
Abstract
Summary
We performed an association study of five candidate genes within chromosome 3p14-25 in 1,080 Chinese female subjects. Polymorphisms in FLNB/CRTAP are associated with bone mineral density (BMD) in Chinese.
Introduction
Chromosomal region 3p14-25 has shown strong evidence of linkage to BMD in genome-wide linkage scans. The variants responsible for this linkage signal, nonetheless, remain obscure.
Methods
Thirty SNPs in five positional and functional candidate genes within 3p14-25 (PPARG, CRTAP, TDGF1, PTHR1, and FLNB) and rs7646054 in the ARHGEF3 gene were genotyped in a case-control cohort of 1,080 Chinese females. Allelic and haplotypic association were tested using logistic regression analysis implemented in PLINK software. Potential transcription factor binding sites were predicted with MatInspector.
Results
Multiple SNPs and haplotypes in FLNB were significantly associated with BMDs, with the strongest association between lumbar spine BMD and rs9828717 (p = 0.005). SNP rs7623768 and the haplotype G-C of rs4076086-rs7623768 in CRTAP were associated with femoral neck BMD (p = 0.009 and p = 0.003, respectively). PTHR1 showed haplotypic associations with lumbar spine and femoral neck BMD (p = 0.02 and p = 0.044, respectively). Nevertheless, the association between rs7646054 in ARHGEF3 and BMD observed in Caucasians was not replicated in our samples. Comparative genomics analysis indicated that rs9828717 is located within a highly conserved region. The minor T allele at rs9828717 may lead to loss of binding site for nuclear factor of activated T cells which binds and triggers the transcriptional program of osteoblasts.
Conclusions
Our data suggest that variants in FLNB and CRTAP at 3p are involved in BMD regulation in southern Chinese.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoporosis is a complex disease characterized by low bone mass and microarchitectural deterioration of bone tissue, leading to enhanced bone fragility and a consequent increase in fracture risk [1]. Assessment of bone mineral density (BMD) is a common approach to evaluate the risk of osteoporosis. BMD is under strong genetic control with heritability ranging from 0.63 to 0.75 at the femoral neck, 0.61 to 0.83 at the lumbar spine, and 0.66 to 0.79 at total hip [2–4]. Recently published genome-wide association studies have revealed a few well-known candidate genes, such as low-density lipoprotein receptor-related protein 5, receptor activator of nuclear factor kappa B ligand (RANKL), osteoprotegerin, estrogen receptor 1, and sclerostin as the causal genes that contribute to BMD variation [5–7]. Since these genes are thought to account for only a small proportion of the total variation in spine and hip BMD, further identification of additional variants remains vital to understand the pathogenesis of osteoporosis.
Chromosomal region 3p14-25 is a susceptible quantitative trait locus (QTL) for BMD regulation that has been identified by four independent linkage studies [8–11] and genome scan meta-analyses [12, 13]. The meta-analysis of published linkage scores in 12,685 individuals from 3,097 families suggested that the summed rank of 3p22.2-p14.1 (bin 3.3) is significantly higher than expected (p = 0.012) [12]. Our recent meta-analysis of genome-wide linkage data, which included 11,842 subjects from 3,045 families, showed that 3p25.3-p22.1 (bin 3.2) had a statistically significant high average rank for lumbar spine BMD in both the whole-sample and female-specific analysis [13].
Mullin et al. [14] recently genotyped 17 SNPs in Rho guanine nucleotide exchange factor 3 (ARHGEF3) and observed the strongest association for rs7646054, which was associated with BMD Z-score at spine (p = 0.006) and femoral neck (p = 0.0007) in postmenopausal Caucasian women. The Rho guanine nucleotide exchange factor 3 specifically activates two members of the RhoGTPase family: RHOA which has been implicated in osteoblast differentiation and RHOB which has a role in cartilage biology [14]. It is unclear whether rs7646054 exerts the same effect in Chinese women who have a different genetic background and lower osteoporosis prevalence compared with Caucasian women [15].
To identify the causal genes contributing to BMD regulation in 3p14-25, a gene-wide and tag SNP-based association study was conducted in 1,080 case-control subjects using both single marker and haplotype approaches on five candidate genes: peroxisome proliferator-activated receptor gamma (PPARG), cartilage-associated protein (CRTAP), teratocarcinoma-derived growth factor 1 (TDGF1), parathyroid hormone receptor type 1 (PTHR1), and filamin B, beta (FLNB). The bone-related traits and phenotypes in knockout mice of these five genes are summarized in Table 1. A SNP rs7646054 in novel ARHGEF3 gene, which was recently reported to be associated with BMD regulation in Caucasians [14], was also examined in our population.
