
Abstract
endo-1,4-β-Glucanase (endo-cellulase, EC 3.2.1.4) is one of the most widely used enzymes in industry. Despite its importance, improved methods for the rapid, selective, quantitative assay of this enzyme have been slow to emerge. In 2014, a novel enzyme-coupled assay that addressed many of the limitations of the existing assay methodology was reported. This involved the use of a bifunctional substrate chemically derived from cellotriose. Reported herein is a much improved version of this assay employing a novel substrate, namely 4,6-O-(3-ketobutylidene)-4-nitrophenyl-β-d-cellopentaoside.

Principle of the CELLG5 assay
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
endo-1,4-β-glucanase (EG, EC 3.2.1.4) finds use in a wide range of industrial applications including finishing and biopolishing in the textiles industry, [1] improving softness and colour brightness of cottons in the detergent industry, [2, 3] and deinking and fiber polishing in the pulp and paper processing industries [4]. It is also employed to hydrolyse β-glucan to increase the digestibility of cereal grain in the animal feed industry [5], to reduce viscosity of cereal feedstock slurries used in the biofuel industry [6] and to reduce wort viscosity in the malting/brewing industries [7, 8].
endo-Glycosyl hydrolases have been traditionally assayed using either reducing sugar [9, 10] or viscometric methods [11]. In the case of endo-1,4-β-glucanase (EC 3.2.1.4, EG), the native substrate cellulose is almost completely insoluble in aqueous media and chemically modified versions of the polysaccharide with improved water solubility, typically containing carboxymethyl functionalisation (CM-Cellulose), have been employed in its place but these methods are labour intensive and time consuming. Dyed polysaccharide substrates were reported in the 1980s [12] as sensitive and selective alternatives to the existing methodology and soon after became commercially available. This class of substrates represented a very significant improvement in the convenience afforded to a user and are limited only by the fact that (a) a filtration step or centrifugation step required in their application precludes their use in automated procedures and (b) an unambiguously chemically defined substrate would be preferable in order to remove the possibility of any batch to batch variability. Cellooligosaccharides, borohydride-reduced cellooligosaccharides [13], and nitrophenyl cellooligosaccharides [14–17] are all useful substrates for the characterisation of EGs in terms of subsite mapping and binding studies but lack general applicability for widespread use as EG substrates in crude samples containing competing hydrolytic enzymes (e.g., β-glucosidase and cellobiohydrolase).
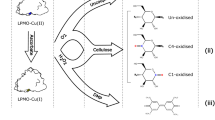
Recently, a coupled enzyme activity assay for EG was reported employing 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellotrioside (1) [18]. The analogous fluorimetric assay employing 4,6-O-benzylidene-4-methylumbelliferyl-β-d-cellotrioside allowing improved sensitivity (2) was reported soon after [19]. The principle of these assays is outlined in Fig. 1. Taking substrate 1 as an example; 0.1 mL of substrate solution containing ancillary β-glucosidase is added to 0.1 mL of enzyme extract containing EG. EG acts to liberate 2-chloro-4-nitrophenyl-β-d-glucopyranoside which is rapidly hydrolysed to 2-chloro-4-nitrophenol (CNP) and glucose by the β-glucosidase present with the substrate. After 10 min, the enzymatic reactions are stopped by the addition of 3 mL of alkaline buffer solution which also ensures complete deprotonation of (and therefore maximum absorption by) CNP.

Overview of the enzyme-coupled assay of EG using substrates 1 and 2 in the presence of ancillary thermostable β-glucosidase
The use of these enzyme-coupled procedures allowed for the rapid and selective assay of EG in a format that was readily suited to automation. However, a number of limitations exist with the use of substrates 1 and 2 in the desired application.
-
1.
The relative rate of hydrolysis of substrates 1 and 2 compared to the rate of hydrolysis of carboxymethyl cellulose (CMC-4M) varied considerably across a number of EGs. This was to be expected as the ability of EG to hydrolyse cellooligosaccharides of DP 3–6 is known to be highly dependent on the source of the enzymes [13]. This variability was undesirable from the perspective of trying to draw relevant information concerning predicted hydrolytic efficacy of EG on the native substrate from results obtained using the synthetic substrates 1 and 2.
-
2.
The use of dimethyl sulfoxide (DMSO) as a co-solvent was necessary to solubilise substrates 1 and 2 as these compounds exhibited somewhat poor water solubility. The solution of substrate 1 contained 33 % v/v DMSO and although this did not inhibit or destabilise the β-glucosidase present as an ancillary enzyme, the resulting 16.5 % v/v DMSO concentration in the assay incubation proved sufficient to inhibit certain EGs.
-
3.
CNP (pK a = 5.45) was originally chosen as a UV-absorbing chromophore over the more commonly employed 4-nitrophenol (NP, pK a = 7.18) because the former provided for the potential to perform kinetic assays by reading the absorbance directly during assay incubations. Although this feature proves advantageous for the assay of those EGs with a suitable pH optimum (>5.5), the lower pK a associated with CNP also significantly increases the leaving group ability of the chromophore making substrate 1 potentially more prone to spontaneous hydrolysis at elevated temperature or in acidic/basic conditions than its NP analogue.
