution in the range MQL—100 ng/g.
The solutions for validation studies were also prepared from individual stock standard solutions on the day of analysis. The resulting solution in acetone contained analytes at concentrations 200 times those of the respective method’s quantification limits. Honey sample used for validation studies was free from contaminations.
Chemometric setup
First, the type of extracting solvent and disperser solvent was chosen. Honey sample free from contaminations was spiked with working standard solution to obtain each OCP level of 25 ng/g honey. Then, volume of extracting solvent and disperser solvent, the amount of salt and pH were optimized by means of Full Factorial Design (orthogonal and non-orthogonal) and Central Composite Design. Considered factors and their two levels, along with the experimental design matrices and the total area for ions of all compounds obtained for each experiment (being average of three repetitions), are presented in Online Resources 1 and 2.
Analytical procedure
0.5 g of homogenized honey sample was dissolved in 3 mL of ultrapure water, the resulting solution was spiked with surrogate standards (deuterized compounds) at 5 ng/g honey and mixed thoroughly. A mixture of 450 μL acetone (disperser solvent) and 100 μL chloroform (extractant) was prepared and rapidly injected into the sample to obtain an emulsion. After 20 s (including 5 s of shaking), the sample was centrifuged (5 min, 4.0 k RPM) and a two-phase solution was obtained. The resulting volume of sediment phase was 80 μL. During the extraction, a precipitate formed between chloroform and aqueous phase, which slightly impeded the collection of the small volume of chloroform. The chloroform phase at the bottom of the conical vial was collected with a microlitre syringe, and 2 μL were injected on GC column.
This is the optimized procedure, validated and applied in real sample analyses.
GC–MS analysis
The analyses were carried out on an Agilent Technologies 7890A gas chromatograph coupled with an Agilent Technologies 5975C mass spectrometer working in selected ion monitoring mode (injection port temperature: 280 °C, interface temperature: 280 °C, MS source temperature: 230 °C, MS quad temperature: 150 °C, gas flow (He): 1 mL/min). A Zebron ZB 5-MS capillary chromatographic column was used (30 m × 0.25 mm, 0.25 μm stationary phase composed of 5% Phenyl-Arylene and 95% Dimethylpolysiloxane + 1 m precolumn (Phenomenex, USA)). The injection volume was 2 μL.
The following temperature gradient was applied to separate the compounds effectively: 80 °C for 1 min, then 15 °C/min to 180 °C, then 10 °C/min to 240 °C, held for 3 min, then 20 °C/min to 300 °C, held for 2 min. The total GC analysis run time was 22 min.
Two ions were used to identify the compound, and the first one, more intensive, was used for quantification. Table 1 shows the relevant information on GC–MS data acquisition.
Method validation
To validate the optimized method, parameters such as linearity over the MQL—100 ng/g range, method detection and quantification limits (MDL and MQL, respectively), recovery at 2 × MQL and 10 × MQL levels, repeatability and intermediate precision (same laboratory, same equipment and materials, same day and different operators) were determined. Samples were run in pentuplicates. MDL and MQL correspond to concentrations producing signal/noise ratios (S/N) of 3 and 10, respectively, after employing the extraction procedure. For the need of validation studies, dissolved samples were spiked with standards, mixed and left for 1 h to allow good integration of analytes with the matrix.
Results and discussion
Extraction optimization
We chose to optimize the extraction by considering four factors that we can control the most easily and which, according to the literature [e.g. 4, 11], have an impact on the recovery: type and volume of extracting solvent (X1, in range of 100–150 μL) and disperser solvent (X2, in range of 300–450 μL), amount of salt (X3, in range of 0–14.3%m/m) and pH (X4, in range of 3.4–7.3).
Honey has a pH of 3.4–4.5, depending on honey type, and it seemed important to check the influence of sample pH on extraction efficiency.