Materials and methods
Subjects
This study included 1,080 southern Chinese female subjects selected from an expanding database of the Hong Kong Osteoporosis Study. Participants were ambulatory subjects recruited at road shows and health talks on osteoporosis since 1998. Women with a history of diseases known to affect bone mass including vitamin D deficiency, hypercalcaemia, primary and secondary hyperparathyroidism, hyper- and hypothyroidism, metabolic and congenital bone diseases, and use of medications that would affect bone metabolism were excluded. A detailed description of subject ascertainment, inclusion, and exclusion criteria has been described previously [4]. BMD was measured by dual energy X-ray absorptiometry (Hologic QDR 4500 plus, Waltham, MA, USA). The in vivo precision of the machine for lumbar spine, femoral neck, and total hip region was 1.2%, 1.5%, and 1.5%, respectively. Subjects with extreme BMD Z-scores at either lumbar spine L1–4 or femoral neck were included in the current study. Subjects with BMD Z-score ≤ −1.28 (lowest tenth percentile of the population) were defined as cases, while those with BMD Z-score ≥ +1 (highest 15th percentile of the population) were defined as controls. All participants gave informed consent, and the study was approved by the Ethics Committee of the University of Hong Kong and conducted according to the Declaration of Helsinki.
There were 457 cases and 254 controls for lumbar spine, 399 cases and 283 controls for femoral neck, and 356 cases and 260 controls for total hip. The Student's t test was applied to compare the characteristics and phenotypes of the cases and controls. Age, height, and weight are potential confounding factors influencing BMD variation. According to our previous heritability estimates for BMD, the proportion of variation explained by age, age2, height, and weight was around 0.3 in women [4]. Factors with significant difference in the cases and controls were employed as covariates in the subsequent analysis.
SNP selection and genotyping
Twenty-seven tag SNPs (tSNPs) from five candidate genes (PPARG, CRTAP, TDGF1, PTHR1, and FLNB) in the chromosomal region 3p14-25 were selected for genotyping based on the genotype data obtained from the Han Chinese panel of the phase II HapMap data [39]. The criterion for tagging was set at r2 > 0.8 and minor allele frequency (MAF) > 0.2. The 27 tag SNPs captured 82.4% of common variants in five genes. SNPs rs709157, rs2177153, and rs1131356 showed significant association with BMD in previous studies and are thus, examined in this study. A total of 30 SNPs were genotyped using high-throughput massArray technology. In the genotyping process, 5% of samples were duplicated for quality check, and the reproducibility rate exceeded 99.8%.
Mullin et al. recently reported strong associations between rs7646054 in ARHGEF3 and BMD Z-scores at the spine and femoral neck in postmenopausal women [14]. We, thus, also genotyped rs7646054 using the TaqMan Genotyping Assay C__29978110_10 (Applied Biosystems, CA, USA) in our case-control samples. Each reaction contained template DNA and a final concentration of 1x TaqMan PCR Master Mix, unlabeled forward and reverse primers, VIC, and 6FAM dye-minor groove binder labeled probe for detection of the two alleles. The polymerase chain reaction program was set at 50°C incubation for 2 min followed by 10 min at 92°C. A two-step reaction was repeated with 40 cycles, with denaturation at 92°C for 15 s and annealing and extension at 50°C for 1 min. Subsequent endpoint reading was performed on the PRISM 7000 Sequence Detection System (Applied Biosystems, CA, USA). The reproducibility and the call rate of the TaqMan assay were 100% and 98.7%, respectively.
Statistical analysis
PLINK, an open source tool set designed for analysis of large data sets in a computationally efficient manner [40], was utilized in quality control filtering, single- and multiple-marker association tests. SNPs missing greater than10%, MAF of less than 1%, or violating the Hardy–Weinberg equilibrium (HWE) (p < 0.001) were excluded from further analysis. Logistic regression for the additive model, with adjustment for covariates, was applied to test the single-marker genotypic association with BMD at different skeletal sites. The Fisher's exact test was employed to execute the basic allelic association test. The variable-size sliding window approach was adopted in haplotype analysis as it includes the SNPs that may fall outside predefined linkage disequilibrium (LD) block and thus, enables the full information on genetic variability to be utilized in haplotype analysis [41, 42]. Another advantage of the variable-size sliding window approach is its greater detection power compared with other association-mapping strategies that employ haplotype block or single-SNP locus [41]. With adjustment of covariates, global omnibus test was conducted on a set of SNPs: in H haplotypes with a frequency more than1%, an H-1 degree of freedom test was performed to compare the alternate model (each haplotype having a unique effect) with the null (no haplotypes having any different effect). When the omnibus test was deemed significant, haplotype-specific test was performed. A conditional haplotype test that controlled for a particular haplotype among a set of haplotypes was also conducted to determine if that particular haplotype alone leads to the significant omnibus association result. Haploview 4.1 [43] was adopted to generate the haplotype block structure for the genotyped markers that passed the quality control requirements. LD is not calculated if markers are greater than 500 kb apart. Statistical power was estimated by the “Case-Control for threshold-selected quantitative traits” module of the web-based Genetic Power Calculator (http://pngu.mgh.harvard.edu/~purcell/gpc/qcc.html) [44].
Bioinformatics analysis
A comparative genomics approach was adopted to determine potential functional elements in the candidate region associated with BMD variation. The chromosomal position of the region was submitted to the VISTA Genome browser. Pre-computed whole-genome alignment among large vertebrates, which had a high sensitivity in covering more than 90% of known exons, was available on the browser with timely update upon the release of new genome assemblies [45].