Each of these limitations was addressed and overcome in the current study.
Materials and methods
Materials
Cellopentaose (Cat. No. O-CPE), cellohexaose (Cat. No. O-CHE), carboxylmethyl cellulose 4M (Cat. No. P-CMC4M), thermostable β-glucosidase (Thermotoga maritima; accession number Q08638, Cat. No. E-BGOSTM) and highly purified endo-1,4-β-glucanases from Trichoderma longibrachiatum (accession number Q12714, Cat. No. E-CELTR), Bacillus amyloliquefaciens (accession number Q8RPQ6, Cat. No. E-CELBA), T. maritima (accession number Q9X273, Cat. No. E-CELTM) and Aspergillus niger (O74705, Cat. No. E-CELAN), were obtained from Megazyme International, Ireland. All other chemicals used in organic synthesis were purchased from Sigma Aldrich, Lennox Laboratory Supplies or Acros Organics and were analytical reagent grade. A Brüker Avance 400 was employed for 1H (400.13 MHz) and 13C (100.61 MHz) NMR spectra. Resonances δ, are in ppm units downfield from an internal reference in C2D6SO (δH = 2.50). Mass spectrometry analysis was performed with a Q-Tof Premier Waters Maldi-quadrupole time-of-flight (Q-Tof) mass spectrometer equipped with Z-spray electrospray ionisation (ESI) and matrix-assisted laser desorption ionisation (MALDI) sources. Silica gel Davisil (Fisher Scientific) was used for column chromatography. All enzyme incubations were performed in duplicate and the mean absorbance value taken. One unit of EG activity is defined as either the amount of enzyme required to release 1 μmol of glucose reducing sugar equivalents per minute from CM-cellulose 4 M (10 mg/mL) under suitable conditions or the amount of enzyme required to release 1 μmol of 4-nitrophenolate/min at 40 °C under the standard CELLG5 assay conditions (“Standard assay of EG using compound 7 in the presence of thermostable β-glucosidase – CELLG5 assay” section). One unit of β-glucosidase activity is defined as the amount of enzyme required to release 1 μmol of 4-nitrophenol/min from 4-nitrophenyl-β-d-glucopyranoside (5 mM) in a suitable buffer at the enzyme’s optimum pH.
Colorimetric substrates
4,6-O-Benzylidene-2-chloro-4-nitrophenyl-β-d-cellopentaoside (3) and 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellohexaoside (4) were synthesized from the corresponding cellooligosaccharides using previously described synthetic methodology [18, 20].
4,6-O-Benzylidene-2-chloro-4-nitrophenyl-β-d-cellopentaoside (3)
All MS data and NMR spectra are included in the Electronic Supplementary Material (ESM).
Yield: 6 % from d-cellopentaose. 1H NMR (400 MHz C2D6SO) δ 3.02–3.20 (m, 4H), 3.28–3.91 (m, 25H), 4.14–4.26 (m, 1H), 4.33 (d, J = 7.03 Hz, 1H (anom)), 4.35 (d, J = 6.77 Hz, 1H (anom)), 4.39 (d, J = 7.78 Hz, 1H (anom)), 4.51 (d, J = 7.78 Hz, 1H (anom)), 4.52–4.57 (m, 1H), 4.66–4.79 (m, 6H), 4.83 (br s, 1H), 5.35 (d, J = 7.78 Hz, 1H (anom)), 5.40–5.50 (m, 4H), 5.57 (d, J = 5.02 Hz, 1H), 5.60 (s, 1H), 5.69 (d, J = 5.27 Hz, 1H), 7.35–7.53 (m, 6H), 8.23 (dd, J = 9.03, 2.76 Hz, 1H), 8.35 (d, J = 2.76 Hz, 1H); 13C NMR (100 MHz, C2D6SO) δ 60.2, 60.5, 60.8, 60.8, 66.4, 68.1, 73.2, 73.3, 73.5, 73.5, 73.6, 74.7, 74.7, 75.3 75.3, 75.3, 75.3, 75.4, 75.4, 75.7, 79.4, 79.9, 80.8, 80.8, 80.9, 99.9, 101.1, 103.2, 103.3, 103.4, 103.5, 116.1, 122.7, 124.7, 126.1, 126.8, 126.8, 128.5, 128.5, 129.4, 138.1, 142.0, 158.1; HRMS ES+ [M + Na]+ Calc. 1094.2732, Found 1094.2781; mp 242–244 °C
4,6-O-Benzylidene-2-chloro-4-nitrophenyl-β-d-cellohexaoside (4)
All MS data and NMR spectra are included in the ESM.