Choice of extracting solvent and disperser solvent
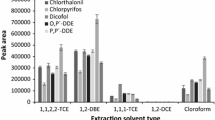
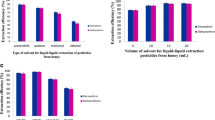
First, a suitable organic extracting solvent was selected. As stated before, this solvent should have a density higher/lower than that of water and a good affinity for pesticides. On the basis of these considerations, two solvents were chosen: 1-undecanol (11-OH) (density lower than that of water, high melting point) and chloroform (density higher than that of water). The disperser had to be miscible both with the organic phase and with water. Acetonitrile, acetone and methanol were tested for this purpose. Extractions were carried out for each pair of solvents. As shown in Fig. 1 the best extraction efficiency was obtained with chloroform and acetone.

Comparison of summary extraction efficiency of analytes using different pairs of extracting and disperser solvents, determined by GC–MS. Extraction conditions: 0.5 g of honey in 3 mL of water + spike with standards at 25 ng/g honey +100 μL of extracting solvent +400 μL of disperser
Full factorial design
In the second step, a full factorial design was employed to determine the most significant factors. A two-level factorial design requires an experiment to be carried out at all possible combinations of the two levels of each factor considered [12–14]. As there are 4 factors, this design consisted of 24 = 16 experiments. The experiments were run in a random manner to minimize the effect of uncontrolled variables. Different factors and their two levels, along with the experimental design matrix and the total area for ions of all compounds obtained for each run, are presented in Online Resource 1.
Subsequently, several multiple linear regressions were calculated with SigmaPlot 12.0 software (Systat Software Inc., USA) in order to build up the best suited model. Both orthogonal and non-orthogonal designs [12–14] were considered. The orthogonal plan does not take into account any replicates except the ones at the centre. A specific aspect of this plan is that the calculated coefficient values and significance sign are always the same whatever the considered interactions in the regression. On the other hand, adding other replicates as in the non-orthogonal plan may facilitate the estimation of pure error.
The strategy for the orthogonal design was as follows:
First, a regression was done by considering X3 as a factor. The determination coefficient, R 2, was about 0.562. Moreover, the X3 effect was found to be significant, and the fact that the F value was superior to the critical F value implied that this model was also statistically significant. Then, X2 was introduced to the model, but as the regression showed it to be insignificant, it was removed. The same conclusion was drawn for X4. Next, X1 was added to X3 and found to be significant, so it was retained in the model. In the next step, second-order interactions were added one by one in the same way as before. X13 and X24 appeared to be significant.
The final effects on the extraction efficiency are best described by the following equation (Eq. 1):
A1 = 1236662, A2 = −256904, A3 = −425277, A4 = 161513, A5 = 146147, with determination coefficient, R 2, equal to 0.914.
The strategy for non-orthogonal design was similar to the one described above for the orthogonal design—a stepwise regression analysis was realized. The final effects on the extraction efficiency in this case were best described by the following equation (Eq. 2):
A1 = 233193, A2 = −263962, A3 = −451158, A4 = 168572, A5 = 120267, with R 2 equal to 0.898.
In order to check if another design fits better to the experiment, a central composite design was considered with factors X1 and X3.
Central composite design (CCD)
In this step, a rotatable, orthogonal CCD was employed to determine the optimum conditions for the critical factors X1 and X3. The CCD consists of a full factorial 2p design to which a 2p star design is added. It is completed by the addition of a centre point, where the experiment is repeated n times [12–14].
This time we decided to change the levels of the salt factor (X3). Indeed, according to the literature [12, 13], the range used previously could have been too wide to detect precisely the influence of salt on the extraction efficiency. Level 1 thus now corresponds to 0.85% of salt (m/m). The levels of studied factors and matrix of the CCD along with the total peak area for ions of all compounds and for each run are presented in Online Resource 2.
The previous regression technique was used to build up the model. This resulted in only X1 being considered a significant factor. The CCD model was statistically significant; however, its R 2 is about 0.612, which is not better than the other designs.
The results obtained for the different plans and different regressions are summarized in Table 2.