The sequence encompassing the significantly associated SNP was scanned against the weight matrices for vertebrates that were publicly available on MatInspector [46]. The optimized matrix threshold of a weight matrix was defined as the threshold that allowed a maximum of three matches in 10 kb of non-regulatory test sequences. The matrix similarity was calculated on-the-run by scanning the imported sequence against the relative frequency of each nucleotide at a particular position in the matrix. Only potential binding sites with: (1) matrix similarity exceeding the optimized threshold; and (2) matrix similarity greater than 0.85 were considered good matches.
Results
Subject characteristics
The characteristics of the subjects are outlined in Table 2. Student's t test was used to compare the mean age, height, weight, and BMD in the case- and control-group, without assuming equal variances. The covariates that showed significant differences between cases and controls were potential confounding factors for BMD variation. These were adjusted in the subsequent analysis as indicated in Table 2.
Quality control
The genomic position, MAF, HWE test statistic, and call rate for each tSNPs that satisfied quality control criteria are listed in Table 3. Two tSNPs (rs4684846 and rs4135280) had call rates less than 90%. One SNP (rs1805192) was monomorphic in our study population. These three SNPs, all located within PPARG, were excluded from further analysis. A SNP in CRTAP (rs4678478) violated the HWE with a p < 0.001 in both the case- and control-group and was also discarded from association analysis.
Single-marker association
The association of each SNP with BMDs at the lumbar spine, femoral neck, and total hip was evaluated using the additive and allelic model. SNPs with p value ≤ 0.05 in the single-marker association test are shown in Table 4. Multiple SNPs (rs9828717, rs1718454, and rs1718456) in FLNB showed significant genotypic association with lumbar spine BMD (p = 0.03–0.005). For femoral neck BMD, significant genotypic association was detected for rs7623768 in CRTAP (p = 0.009) and rs1718456 in FLNB (p = 0.027). Significant association with total hip BMD was only observed for multiple SNPs in FLNB: rs9828717, rs1718454, and rs9822918 (p = 0.016–0.048).
Haplotype analysis
SNPs with MAF of less than 0.1 were removed from the haplotype analysis to reduce the type I error rate. Table 5 lists the combinations of SNPs most significantly associated with BMD in each gene resulting in p < 0.05 in the covariate-adjusted omnibus test. Only haplotypes with frequency of greater than 0.05 are shown in the table.
The global omnibus test revealed that a region rs724448–rs2242116 within the PTHR1 gene, which was in strong LD (r2 = 0.96), was significantly associated with lumbar spine and femoral neck BMD after adjustment of height and weight (p = 0.02 and p = 0.044, respectively).
FLNB showed regional associations with BMDs at all three measured skeletal sites. The region rs9828717–rs1718456–rs1718481–rs1718454 was significantly associated with lumbar spine BMD (p = 0.003). However, none of the common haplotypes are associated with lumbar spine BMD. The region rs1718456–rs1718481–rs1718454 was significantly associated with femoral neck BMD (p = 0.03). The T–A–C haplotype was associated with lower BMD status (p = 0.013, odds ratio (OR) = 1.52) while the C–A–C haplotype was associated with higher BMD status (p = 0.0145, OR = 0.41). The global omnibus test was no longer significant after controlling for either of the haplotypes, indicating their potential role in regulation of femoral neck BMD. The region rs1718454–rs9822918 was significantly associated with total hip BMD (p = 0.027). The C–T and T–G haplotype were correspondingly associated with the increased (p = 0.006, OR = 1.69) and reduced risk of low BMD (p = 0.025, OR = 0.66).
The global omnibus test demonstrated that the region rs4076086–rs7623768 in CRTAP was significantly associated with femoral neck (p = 0.028) and total hip BMD (p = 0.015). According to the haplotype-specific and conditional haplotype test, G–C was potentially the haplotype that conferred a protective effect on femoral neck (p = 0.003, OR = 0.43) and total hip (p = 0.007, OR = 0.44) BMD.
rs7646054 in ARHGEF3 and BMD
Mullin et al. [14] recently reported a significant association between rs7646054 and BMD Z-score in postmenopausal women: subjects homozygous for the G allele had lower BMD than subjects heterozygous or homozygous for the A allele. The same model (AA + AG vs GG) was, therefore, adopted in the analysis of this SNP using logistic regression implemented in SPSS. No association was observed between rs7646054 and BMD Z-score at the lumbar spine, femoral neck, or total hip in the whole study population, nor in the 533 postmenopausal case-controls (results not shown).
Bioinformatics analysis
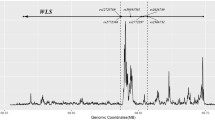
Since four of the five SNPs genotyped within intron 1 of FLNB showed significant associations with BMD in the single-marker test, the chromosomal position of intron 1 (Chr3:57,969,624-58,037,812) was submitted to VISTA genome browser to determine the presence of any potential conserved elements. RankVISTA for multiple alignment shows that intron 1 of FLNB in humans is a conserved noncoding sequence among five other species, including rhesus, dog, horse, mouse, and rat (Fig. 1). It is worth noting that rs9828717 is located within a highly conserved region with an alignment p value of 2.4 × 10−16. Prediction of potential transcription factor binding sites with MatInspector revealed that the minor T allele at rs9828717 may lead to the loss of binding site for nuclear factor of activated T cells (NFAT). The similarity score for the major C allele with NFAT matrix was 0.96.