Yield: 4 % from d-cellohexaose. 1H NMR (400 MHz C2D6SO) δ 3.01–3.18 (m, 5H), 3.23–4.02 (m, 30H), (m, 16H), 5.22–5.82 (m, 9H), 7.22–7.60, (m, 6H), 8.23 (dd, J = 9.03, 2.76 Hz, 1H), 8.35 (d, J = 2.76 Hz, 1H); 13C NMR (100 MHz, C2D6SO) δ 59.8, 60.1, 60.4, 60.4, 60.4, 66.0, 67.7, 72.8, 72.9, 73.0, 73.0, 73.0, 73.2, 74.3, 74.3, 74.8, 74.8, 74.8, 74.9, 74.9, 74.9, 75.0, 75.0, 75.3, 79.0, 79.4, 80.4, 80.4, 80.5, 80.5, 99.5, 100.7, 102.8, 102.9, 102.9, 102.9, 103.1, 115.7, 122.3, 124.3, 125.7, 126.4, 126.4, 128.1, 128.1, 129.0, 137.7, 141.6, 157.7; HRMS ES+ [M + Na]+ Calc. 1256.3260, Found 1256.3320; mp 255–258 °C
4,6-O-(3-ketobutylidene)-4-nitrophenyl-β-d-cellopentaoside (7)
4-Nitrophenyl-β-d-cellopentaoside was prepared from cellopentaose following the previously described synthetic methodology [20]. The product obtained was carried into the 4,6-O-(3-ketobutylidene) acetal formation reaction without purification according to the following procedure. To a solution of crude 4-nitrophenyl-β-d-cellopentaoside (2 g, 2.11 mmol) and p-toluenesulfonic acid monohydrate (118 mg, 0.62 mmol) under an argon atmosphere in anhydrous DMSO (8 mL), was added 4-methoxy-3-butene-2-one (420 μL, 4.14 mmol) via syringe. The reaction was heated to 50 °C and stirred for 5 h at reduced pressure (30 mbar), then cooled to room temperature. Triethylamine (129 μL, 0.93 mmol) was added via syringe to the reaction mixture which was then precipitated by adding it slowly to EtOH (8 mL). The resulting solid was first absorbed onto silica gel and semi-purified by flash chromatography. 4,6-O-(3-ketobutylidene)-4-nitrophenyl-β-d-cellopentaoside was obtained as a white solid in 5 % overall yield from d-cellopentaose following repeated recrystallisations from EtOH/H2O.
All MS data and NMR spectra are included in the ESM.
1H NMR (400 MHz C2D6SO) δ 2.12 (s, 3H), 2.65–2.83 (m, 2H), 2.95–3.93 (m, 29H), 3.96–4.13 (m, 1H), 4.24–4.58 (m, 5H), 4.63–4.89 (m, 7H), 4.97, (br s, 1H), 5.11–5.26 (m, 1H), 5.27–5.58 (m, 6H), 7.24 (d, J = 7.78 Hz, 2H), 8.22 (d, J = 7.78 Hz, 2H); 13C NMR (100 MHz, C2D6SO) δ 30.7, 48.1, 59.9, 60.2, 60.4, 60.4, 65.9, 67.4, 72.8, 73.0, 73.2, 73.1, 73.1, 74.4, 74.4, 74.8, 74.9, 74.9, 75.0, 75.0, 75.0, 75.3, 79.1, 79.6, 80.1, 80.4, 80.5, 98.4, 99.5, 102.9, 103.0, 103.0, 103.2, 116.7, 116.7, 125.9, 125.9, 141.9, 162.4, 204.7; HRMS ES+ [M + Na]+ Calc. 1040.3070, Found 1040.3083; mp 259–263 °C
Preparation of assay reagents containing ancillary β-glucosidase
A solution of compounds 1, 3, 4, 5 and 6 (1 mL, 10 mM in DMSO) was diluted with H2O (2 mL) to give 3.3 mM solutions (3 mL, 33 % v/v DMSO/H2O). Thermostable β-glucosidase (100 μL, 3.2 M (NH4)2SO4, 400 U/mL) was then added and mixed by swirling to give a final concentration of 13.3 U/mL. Note that for compounds 5 and 6, the 3.3 mM solution should be prepared immediately before use from solutions pre-equilibrated to 40 °C and then maintained at 40 °C to avoid rapid precipitation of the substrate.
Compound 7 was handled as described above to give a 3.3 mM solution in 10 % v/v DMSO/H2O containing 20 U/mL thermostable β-glucosidase. This solution is termed the CELLG5 reagent.
Enzymatic assays
Enzymatic hydrolysis of a series of 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellooligosaccharides (DP2-6)
The relative rates of hydrolysis of 1, 3, 4, 5 and 6 by A. niger EG (Fig. 2) were determined by the incubation of substrate solutions (0.1 mL, 3.3 mM, 33 % v/v DMSO/H2O) containing thermostable β-glucosidase (1.33 U) with a solution of A. niger EG (0.1 mL, 17.6 mU on CMC-4M, 14.8 mU on CELLG5) at 40 °C. Reaction was terminated at 0, 3, 6, 9 and 12 min by adding 3 mL of 2 % w/v Tris solution (pH 9) with mixing. The absorbance values were read at 400 nm against the time zero reading for the respective substrate.