It should be kept in mind that the extraction of pesticides from honey involves a very complex matrix. Hence, we cannot expect to obtain as good a value of R 2 as with pure compounds in a simple matrix such as water.
Method validation
Linearity, MDL and MQL
The linearity of the developed method was examined and confirmed in the range of MQL—100 ng/g honey. Each of the solutions had a constant deuterized compound concentration of 5 ng/g honey.
Table 3 lists the determination coefficient, the method detection limit (MDL) and quantification limit (MQL) for each compound, along with Maximum Residue Limits (MRL) established by EU [15]. Good linearity was achieved for all the analytes.
Preconcentration factors, repeatability and intermediate precision
Preconcentration factors were calculated for analytes at 2MQL level, whereas repeatability and intermediate precision were determined for two concentrations of analytes: 2 times respective MQLs and 10 times respective MQLs. Intermediate precision was determined by performing same experiments by two operators. Surrogate standards of 2,4′-D8-DDE and 2,4′-D8-DDT were added to each sample at the levels of 5 ng/g honey. The aim of adding surrogate standards and evaluating their recovery is important from real sample analysis point of view. Samples were prepared and analysed in pentuplicates. The results are presented in Table 4.
In microextraction methods, repeatability (in terms of coefficient of variation—CV) of obtained results and preconcentration factor are more important than the actual recovery.
Preconcentration factor was calculated as described elsewhere [5]. In general, obtained PF values were between 13.2 and 30.5, which is not very high, compared to typical PF values of DLLME methods [11], but it still provides MQLs well below MRLs and that, in turn, enables detection and quantitative analysis of OCPs at levels not exceeding MRLs for honey. The reason for relatively low PF values was relatively high volume [11] of extracting solvent used.
CV values were in the range of 6.5–14.6% for most analytes at two concentration levels, and the highest value of coefficient of variation (CV = 17.1%) was for 4,4′-DDT at the 2MQL level.
In general, the repeatability decreases (higher CV) with decrease in the examined concentration: this is a well-known fact described by Horwitz and discussed by others [e.g., 16].
Intermediate precision of the method was acceptable for all analytes; better intermediate precision was achieved for higher concentration levels.
Comparison with available methods and application to real samples
The method developed here was compared with other methods enabling the determination of OCPs in honey reported in the last 10 years (Table 5).
Compared to other methods the proposed method is quick and easy to use, and consumes only minute amounts of organic solvents. It enables the quantitative determination of OCPs at levels below EU-established MRLs.
19 honey samples from apiaries in Poland were analysed for the presence of OCPs using this method. The results of analyses for OCPs in honey are summarized in Table 6, and an example SIM chromatogram of a honey sample with detected lindane residue is shown in Fig. 2. The greatest contamination was due to 4,4′-DDT, which was present in almost 80% of the samples. In general, residue levels in honey were quite low. They were probably introduced into honey by bees that fed on nectar from contaminated flowers. Pesticides are thus carried along with nectar and pollen into the hives.

SIM chromatogram of a multi-flower honey sample with detected lindane residue (RT = 10.602 min)
Conclusions
A new rapid, easy and sensitive method allowing the determination of OCPs in honey was developed. Employing experimental designs enabled the optimization of the dispersive liquid–liquid microextraction conditions. DLLME consumes minute amounts of organic solvents, and GC–MS enables the qualitative and quantitative determination of pesticide residues.
Our method is characterized by MDL and MQLs comparable to or lower than those of other methods recently reported for determining OCP levels in honey (Table 6); it also consumes significantly smaller amounts of organic solvents.
Analysis of metrological parameters, such as MQL and CV, and the method’s good linearity over the investigated range lead to the conclusion that it can be successfully used in analytical practice, as it allows the unequivocal determination of OCP residues below EU MRLs. The method was successfully used to detect and quantitatively determine OCP residues in real honey samples. 4,4′-DDT residues and its metabolites were detected in most of the samples (ca. 80%), endrin and lindane were detected in 21% of samples, and aldrin was present in 10% of honey samples.