VISTA browser plot of the comparative analysis for intron 1 in FLNB (Chr3:57,969,624-58,037,812 on the human March 2006 genome). The position of rs9828717 was indicated by the red arrow
Discussion
In the present study, we tested associations between common variants in five candidate genes in 3p14-25 (FLNB, PPARG, TDGF1, CRTAP, and PTHR1) and BMD in 1,080 southern Chinese women. Among these candidate genes, FLNB showed the strongest and most consistent association with BMD in both single-marker and haplotype analysis. At the SNP level, rs9828717, rs1718456, rs1718454, and rs9822918 were significantly associated with lumbar spine, femoral neck, or total hip BMD (p = 0.005–0.029). At the haplotype level, the strongest association was observed with total hip BMD for the haplotype C–T of rs1718454–rs9822918 (p = 0.006, OR = 1.69). Additionally, SNP rs7623768 and the haplotype G–C of rs4076086–rs7623768 in CRTAP is associated with femoral neck BMD (p = 0.009 and p = 0.003, respectively). PTHR1 showed haplotypic associations with lumbar spine and femoral neck BMD (p = 0.02 and p = 0.044, respectively).
Mutations in FLNB have been observed in a number of human skeletal disorders, including boomerang dysplasia [16], Larson syndrome [17, 18], spondylocarpotarsal synostosis [18, 19], and atelosteogenesis I and III [18, 20]. Together with the intense and uniform FLNB expression detected throughout the growth plate in normal mouse embryos in resting, proliferating, and prehypertrophic and hypertrophic chondrocytes, it is thought that FLNB plays a central role in skeletogenesis and joint formation [18]. Interestingly, a number of mutations that lead to the broad phenotypic spectrum are located within the actin-binding domain of FLNB. A functional actin cytoskeleton may be important for many normal morphogenetic processes, including skeletogenesis [16].
The phenotypes of FLNB-deficient mice also revealed the importance of the gene in skeletogenesis. FLNB −/− mice have vertebral fusions and abnormalities and decreased hyaline cartilage in the vertebral, carpal, and tarsal bones (Table 1) similar to the human clinical malformations seen in vertebral segmentation, joint formation, and skeletogenesis in the syndromes of spondylocarpotarsal syndrome [22, 23], atelosteogenesis I and III [23], Larsen syndrome [23], and boomerang dysplasia [23]. Scoliotic and kyphotic abnormalities of the vertebral column in FLNB −/− mice resemble those observed in human boomerang dysplasia [23].
In addition to these monogenic bone diseases, FLNB is also associated with human BMD measured at various sites. SNPs rs9822918 and rs2177153 were associated with age-corrected BMD at both the femoral neck (p = 0.02–0.0002) and total hip (p = 0.02–0.0006) in 771 women from the GENOS sib-pairs study [21]. Such association was replicated in a population-based cohort of 1,192 unrelated Caucasian women from the CAIFOS (CAlcium Intake Fracture Outcome Study)/CARES (Caring for Adults Recovering from the Effects of Stroke) study [21]. Both rs9822918 and rs2177153 were included in our present study. In our cohort, rs9822918 was also significantly associated with total hip BMD (p = 0.017, OR = 1.55). No association was nevertheless observed for rs2177153 (p > 0.05). The large discrepancy between the MAF of rs2177153 in Caucasian (MAF = 0.292 from HapMap) and southern Chinese women (MAF = 0.02 from the present study) may explain the association difference.
Kiel et al. [47] used the Affymetrix 100K SNP GeneChip marker set in the Framingham Heart Study to examine genetic associations with BMD. Two SNPs in FLNB were included in the 100K marker set. According to the results available at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000007.v3.p2, rs1658397 was significantly associated with lumbar spine BMD using the additive generalized estimating equation model (p = 0.0005) while rs6445945 demonstrated only a modest association (p = 0.03) in all the 1,141 phenotyped individuals. Both rs1658397 and rs6445945 are located within the BMD-associated rs9828717–rs1718456–rs1718481–rs1718454–rs9822918 locus. Nevertheless, HapMap phase II data revealed a large discrepancy in the MAF of these two markers between different ethnic groups. The frequency of the minor allele C of rs1658397 is 0.325 and 0.044 in Europeans and Han Chinese, respectively. With a MAF of 0.4 in the European population, rs6445945 is monomorphic in the Han Chinese. Thus, other variants within the locus may affect BMD regulation in the southern Chinese population. In our study, association was more significant at haplotype level than single-marker level, presumably implying that the real causal variant is located within this locus but was not tagged. Another possibility is that overall variation in this locus may influence BMD regulation. We have recently demonstrated that multiple genes at 1p36 contribute to osteoporosis susceptibility in Chinese [48]. Resequencing and genotyping with higher marker density in the FLNB gene may provide more evidence of a regional association with BMD.