Incubation of compounds 1, 3, 4, 5 and 6 (0.1 mL, 3.3 mM, 33 % v/v DMSO/H2O) containing thermostable β-glucosidase (1.33 U) by A. niger EG (0.1 mL, 17.6 mU, 100 mM sodium acetate, pH 4.5). Reactions were stopped at 3, 6, 9 and 12 min by the addition of Tris buffer solution (3.0 mL, pH 9) and absorbances read at 400 nm
CELLG5 assay development
To determine the effect of substrate concentration (Fig. 3) on the rate of hydrolysis of 7 by T. longibrachiatum EG and A. niger EG, 0.1 mL of 7 (0.22–3.00 mM) in the presence of thermostable β-glucosidase (2 U), was incubated with either 0.1 mL of T. longibrachiatum EG (15.3 mU on CMC-4M, 14.6 mU on CELLG5) and A. niger EG (22.0 mU on CMC-4M, 17.4 mU on CELLG5) in 100 mM sodium acetate buffer (pH 4.5) at 40 °C. The reaction was terminated after fixed time intervals by adding 3 mL of 2 % w/v Tris solution (pH 9.0) and absorbance measured at 400 nm. K M values were calculated using Prism Version 6.04.

Effect of substrate 7 concentration on reaction kinetics over 8 min in the presence of a T. longibrachiatum EG (15.3 mU/assay on CMC-4M, 14.6 mU/assay on CELLG5) and b A. niger EG (22.0 mU/assay on CMC-4M, 17.4 mU/assay on CELLG5) plus β-glucosidase (2 U/assay) in pH 4.5 100 mM sodium acetate at 40 °C. Determination of the Michaelis-Menten K M constant for c T. longibrachiatum EG and d A. niger EG on substrate 7
The effect of β-glucosidase concentration (Fig. 4) on the rate of increase in absorbance at 400 nm was determined by preparing solutions containing 7 (3.3 mM) and β-glucosidase (1.7–20 U/mL) and was determined as follows: aliquots (0.1 mL) of the reagent solutions were incubated with T. longibrachiatum EG (0.1 mL, 100 mM sodium acetate, 18.9 mU on CMC-4M, 18.0 mU on CELLG5) at 40 °C and reaction terminated after 0, 3, 6, and 9 min by adding 3 mL of 2 % w/v Tris solution (pH 9.0).

Effect of β-glucosidase concentration on reaction kinetics over 9 min in the presence of T. longibrachiatum EG (18.9 mU/assay on CMC-4M, 18.0 mU/assay on CELLG5) and substrate 7 (1.67 mM) in pH 4.5 100 mM sodium acetate at 40 °C
Standard assay of EG using compound 7 in the presence of thermostable β-glucosidase – CELLG5 assay
A pre-equilibrated aliquot of EG (0.1 mL, 0–20 mU on CMC-4M, 100 mM suitable pH buffer) was added to a pre-equilibrated aliquot of compound 7 solution (0.1 mL, 3.3 mM, 10 % v/v DMSO/H2O) containing thermostable β-glucosidase (2 U) and the mixture was incubated at 40 °C for exactly 10 min. The reaction was terminated by adding 3 mL of 2 % w/v Tris solution (pH 9). The tube contents were well mixed and the absorbance measured at 400 nm against a reagent blank. One unit of activity is the amount of enzyme required to release 1 μmol of 4-nitrophenolate/min at 40 °C.
Comparison of CELLG5 with CELLG3
Standard curves for the action of T. longibrachiatum, A. niger, B. amyloliquefaciens, and T. maritima EGs on CELLG5 (Fig. 5) were determined by performing the standard CELLG5 assay on these enzymes using the following EG concentrations and buffers: T. longibrachiatum (0–18.0 mU/assay on CMC-4M, 0–17.1 mU/assay on CELLG5, 100 mM sodium acetate, pH 4.5), A. niger (0–22.0 mU/assay on CMC-4M, 0–17.4 mU/assay on CELLG5, 100 mM sodium acetate, pH 4.5), B. amyloliquefaciens (0–17.0 mU/assay on CMC-4M, 0–15.4 mU/assay on CELLG5, 100 mM sodium phosphate, pH 6), T. maritima (0–15.0 mU/assay on CMC-4M, 0–16.1 mU/assay on CELLG5, sodium phosphate, pH 6).

Standard curves relating absorbance increase at 400 nm observed with CELLG3 and CELLG5 reagents to T. longibrachiatum, A. niger, T. maritima and B. amyloliquefaciens EG activity on CMC-4M at 40 °C in pH 4.5 100 mM sodium acetate
To determine the effect of reducing the level of DMSO co-solvent required (Fig. 6), the standard CELLG5 assay was performed using A. niger EG (17.6 mU/assay on CMC-4M, 14.8 mU/assay on CELLG5) while varying the level of DMSO present in the CELLG5 reagent from 5–30 % v/v.

Effect of DMSO concentration on reaction kinetics over 10 min in the presence of A. niger EG (17.6 mU/assay on CMC-4M, 14.8 mU/assay on CELLG5), substrate 7 (1.67 mM) and thermostable β-glucosidase (2 U/assay) in pH 4.5 100 mM sodium acetate at 40 °C
To determine the thermal stability (Fig. 8a) observed for CELLG5 and CELLG3 a solution of substrate 1 or 7 (0.1 mL, 3.3 mM in 33 % v/v DMSO/H2O) was added to buffer solution (0.1 mL, 100 mM sodium acetate, pH 4.5) and incubated for 10 min at 60 °C, 70 °C, 80 °C and 90 °C. The incubation was stopped by the addition of 3 mL of 2 % w/v Tris solution (pH 9). The tube contents were well mixed and the absorbance measured at 400 nm against a reagent blank.
