
Abstract
Aims/hypothesis
Lack of insulin and infection/inflammation are the two most common causes of diabetic ketoacidosis (DKA). We used insulin withdrawal followed by insulin administration as a clinical model to define effects on substrate metabolism and to test whether increased levels of counter-regulatory hormones and cytokines and altered adipose tissue signalling participate in the early phases of DKA.
Methods
Nine individuals with type 1 diabetes, without complications, were randomly studied twice, in a crossover design, for 5 h followed by 2.5 h high-dose insulin clamp: (1) insulin-controlled euglycaemia (control) and (2) after 14 h of insulin withdrawal in a university hospital setting.
Results
Insulin withdrawal increased levels of glucose (6.1 ± 0.5 vs 18.6 ± 0.5 mmol/l), NEFA, 3-OHB (127 ± 18 vs 1837 ± 298 μmol/l), glucagon, cortisol and growth hormone and decreased HCO3− and pH, without affecting catecholamine or cytokine levels. Whole-body energy expenditure, endogenous glucose production (1.55 ± 0.13 vs 2.70 ± 0.31 mg kg−1 min−1), glucose turnover, non-oxidative glucose disposal, lipid oxidation, palmitate flux (73 [range 39–104] vs 239 [151–474] μmol/min), protein oxidation and phenylalanine flux all increased, whereas glucose oxidation decreased. In adipose tissue, Ser473 phosphorylation of Akt and mRNA levels of G0S2 decreased, whereas CGI-58 (also known as ABHD5) mRNA increased. Protein levels of adipose triglyceride lipase (ATGL) and hormone-sensitive lipase phosphorylations were unaltered. Insulin therapy decreased plasma glucose concentrations dramatically after insulin withdrawal, without any detectable effect on net forearm glucose uptake.
Conclusions/interpretation
Release of counter-regulatory hormones and overall increased catabolism, including lipolysis, are prominent features of preacidotic ketosis induced by insulin withdrawal, and dampening of Akt insulin signalling and transcriptional modulation of ATGL activity are involved. The lack of any increase in net forearm glucose uptake during insulin therapy after insulin withdrawal indicates muscle insulin resistance.
Trial registration
ClinicalTrials.gov NCT02077348
Funding
This study was supported by Aarhus University and the KETO Study Group/Danish Agency for Science Technology and Innovation.
Similar content being viewed by others


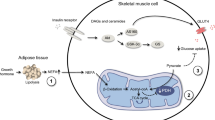

Introduction
Diabetic ketoacidosis (DKA) is one of the most common complications of type 1 diabetes and is one of the most serious acute medical conditions in the field of endocrinology, imposing substantial healthcare challenges and costs [1]. The in-hospital mortality rate is low (<1%) but overall mortality is increased in people >60 years of age, individuals with comorbidity [1,2,3], socioeconomic and psychosocial burdens [4] and in those from developing areas [5]. DKA and diabetic coma account for the greatest proportion of excess mortality (~25%) in individuals with type 1 diabetes aged <50 years [2, 6] and it is likely that a high mortality outside of hospital contributes to this.
The causes of DKA differ among countries, with lack of insulin and infection/inflammation in general accounting for the majority of all cases [7]. Given the clinical magnitude of DKA, relatively few studies have examined the chain of events leading to, and the mechanisms initiating, the metabolic disarray. DKA is characterised by relative or absolute insulin deficiency and increased levels of counter-regulatory hormones, such as glucagon, adrenaline (epinephrine), cortisol and growth hormone, and proinflammatory cytokines [1, 7]. These hormonal changes lead to increased endogenous glucose production (EGP), decreased glucose disposal in skeletal muscle, increased lipolysis and ketogenesis, eventually generating hyperglycaemia, ketonaemia, dehydration, electrolyte de-arrangement and metabolic acidosis. This concept is primarily based on observational studies showing increased levels of cytokines, stress hormones and NEFA in individuals admitted to hospital with fulminant DKA, leaving it open to discussion that these changes could be consequences of (rather than causative agents behind) the metabolic disarrangement [8]. Experimental studies using insulin withdrawal or deficiency models to assess the precipitating events in DKA in type 1 diabetes have shown increased lipolysis, increased NEFA availability and increased levels of glucagon to be important triggers of ketogenesis and that glucose and amino acid flux are increased [9,10,11]. It has been reported that energy expenditure is increased during insulin deficiency in type 1 diabetes [12, 13]. Generally, proinflammatory cytokines have not been measured and levels of cortisol, adrenaline and growth hormone, when measured, have not been increased. To our knowledge, no controlled clinical experimental studies have examined the effects of insulin treatment under conditions of incipient ketoacidosis after insulin withdrawal.