The strongest association was observed for rs9828717 with lumbar spine BMD. Comparative genomics analyses indicated that the rs9828717 is located within a conserved noncoding sequence. Prediction of potential transcription factor binding sites shows that the minor T allele at rs9828717 may abolish the binding site of NFAT that the major C allele possesses. NFAT is a family of transcription factors with activity inhibited by calcineurin inhibitors. Bone loss has been observed in both humans [49] and rats [50] treated with calcineurin inhibitors. Such bone loss is attributable to the suppressive effects of calcineurin inhibitors on osteoblast differentiation and osteoblastic bone formation [51]. This has outweighed its inhibition of osteoclastogenesis by suppressing NFAT induction by RANKL [52]. In addition, NFATc2 knockout mice suffered from a reduction of trabecular bone volume caused by the downregulation of markers for osteoblastic bone formation [51]. The regulatory role of NFAT in osteoblastogenesis is in line with our association result that the minor T allele increases the risk of low BMD, as NFAT fails to bind and trigger the transcriptional program of osteoblasts.
CRTAP is expressed in both osteoblasts and osteoclasts. CRTAP shares homology with a family of putative prolyl 3-hydroxylases and can form a complex with cyclophilin B and prolyl 3-hydroxylase 1 which is crucial for bone development and collagen helix formation [53]. Loss of CRTAP in mice causes osteochondrodysplasia which is characterized by severe osteoporosis due to deficient bone formation [35]. Loss of CRTAP in humans is associated with recessive osteogenesis imperfecta [35]. In the current study, rs7623768 in CRTAP is significantly associated with femoral neck BMD (p = 0.009), and the haplotype G–C of rs4076086–rs7623768 is consistently associated with femoral neck BMD (p = 0.003) and total hip BMD (p = 0.007). We recently demonstrated that variants of the sclerostin gene that cause sclerosteosis and van Buchem disease are also associated with osteoporosis [54]. Association of CRTAP polymorphisms with femoral neck BMD further supports previous observations that genes associated with monogenic bone diseases also contribute to BMD variation and osteoporosis risk in the general population.
PTHR1 is a member of the superfamily of G-protein-coupled receptors. The gain-of-function mutations in the PTHR1 gene cause Jansen's metaphyseal chondrodysplasia that is characterized by growth plate abnormalities and increased bone resorption, while loss-of-function mutations in PTHR1 cause Blomstrand chondrodysplasia which is characterized by advanced endochondral bone maturation and increased BMD. In the current study, PTHR1 showed haplotypic association with lumbar spine and femoral neck BMD (p = 0.02 and p = 0.044, respectively), although no association was observed between BMD and individual SNP in PTHR1. It is worth noting that two previous studies also reported the association of BMD with haplotypes but not single SNPs in this region of PTHR1 [29, 31]. It is likely that untyped common variant or multiple rare variants are responsible for the observed association. Because SNPs in this region of PTHR1 are in strong LD, it is difficult to clearly define the primary associated variant(s) by population genetics approaches. Functional assessment of the variants via computational methods, laboratory assays, or model systems will be required to determine variant(s) responsible and the mechanism of the observed association.
The strength of our study is that the selected sampling strategy can substantially increase power over random sampling for detection of allelic association [55]. Assuming a marker is in complete LD (D′ = 1) with a QTL or the causal allele accounting for 1% of BMD variation and the MAFs of the marker and QTL are both 0.1, more than 98% power can be achieved to detect the additive genetic effects of the marker at a significance level of α = 0.05 in the whole study population. Making the same assumptions with use of the same parameters, the power was 87%, 77%, and 73% for lumbar spine, femoral neck, and total hip BMD, respectively, in the postmenopausal women subgroup. Based on the power calculation, our study should have sufficient power to detect any association between a marker and BMD. Nonetheless, this study failed to replicate the association between rs7646054 in ARFGEH3 and BMD in postmenopausal women recently observed by Mullin et al. [14]. The limitation of this study was that some susceptibility genes/variants may have been missed in this well-replicated region, because a candidate gene approach was used. Recent genome-wide association studies demonstrated that many associations implicate non-protein-coding regions [5–7]. Another limitation of this study was no correction for multiple testing. Although smaller p values generally provide greater support for a true association, it is the consistency and strength of the association across one or more replication studies, rather than the strength of the p value in a single study, that is critical to exclude false-positive association. Thus, we mainly evaluated the significance of our association in relation to previous replication. Since our design and choice of SNPs was based on evidence drawn from previous linkage and functional studies, our success to replicate the association of some of the SNPs provides evidence that these associations are likely to be valid.
In conclusion, our results suggest that FLNB and CRTAP are promising susceptibility genes for BMD regulation within 3p14-25 in the southern Chinese women. Further replication and functional studies are required to elucidate their role in bone remodeling.