To determine the alkaline stability (Fig. 8b) observed for CELLG5 and CELLG3, a typical assay solution was prepared by adding a solution of compound 1 or 7 (0.1 mL, 3.3 mM in 33 % v/v DMSO/H2O) to buffer solution (0.1 mL, 100 mM sodium acetate, pH 4.5). To this assay solution was added a range of stop solutions (3 mL, 100 mM sodium carbonate, pH 11, 12 and 13) which were adjusted to the required pH using 4 M NaOH. The absorbance at 400 mM was recorded continuously over 30 min.
Long-term CELLG5 reagent stability
CELLG5 reagent was stored at −20 °C, 4 °C and 25 °C. The standard CELLG5 assay was performed using each of these reagents with a standardised solution of T. longibrachiatum EG (172 mU/mL on CMC-4M, 164 mU/mL on CELLG5) over a period of 3 months. Blank and reaction absorption values were monitored over this time period.
Repeatability and reproducibility using CELLG5
The standard CELLG5 assay was performed on three samples containing T. longibrachiatum EG (58, 86 and 172 mU/mL on CMC-4M; 55, 82 and 164 mU/mL on CELLG5) by two analysts on two consecutive days. The mean and standard deviation of the blank reaction value was determined from 10 replicates.
Automated CELLG5 assay
A standard curve for the action of T. longibrachiatum EG was created for the Chemwell-T automated CELLG5 assay (Fig. 9) by the incubation of CELLG5 reagent (25 μL, 3.3 mM, 10 % v/v DMSO/H2O) containing thermostable β-glucosidase (0.5 U) with a solution of T. longibrachiatum EG (25 μL, 2.1–11.1 on CMC-4M, 1.9–9.5 mU on CELLG5, 100 mM sodium acetate, pH 4.5) at 37 °C for 10 min. The reaction was terminated by the addition of stop buffer (300 μL, 500 mM sodium carbonate, pH 12) and the absorbance was read at 400 nm.
Standard assay for EG using CM-cellulose 4 M (10 mg/mL) by reducing sugar determination [9, 10, 13]
To a pre-equilibrated aliquot of CM-cellulose 4 M (0.5 mL, 10 mg/mL) in 100 mM sodium acetate buffer (pH 4.5) or 100 mM sodium phosphate (pH 6.0) a pre-equilibrated aliquot of EG was added. The resulting solution was mixed on a vortex stirrer and incubated at 40 °C. Incubations were terminated by addition of Nelson-Somogyi solution C (0.5 mL) at 0, 3, 6, 9 and 12 min. The colour was developed according to the Nelson-Somogyi procedure and incubation tubes centrifuged at 1500×g for 10 min to remove insoluble CM-cellulose 4 M. The absorbance was measured at 520 nm against a substrate/reagent blank and a glucose standard solution (50 μg) was included. One unit of activity is the amount of enzyme required to release 1 μmol of glucose reducing sugar equivalents/min at 40 °C and the pH used in the particular assay.
Results and discussion
Colorimetric endo-cellulase substrate requirements
In order to fully probe the effect of varying the chain length of the cellooligosaccharide core for this class of cellulase substrates, two novel substrates 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellopentaoside (3) and 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellohexaoside (4) were prepared using methodology described previously [18]. These substrates were used to assay A. niger EG under the conditions described in the “Enzymatic hydrolysis of a series of 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellooligosaccharides (DP2-6)” section alongside compounds 1, 5 and 6 which had been prepared and fully characterised previously [18]. The results shown in Fig. 2 highlight the need for the target substrate to contain a cellooligosaccharide core of at least DP5 to ensure near maximal hydrolytic activity. From this experiment it also emerged that substrates 3 and 4 exhibited much lower water solubility than their shorter chain analogues. The hexasaccharide 4 in particular needed to be used immediately once the substrate solution had been prepared as extensive precipitation would occur within 10–20 min even at 40 °C. Considering this data, it was decided to synthesise 4,6-O-(3-ketobutylidene)-4-nitrophenyl-β-d-cellopentaoside (7) as a new substrate for EG. The 3-ketobutylidene acetal had been employed by Ishimaru and co-workers [21–23] for a similar blocking role in an analogous maltooligosaccharide substrate in order to achieve higher water solubility than that observed for the benzylidene acetal. The synthesis proved relatively straightforward and gratifyingly, compound 7 exhibited much improved water solubility over its pentasaccharide analogue 3 requiring only 10 % v/v DMSO as a co-solvent to prevent precipitation at 3.3 mM.
Formulation of CELLG5 reagent
The K M for compound 7 was calculated as 0.19 mM and 0.91 mM for T. longibrachiatum EG and A. niger EG respectively as described in “CELLG5 assay development” section and shown in Fig. 3. The optimum level of ancillary β-glucosidase required to ensure maximum sensitivity in the assay of T. longibrachiatum EG was determined empirically to be 2 U/assay as described in “CELLG5 assay development” section and shown in Fig. 4. This β-glucosidase content was used in all subsequent assays and the term CELLG5 is used from here on to describe an assay reagent composed of a solution of compound 7 (3.3 mM in 10 % v/v DMSO) containing 20 U/mL thermostable β-glucosidase. The term CELLG3 is used to describe a solution of compound 1 (3.3 mM in 33 % v/v DMSO) containing 6.7 U/mL thermostable β-glucosidase which had been shown previously to be sufficient to ensure maximum assay sensitivity [18].