Even though organochlorine pesticides have been banned for quite a long time, they are still present in the environment owing to their persistence. Honey can be used as an indicator of environmental contamination with different pollutants, such as OCPs. In the light of the average level of honey consumption in the EU of 0.63 kg/year/capita and the span of 0.36–1.57 kg/y/capita for EU Member States [26], the levels of detected OCPs in analysed honey do not pose a threat to human health.
References
Fernández M, Picó Y, Mañs J (2002) J Food Prot 65:1502
Bogdanov S (2006) Apidologie 37:1
Picó Y, Barceló D (2008) TrAC 27:821
Kokosa JM, Przyjazny A, Jeannot M (2009) Solvent Microextraction: Theory and Practice. Wiley, New Jersey
Rezaee M, Assadi Y, Hosseini MRM, Aghaee E, Ahmadi F, Berijani S (2006) J Chromatogr A 1116:1
Leong MI, Huang SD (2008) J Chromatogr A 1211:8
Chen H, Ying J, Chen H, Huang J, Liao L (2008) Chromatographia 68:629
Chen H, Chen H, Ying J, Huang J, Liao L (2009) Anal Chim Acta 632:80
Chen H, Chen H, Liao L, Ying J, Huang J (2010) J Chromatogr Sci 48:450
Wang Y, You J, Ren R, Xiao Y, Gao S, Zhang H, Yu A (2010) J Chromatogr A 1217:4241
Rezaee M, Yamini Y, Faraji M (2010) J Chromatogr A 1217:2342
Brereton RG (2003) Chemometrics: Data Analysis for the Laboratory and Chemical Plant. Wiley, West Sussex
Stoyanov K, Walmsley AD (2006) Response-surface modeling and experimental design. In: Gemperline P (ed) Practical guide to chemometrics. CRC Press, Boca Raton
Miller JN, Miller JC (2005) Statistics and chemometrics for analytical chemistry. Pearson Education Ltd., Essex
[15] European Commission Regulation No 396/2005 with annexes, O.J. L 70, 1 16.3.2005
Thompson M (ed) (2004) AMC technical brief no. 17. Royal Society of Chemistry
Erdoğrul Ö (2007) Food Control 18:866
Blasco C, Lino CM, Picó Y, Pena A, Font G, Silveira MIN (2004) J Chromatogr A 1049:155
Mukherjee I (2009) Bull Environ Contam Toxicol 83:818
Herrera A, Pérez-Arquillué C, Conchello P, Bayarri S, Lázaro R, Yagüe C, Ariño A (2005) Anal Bioanal Chem 381:695
Yavuz H, Guler GO, Aktumsek A, Cakmak YS, Ozparlak H (2010) Environ Monit Assess 168:277
Blasco C, Fernández M, Pena A, Lino C, Silveira MI, Font G, Picó Y (2003) J Agric Food Chem 51:8132
Albero B, Sánchez-Brunete C, Tadeo JL (2004) J Agric Food Chem 52:5828
Rissato SR, Galhiane MS, de Almeida MV, Gerenutti M, Apon BM (2007) Food Chem 101:1719
Tahboub YR, Zaater MF, Barri TA (2006) Anal Chim Acta 558:62
Eurostat http://epp.eurostat.ec.europa.eu. Accessed on 16 Aug 2011
Acknowledgments
The financial support of Polish Ministry of Science and Higher Education (grant no. NN312 427037) is gratefully acknowledged. One of the authors (E.P.) wishes to express her thanks for a Socrates–Erasmus mobility grant, which enabled her to undergo research training at the Gdańsk University of Technology (Poland). The authors would also like to thank the anonymous reviewers for their comments and suggestions that helped improve the quality of the paper.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Kujawski, M.W., Pinteaux, E. & Namieśnik, J. Application of dispersive liquid–liquid microextraction for the determination of selected organochlorine pesticides in honey by gas chromatography–mass spectrometry. Eur Food Res Technol 234, 223–230 (2012). https://doi.org/10.1007/s00217-011-1635-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-011-1635-1