References
World Health Organization (1994) Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO Study Group. World Health Organ Tech Rep Ser 843:1–129
Huang QY, Kung AWC (2006) Genetics of osteoporosis. Mol Genet Metab 88:295–306
Deng FY, Lei SF, Li MX, Jiang C, Dvornyk V, Deng HW (2006) Genetic determination and correlation of body mass index and bone mineral density at the spine and hip in Chinese Han ethnicity. Osteoporos Int 17:119–124
Ng MY, Sham PC, Paterson AD, Chan V, Kung AW (2006) Effect of environmental factors and gender on the heritability of bone mineral density and bone size. Ann Hum Genet 70:428–438
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Center JR, Nguyen TV, Bagger Y, Gulcher JR, Eisman JA, Christiansen C, Sigurdsson G, Kong A, Thorsteinsdottir U, Stefansson K (2008) Multiple genetic loci for bone mineral density and fractures. N Engl J Med 358:2355–2365
Richards JB, Rivadeneira F, Inouye M, Pastinen TM, Soranzo N, Wilson SG, Andrew T, Falchi M, Gwilliam R, Ahmadi KR, Valdes AM, Arp P, Whittaker P, Verlaan DJ, Jhamai M, Kumanduri V, Moorhouse M, van Meurs JB, Hofman A, Pols HA, Hart D, Zhai G, Kato BS, Mullin BH, Zhang F, Deloukas P, Uitterlinden AG, Spector TD (2008) Bone mineral density, osteoporosis, and osteoporotic fractures: a genome-wide association study. Lancet 371:1505–1512
Styrkarsdottir U, Halldorsson BV, Gretarsdottir S, Gudbjartsson DF, Walters GB, Ingvarsson T, Jonsdottir T, Saemundsdottir J, Snorradóttir S, Center JR, Nguyen TV, Alexandersen P, Gulcher JR, Eisman JA, Christiansen C, Sigurdsson G, Kong A, Thorsteinsdottir U, Stefansson K (2009) New sequence variants associated with bone mineral density. Nat Genet 41:15–17
Duncan EL, Brown MA, Sinsheimer J, Bell J, Carr AJ, Wordsworth BP, Wass JA (1999) Suggestive linkage of the parathyroid receptor type 1 to osteoporosis. J Bone Miner Res 14:1993–1999
Wilson SG, Reed PW, Bansal A, Chiano M, Lindersson M, Langdown M, Prince RL, Thompson D, Thompson E, Bailey M, Kleyn PW, Sambrook P, Shi MM, Spector TD (2003) Comparison of genome screens for two independent cohorts provides replication of suggestive linkage of bone mineral density to 3p21 and 1p36. Am J Hum Genet 72:144–155
Xiao P, Shen H, Guo YF, Xiong DH, Liu YZ, Liu YJ, Zhao LJ, Long JR, Guo Y, Recker RR, Deng HW (2006) Genomic regions identified for BMD in a large sample including epistatic interactions and gender-specific effects. J Bone Miner Res 21:1536–1544
Streeten EA, McBride DJ, Pollin TI, Ryan K, Shapiro J, Ott S, Mitchell BD, Shuldiner AR, O'Connell JR (2006) Quantitative trait loci for BMD identified by autosome-wide linkage scan to chromosomes 7q and 21q in men from the Amish family osteoporosis study. J Bone Miner Res 21:1433–1442
Lee YH, Rho YH, Choi SJ, Ji JD, Song GG (2006) Meta-analysis of genome-wide linkage studies for bone mineral density. J Hum Genet 51:480–486
Ioannidis JP, Ng MY, Sham PC, Zintzaras E, Lewis CM, Deng HW, Econs MJ, Karasik D, Devoto M, Kammerer CM, Spector T, Andrew T, Cupples LA, Duncan EL, Foroud T, Kiel DP, Koller D, Langdahl B, Mitchell BD, Peacock M, Recker R, Shen H, Sol-Church K, Spotila LD, Uitterlinden AG, Wilson SG, Kung AW, Ralston SH (2007) Meta-analysis of genome-wide scans provides evidence for sex- and site-specific regulation of bone mass. J Bone Miner Res 22:173–183
Mullin BH, Prince RL, Dick IM, Hart DJ, Spector TD, Dudbridge F, Wilson SG (2008) Identification of a role for the ARHGEF3 gene in postmenopausal osteoporosis. Am J Hum Genet 82:1262–1269
Dvornyk V, Liu XH, Shen H, Lei SF, Zhao LJ, Huang QR, Qin YJ, Jiang DK, Long JR, Zhang YY, Gong G, Recker RR, Deng HW (2003) Differentiation of Caucasians and Chinese at bone mass candidate genes: implication for ethnic difference of bone mass. Ann Hum Genet 67:216–227
Bicknell LS, Morgan T, Bonafe L, Wessels MW, Bialer MG, Willems PJ, Cohn DH, Krakow D, Robertson SP (2005) Mutations in FLNB cause boomerang dysplasia. J Med Genet 42:e43
Bicknell LS, Farrington-Rock C, Shafeghati Y, Rump P, Alanay Y, Alembik Y, Al-Madani N, Firth H, Karimi-Nejad MH, Kim CA, Leask K, Maisenbacher M, Moran E, Pappas JG, Prontera P, de RT, Fryns JP, Sweeney E, Fryer A, Unger S, Wilson LC, Lachman RS, Rimoin DL, Cohn DH, Krakow D, Robertson SP (2007) A molecular and clinical study of Larsen syndrome caused by mutations in FLNB. J Med Genet 44:89–98