Investigation into the use of CELLG5 with EGs from various sources
The CELLG5 reagent was employed to assay four EGs from T. longibrachiatum, A. niger, B. amyloliquefaciens and T. maritima. These EGs were also assayed on CMC-4M using the Nelson-Symogyi reducing sugar method described in “Standard assay for EG using CM-cellulose 4 M (10 mg/mL) by reducing sugar determination [9, 10, 13]” section. The standard curves for these four EGs on the CELLG5 reagent are outlined in Fig. 5. Also included in this figure are the standard curves for two of the four EGs on the CELLG3 reagent which were used to draw a comparison between the two reagents. It is evident that there is very marginal difference observed in terms of hydrolytic rate between CMC-4M and the small molecule substrate in the case of CELLG5 while a stark difference of up to 20-fold is observed in the case of CELLG3. This experiment highlights the fact that the increase in DP from three to five corresponding to the evolution of CELLG3 into CELLG5 has apparently conferred a universal applicability of the latter across a wide range of EGs. Figure 5 clearly shows how activity units calculated through the reducing sugar assay on CMC-4M are almost interchangeable with the activity units calculated from the absorbance obtained in the CELLG5 assay. While it is possible that other EGs may exist that require even longer cellooligosaccharide substrates to achieve maximum hydrolytic activity it is noteworthy that the A. niger EG employed in this study was previously found to be the most recalcitrant of a series of 5 EGs from various sources towards the hydrolysis of short-chain cellooligosaccharides [13].
Comparison of CELLG5 reagent with CELLG3 reagent
Substrate solubility
The exchange of the benzylidene acetal that was employed with CELLG3 for the 3-ketobutylidene acetal in CELLG5 allowed a reduction in the DMSO co-solvent required to fully solubilise the reagent. Figure 6 shows the effect of varying the DMSO content in the reagent solution from 5–30 % v/v, an experiment that would not have been possible with CELLG3. It is evident that DMSO inhibits the hydrolytic activity of A. niger EG significantly with a 50 % increase in hydrolysis observed when the incubation is conducted in 5 % v/v DMSO as opposed to 30 % v/v DMSO. The same experiment was performed using T. longibrachiatum EG and no inhibitory effect from DMSO was observed (data not shown).
Colorimetric moiety
The use of NP over CNP as a chromophore in these applications is beneficial for a number of reasons. The extinction coefficient of NP (18.1 × 103 L mol−1 cm−1) is ~10 % higher than that of CNP (16.6 × 103 L mol−1 cm−1) which produces a corresponding increase in sensitivity at equimolar concentrations. NP functionalised substrates are also expected to exhibit improved thermal and alkaline stability over CNP analogues owing to the difference in the pK a of their chromophores (CNP = 5.45, NP = 7.18). CNP was employed as the chromophore in the CELLG3 substrate over NP for two major reasons: (a) It’s relatively low pK a provided for the possibility of performing kinetic assays of EG and (b) the increased leaving group ability of the chromophore accelerated the β-glucosidase mediated hydrolysis of the 2-chloro-4-nitrophenol-β-d-glucopyranoside that was formed as an intermediate in the assay reaction (Fig. 7a) therefore allowing a reduction in the quantity of β-glucosidase that was required to achieve maximum sensitivity. It was hypothesised that the latter benefit would not apply in the case of CELLG5 as many EGs would preferentially cleave between the second and third glucopyranosyl residues from the reducing end (Fig. 7b), thereby releasing 4-nitrophenyl-β-d-cellobioside as an intermediate. The rate-determining step in the hydrolysis of 4-nitrophenyl-β-d-cellobioside by β-glucosidase is the cleavage of the non-reducing end β-d-glucopyranosyl residue and so there would be no advantage in increasing the lability of the chromophore in this case. Given that the ability to perform kinetic assays for some EGs could be considered a niche application, it was decided that the overall advantages associated with the use of NP outweighed this.

Proposed hydrolysis points within CELLG3 and CELLG5 substrates generating alternative β-glucosidase substrates
Substrate stability
Figure 8 demonstrates the improved thermal and alkaline stability afforded to CELLG5 over CELLG3 in replacement of the CNP chromophore with NP. While the thermal stability observed for compound 1 is sufficient for the vast majority of applications (the absorbance increase observed following a 10-min incubation at 90 °C corresponds to substrate hydrolysis of <5 %), its alkaline lability is a potential issue. There is a growing body of EGs reported in the literature with alkaline pH optima [24–28]. Assays of these EGs need to be stopped with the addition of strongly alkaline buffers as these enzymes can exhibit significant activity above pH 11. A stop solution with pH > 12 for example would clearly be unsuitable for use with CNP containing substrates such as CELLG3 as spontaneous hydrolysis occurs readily under these conditions as can be seen from Fig. 8b. This spontaneous hydrolysis would lead to an increased absorbance at 400 nm and therefore an overestimation of EG activity. No absorbance increase was detected upon incubation of the CELLG5 reagent under all pH conditions investigated and the minimal hydrolysis that is observed following incubation at 100 °C for 10 min is irrelevant given that the maximum possible assay temperature is dictated by the thermostability limit of the ancillary β-glucosidase (90 °C).

Spontaneous hydrolysis of CELLG3 and CELLG5 substrates as observed by absorbance increase at 400 nm under a various temperature conditions: 10 min incubations of assay reagent (0.1 mL) with buffer (0.1 mL, 100 mM sodium acetate) at 40, 50, 60, 70, 80, 90 and 100 °C stopped by the addition of Tris buffer solution (0.1 mL, pH 9) and b various pH conditions: 20 min incubations of assay reagent (0.1 mL), buffer (0.1 mL, 100 mM sodium acetate, pH 4.5) and sodium carbonate buffer solution (0.1 mL, 100 mM, pH 12 and 13) at 40 °C
The long-term stability of the CELLG5 reagent was also assessed at various temperatures. Solutions maintained at −20 and 4 °C for 3 months prior to their assay using a T. longibrachiatum EG standard solution behaved exactly the same as freshly prepared CELLG5 reagent while a solution kept at 25 °C lost efficacy after 2 weeks, presumably due to the loss of the ancillary β-glucosidase activity.
Reproducibility and repeatability of CELLG5 assay
The repeatability and reproducibility of the assay was determined by having two analysts perform a series of assays on three different samples ranging in activity from 55–164 mU/mL over two consecutive days and the results are outlined in Table 1. Ten replicates were used to determine the standard deviation of the blank sample solution absorbance (σ = 0.002).
The limit of detection (LOD) was calculated as 3.5 × 10−4 U/mL (3 × σ, which corresponds to an absorbance of 0.007) and the limit of quantification (LOQ) was calculated as 4.1 × 10−3 U/mL (10 × σ of the blank sample solution absorbance, which corresponds to an absorbance of 0.023).
CELLG5 automated assay format
As stated earlier, one of the major advantages of the enzyme-coupled assay format described here over traditional assay methods is the ability to perform high-throughput automated assays using autoanalyser systems. The CELLG5 reagent is readily adapted to this application by varying slightly the ratios of CELLG5 reagent, analyte sample size and stop solution as described in “Automated CELLG5 assay” section. Figure 9 shows the standard curve for the automated analysis of T. longibrachiatum cellulase on a Chemwell®-T system. This provides a platform for high-throughput analysis with only 25 μL of CELLG5 reagent required per assay, increasing the number of assays that can be performed by a factor of 4 over the manual assay format.

Standard curve relating absorbance increase at 400 nm observed with CELLG5 reagent under Chemwell®-T assay format (Automated CELLG5 assay Section) to T. longibrachiatum EG activity on CMC-4M at 37 °C in pH 4.5 100 mM sodium acetate
Conclusions
A novel substrate, namely 4,6-O-(3-ketobutylidene)-4-nitrophenyl-β-d-cellopentaoside (7), has been synthesised and combined with a thermostable β-glucosidase in a coupled enzyme assay format for the measurement of EG activity. This reagent, termed CELLG5, displays marked advances on its forerunner, CELLG3, which contained 4,6-O-benzylidene-2-chloro-4-nitrophenyl-β-d-cellotrioside (1). It has been shown to be more generally applicable to a wide range of EGs from various sources and to be more stable to high temperature and high pH conditions. Its higher water solubility allows for a reduction in the quantity of DMSO needed for complete dissolution which assists in curtailing any inhibitory effects caused by the co-solvent. A reproducibility/repeatability study has been performed and the assay shown to be extremely robust and also highly sensitive with impressive LOD (<0.5 mU/mL) and LOQ (<5 mU/mL) values. An automated version of the assay has also been developed for high-throughput applications.
Given the importance of EG as an industrial biocatalyst in a wide range of industries, there is a growing global need for novel EGs offering improved activity and stability. The directed evolution projects needed to unlock improved mutant enzymes rely heavily on high-throughput solution-based screening assays. The lack of a rapid, specific, automatable assay for this enzyme has restrained research and development in this area over the past number of decades. It is the opinion of the authors that this assay will find widespread use both in academia and industry.
References
Gusakov AV, Berlin AG, Popova NN, et al. A comparative study of different cellulase preparations in the enzymatic treatment of cotton fabrics. Appl Biochem Biotechnol. 2000;88:119–26.
Maurer KH. Development of new cellulases. In: Van Ee JH, Misset O, editors. Enzymes in detergency. New York: Marcel Dekker; 1997. p. 175–202.
Singh A, Kuhad RC, Ward OP. Industrial application of microbial cellulases. In: Kuhad RC, Singh A, editors. Lignocellulose biotechnology: future prospects. New Delhi: I. K. International; 2007. p. 345–58.
Buchert J, Suurnakki A, Tenkanen M, et al. Enzymatic characterization of pulps. In: Jeffries TW, Viikari L, editors. Enzymes for pulp and paper processing. Washington: ACS Chemical Society; 1996. p. 38–43.