Krakow D, Robertson SP, King LM, Morgan T, Sebald ET, Bertolotto C, Wachsmann-Hogiu S, Acuna D, Shapiro SS, Takafuta T, Aftimos S, Kim CA, Firth H, Steiner CE, Cormier-Daire V, Superti-Furga A, Bonafe L, Graham JM Jr, Grix A, Bacino CA, Allanson J, Bialer MG, Lachman RS, Rimoin DL, Cohn DH (2004) Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation, and skeletogenesis. Nat Genet 36:405–410
Mitter D, Krakow D, Farrington-Rock C, Meinecke P (2008) Expanded clinical spectrum of spondylocarpotarsal synostosis syndrome and possible manifestation in a heterozygous father. Am J Med Genet 146:779–783
Farrington-Rock C, Firestein MH, Bicknell LS, Superti-Furga A, Bacino CA, Cormier-Daire V, Le MM, Baumann C, Roume J, Rump P, Verheij JB, Sweeney E, Rimoin DL, Lachman RS, Robertson SP, Cohn DH, Krakow D (2006) Mutations in two regions of FLNB result in atelosteogenesis I and III. Hum Mutat 27:705–710
Wilson SG, Mullin BH, Jones MR, Dick IM, Dudbridge F, Spector TD, Prince RL (2007) Variation in the FLNB gene regulates bone density in two populations of Caucasian women. J Bone Miner Res 22(suppl.1):S57
Farrington-Rock C, Kirilova V, Llard-Telm L, Borowsky AD, Chalk S, Rock MJ, Cohn DH, Krakow D (2008) Disruption of the FLNB gene in mice phenocopies the human disease spondylocarpotarsal synostosis syndrome. Hum Mol Genet 17:631–641
Zhou X, Tian F, Sandzen J, Cao R, Flaberg E, Szekely L, Cao Y, Ohlsson C, Bergo MO, Boren J, Akyurek LM (2007) Filamin B deficiency in mice results in skeletal malformations and impaired microvascular development. Proc Natl Acad Sci USA 104:3919–3924
Rhee EJ, Oh KW, Lee WY, Kim SY, Oh ES, Baek KH, Kang MI, Kim SW (2005) The effects of C16-->T polymorphisms in exon 6 of peroxisome proliferator-activated receptor-gamma gene on bone mineral metabolism and serum osteoprotegerin levels in healthy middle-aged women. Am J Obstet Gynecol 192:1087–1093
Rhee EJ, Oh KW, Yun EJ, Jung CH, Park CY, Lee WY, Oh ES, Baek KH, Kang MI, Park SW, Kim SW (2007) The association of Pro12Ala polymorphism of peroxisome proliferator-activated receptor-gamma gene with serum osteoprotegerin levels in healthy Korean women. Exp Mol Med 39:696–704
Ogawa S, Urano T, Hosoi T, Miyao M, Hoshino S, Fujita M, Shiraki M, Orimo H, Ouchi Y, Inoue S (1999) Association of bone mineral density with a polymorphism of the peroxisome proliferator-activated receptor gamma gene: PPARgamma expression in osteoblasts. Biochem Biophys Res Commun 260:122–126
Kawaguchi H (2006) Molecular backgrounds of age-related osteoporosis from mouse genetics approaches. Rev Endocr Metab Disord 7:17–22
Liu PY, Zhang YY, Lu Y, Long JR, Shen H, Zhao LJ, Xu FH, Xiao P, Xiong DH, Liu YJ, Recker RR, Deng HW (2005) A survey of haplotype variants at several disease candidate genes: the importance of rare variants for complex diseases. J Med Genet 42:221–227
Vilariño-Güell C, Miles LJ, Duncan EL, Ralston SH, Compston JE, Cooper C, Langdahl BL, Maclelland A, Pols HA, Reid DM, Uitterlinden AG, Steer CD, Tobias JH, Wass JA, Brown MA (2007) PTHR1 polymorphisms influence BMD variation through effects on the growing skeleton. Calcif Tissue Int 81:270–278
Scillitani A, Jang C, Wong BY, Hendy GN, Cole DE (2006) A functional polymorphism in the PTHR1 promoter region is associated with adult height and BMD measured at the femoral neck in a large cohort of young Caucasian women. Hum Genet 119:416–421
Zhang YY, Liu PY, Lu Y, Xiao P, Liu YJ, Long JR, Shen H, Zhao LJ, Elze L, Recker RR, Deng HW (2006) Tests of linkage and association of PTH/PTHrP receptor type 1 gene with bone mineral density and height in Caucasians. J Bone Miner Metab 24:36–41
Duchatelet S, Ostergaard E, Cortes D, Lemainque A, Julier C (2005) Recessive mutations in PTHR1 cause contrasting skeletal dysplasias in Eiken and Blomstrand syndromes. Hum Mol Genet 14:1–5
Karaplis AC, He B, Nguyen MT, Young ID, Semeraro D, Ozawa H, Amizuka N (1998) Inactivating mutation in the human parathyroid hormone receptor type 1 gene in Blomstrand chondrodysplasia. Endocrinology 139:5255–5258