Campbell GL, Bedford MR. Enzyme applications for monogastric feeds: a review. Can J Anim Sci. 1992;72:449–66.
Maki M, Leung KT, Qin W. The prospects of cellulase-producing bacteria for the bioconversion of lignocellulosic biomass. Int J Biol Sci. 2009;5(5):500–16.
Bamforth CW. Current perspectives on the role of enzymes in brewing. J Cereal Sci. 2009;50(3):353–7.
Oksanen J, Ahvenainen J, Home S. Microbial cellulase for improving filterability of wort and beer. In: Wijngaarden MV, editor. Proceedings of the 20th European Brewery chemistry congress. Oxford: Oxford University Press; 1985. p. 419–25.
Nelson N. A photometric adaptation of the Somogyi method for the determination of glucose. J Biol Chem. 1944;153:375–80.
Somogyi M. Notes on sugar determination. J Biol Chem. 1952;195:19–23.
Manning K. Improved viscometric assay for cellulase. J Biochem Biophys Methods. 1981;5(4):189–202.
McCleary BV. New chromogenic substrates for the assay of alpha-amylase and (1–4)-β-d-glucanase. Carbohydr Res. 1980;86:97–104.
McCleary BV, McKie V, Draga A. Cellulases. In: Gilbert H, editor. Methods in enzymology. Cambridge: Academic; 2012. p. 1–17.
Bhat KM, Hay AJ, Claeyssens M, et al. Study of the mode of action and site-specificity of the endo-(1–4)-beta-d-glucanases of the fungus Penicillium pinophilum with normal, 1-3H-labelled, reduced and chromogenic cello-oligosaccharides. Biochem J. 1990;266:371–8.
Claeyssens M, Aerts G. Characterisation of cellulolytic activities in commercial Trichoderma reesei preparations: an approach using small, chromogenic substrates. Bioresour Technol. 1992;39:143–6.
Rahman MM, Bhuiyan SH, Nirasawa S, et al. Characterisation of an endo-β-1,4-glucanase of Thermatoga maritima expressed in Escherichia coli. J Appl Glycosci. 2002;49:487–95.
Claeyssens M, Henrissat B. Specificity mapping of cellulolytic enzymes: classification into families of structurally related proteins confirmed by biochemical analysis. Protein Sci. 1992;1:1293–7.
McCleary BV, Mangan D, Daly R, et al. Novel substrates for the measurement of endo-1,4-β-glucanase (endo-cellulase). Carbohydr Res. 2014;385:9–17.
Mangan D, McCleary BV, Liadova A, et al. Quantitative fluorometric assay for the measurement of endo-1,4-β-glucanase. Carbohydr Res. 2014;395:47–51.
Planas A, Abel M, Millet Ó, et al. Synthesis of aryl 3-O-beta-cellobiosyl-beta-d-glucopyranosides for reactivity studies of 1,3-1,4-beta-glucanases. Carbohydr Res. 1998;310:53–64.
Teshima S, Hayashi Y, Emi S, et al. Determination of alpha-amylase using a new blocked substrate (3-ketobutylidene beta-2-chloro-4-nitrophenyl-maltopentaoside). Clin Chim Acta. 1991;199(1):23–31.
Ishimaru K, Kamezono Y, Teshima S, et al. Enzymatic synthesis of 2-chloro-4-nitrophenyl 4,6-O-3-ketobutylidene beta-maltopentaoside, a substrate for alpha-amylase. Biosci Biotechnol Biochem. 1992;56(10):1552–6.
Ishimaru K, Kamezono Y, Hanayama N. New method for preparing 3-ketobutylidene 2-chloro-4-nitrophenyl β-maltopentaoside. Biosci Biotechnol Biochem. 1992;56(7):1136–7.
Vega K, Villena GK, Sarmiento VH, et al. Production of alkaline cellulase by fungi isolated from an undisturbed rain forest of Peru. Biotechnol Res Int. 2012. doi:10.1155/2012/934325
Ito S. Alkaline cellulases from alkaliphilic Bacillus: enzymatic properties, genetics, and application to detergents. Extremophiles. 1997;1(2):61–6.
Deka D, Jawed M, Goyal A. Purification and characterization of an alkaline cellulase produced by Bacillus subtilis (AS3). Prep Biochem Biotechnol. 2013;43(3):256–70.
Shirai T, Ishida H, Noda J, et al. Crystal structure of alkaline cellulase K: insight into the alkaline adaptation of an industrial enzyme. J Mol Biol. 2001;310(5):1079–87.
Hakamada Y, Koike K, Yoshimatsu T, et al. Thermostable alkaline cellulase from an alkaliphilic isolate. Bacillus sp. KSM-S237. Extremophiles. 1997;1(3):151–6.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
All authors were full time employees of Megazyme International throughout this research project. K-CELLG5 and K-CELLG3 are now commercially available assay kits for the measurement of endo-1,4-β-glucanase (endo-cellulase) activity.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1410 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mangan, D., Cornaggia, C., McKie, V. et al. A novel automatable enzyme-coupled colorimetric assay for endo-1,4-β-glucanase (cellulase). Anal Bioanal Chem 408, 4159–4168 (2016). https://doi.org/10.1007/s00216-016-9507-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-016-9507-y