Barnes AM, Chang W, Morello R, Cabral WA, Weis M, Eyre DR, Leikin S, Makareeva E, Kuznetsova N, Uveges TE, Ashok A, Flor AW, Mulvihill JJ, Wilson PL, Sundaram UT, Lee B, Marini JC (2006) Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta. N Engl J Med 355:2757–2764
Morello R, Bertin TK, Chen Y, Hicks J, Tonachini L, Monticone M, Castagnola P, Rauch F, Glorieux FH, Vranka J, Bachinger HP, Pace JM, Schwarze U, Byers PH, Weis M, Fernandes RJ, Eyre DR, Yao Z, Boyce BF, Lee B (2006) CRTAP is required for prolyl 3- hydroxylation and mutations cause recessive osteogenesis imperfecta. Cell 127:291–304
Bodian DL, Chan TF, Poon A, Schwarze U, Yang K, Byers PH, Kwok PY, Klein TE (2009) Mutation and polymorphism spectrum in osteogenesis imperfecta type II: implications for genotype–phenotype relationships. Hum Mol Genet 18:463–471
Baldridge D, Schwarze U, Morello R, Lennington J, Bertin TK, Pace JM, Pepin MG, Weis M, Eyre DR, Walsh J, Lambert D, Green A, Robinson H, Michelson M, Houge G, Lindman C, Martin J, Ward J, Lemyre E, Mitchell JJ, Krakow D, Rimoin DL, Cohn DH, Byers PH, Lee B (2008) CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Hum Mutat 29:1435–1442
Huang QY, Li GH, Cheung WM, Song YQ, Kung AW (2008) Prediction of osteoporosis candidate genes by computational disease–gene identification strategy. J Hum Genet 53:644–655
Consortium International HapMap (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Guo Y, Li J, Bonham AJ, Wang Y, Deng H (2008) Gains in power for exhaustive analyses of haplotypes using variable-sized sliding window strategy: a comparison of association-mapping strategies. Eur J Hum Genet (in press)
Li Y, Sung WK, Liu JJ (2007) Association mapping via regularized regression analysis of single-nucleotide polymorphism haplotypes in variable-sized sliding windows. Am J Hum Genet 80:705–715
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Purcell S, Cherny SS, Sham PC (2003) Genetic power calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19:149–150
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:W273–W279
Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T (2005) MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942
Kiel DP, Demissie S, Dupuis J, Lunetta KL, Murabito JM, Karasik D (2007) Genome-wide association with bone mass and geometry in the Framingham heart study. BMC Med Genet 8:S14
Huang QY, Li GH, Kung AW (2009) Multiple osteoporosis susceptibility genes on chromosome 1p36 in Chinese. Bone 44:984–988
Rodino MA, Shane E (1998) Osteoporosis after organ transplantation. Am J Med 104:459–469
Cvetkovic M, Mann GN, Romero DF, Liang XG, Ma Y, Jee WS, Epstein S (1994) The deleterious effects of long-term cyclosporine A, cyclosporine G, and FK506 on bone mineral metabolism in vivo. Transplantation 57:1231–1237
Koga T, Matsui Y, Asagiri M, Kodama T, de CB, Nakashima K, Takayanagi H (2005) NFAT and Osterix cooperatively regulate bone formation. Nat Med 11:880–885
Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, Wagner EF, Mak TW, Kodama T, Taniguchi T (2002) Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 3:889–901
Marini JC, Cabral WA, Barnes AM, Chang W (2007) Components of the collagen prolyl 3-hydroxylation complex are crucial for normal bone development. Cell Cycle 6:1675–1681
Huang QY, Li GH, Kung AW (2009) The −9247 T/C polymorphism in the SOST upstream regulatory region that potentially affects C/EBPalpha and FOXA1 binding is associated with osteoporosis. Bone 45:289–294
Abecasis GR, Cookson WO, Cardon LR (2001) The power to detect linkage disequilibrium with quantitative traits in selected samples. Am J Hum Genet 68:1463–1474
Acknowledgments
This project is supported by Hong Kong Research Grant Council (HKU7514/06M), seed funding for basic research, the University of Hong Kong, and the Bone Health Fund. Qing-Yang Huang is partially supported by the KC Wong Education Foundation.
Conflicts of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 2.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by-nc/2.0/.
About this article
Cite this article
Li, G.H.Y., Kung, A.W.C. & Huang, QY. Common variants in FLNB/CRTAP, not ARHGEF3 at 3p, are associated with osteoporosis in southern Chinese women. Osteoporos Int 21, 1009–1020 (2010). https://doi.org/10.1007/s00198-009-1043-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-009-1043-6