A recent experimental study using combined insulin deficiency (15% of basal insulin) and lipopolysaccharide (LPS) administration as a model reported release of proinflammatory cytokines and stress hormones and increased lipolysis as precipitating events in the early phases of DKA. These changes were associated with decreased mRNA levels of G0S2 and increased mRNA levels of CGI-58 (also known as ABHD5, encoding a potent stimulator of adipose triglyceride lipase [ATGL] activity), compatible with latent ATGL stimulation in adipose tissue [14]. The design did not allow for differentiation between the effects of insulin deficiency and the effects of acute LPS-induced inflammation.
The present study was designed to define the effects of insulin deprivation per se in the early precipitating phases of DKA and the metabolic impact of insulin treatment. We specifically sought to define the possible roles of increased levels of proinflammatory cytokines and stress hormones and to identify any precipitating intracellular adipocyte signalling event participating in this scenario, focusing on modulation of insulin signalling and ATGL. In addition, we examined the metabolic effects of insulin treatment in the early stages of DKA.
Methods
Participants, study design and protocol
A randomised crossover study was conducted involving nine male volunteers over two study days. Inclusion criteria were as follows: type 1 diabetes, C-peptide negative, age >18 and <65 years, BMI 19–26 kg/m2 and written and oral informed consent. Exclusion criteria were the presence of diabetic complications and other known diseases including ischaemic heart disease, cardiac arrhythmias and epilepsy. Prior to the studies, the participants underwent a medical interview, physical examination and blood tests. Twelve volunteers were examined, ten were found eligible for participation and nine participated (two were disqualified because C-peptide levels were 156 and 426 pmol/l and one decided not to participate).
Volunteers were randomly allocated to receive two interventions: (1) insulin and euglycaemia control (control) and (2) insulin withdrawal. Study days were separated by a minimum of 3 weeks.
Volunteers were instructed not to perform strenuous physical exercise 48 h prior to each study day. They were admitted to the medical research facility at 21:30 hours on the day before the study day and fasted until the end of the study day. Long- and intermediate-acting insulin treatments were paused and only short-acting insulin was given on the day prior to the study. Between 21:30 hours and 22:00 hours, two i.v. catheters were placed in the right arm of each participant—one into an antecubital vein for insulin infusion (if the intervention was ‘control’) and another in a hand vein for blood ketone and glucose monitoring (FreeStyle Precision; Abbott Diabetes Care, Copenhagen, Denmark). Insulin (Actrapid; Novo Nordisk, Copenhagen, Denmark) was dissolved in a saline (154 mmol/l NaCl) bag, with 10 ml of the volunteers’ own blood, and administered from 22:00 hours based on hourly blood glucose measurements. During insulin withdrawal, the volunteers were insulin deprived (i.e. they did not receive any insulin) and the antecubital vein was kept patent with a slow saline infusion. The volunteers were supervised by a physician during their stay at the research facility for safety reasons and for glucose management overnight. At 06:00 hours on the study day, a third i.v. catheter was inserted in a retrograde fashion into an antecubital vein of the left arm and the volunteer’s right hand was placed in a heating pad for arterial blood sampling. Studies commenced at 06:45 hours (t = 0 min) and volunteers were examined for 7.5 h. After 5 h (basal period t = 300 min), an insulin infusion was administered for 2.5 h (until t = 450 min) with or without glucose infusion depending on plasma glucose measurements. For the first 30 min of the period, the insulin infusion rate was 3 mU kg−1 min−1; thereafter the infusion rate was reduced to 1.5 mU kg−1 min−1.
Abdominal subcutaneous fat biopsy
Fat biopsies were obtained under sterile conditions 15 min after application of local anaesthesia (10 ml of 1% wt/vol. lidocaine). Volunteers were biopsied from the right side at t = 0 min and from the left at t = 270 min. Biopsies were processed as described previously [14].
Western blotting
Western blotting analyses were performed using 4–15% Criterion XT Bis-Tris gels (Bio-Rad, Hercules, CA, USA) as described previously [15]. Stain-free protein technology was used to demonstrate equal loading [16]. Primary antibodies are listed in electronic supplementary materials (ESM) Methods.
mRNA
was isolated using TRIzol (Gibco BRL; Life Technologies, Roskilde, Denmark) and quantitative PCR (qPCR) was performed with a LightCycler 480 (Roche, Copenhagen Denmark). ATGL (also known as PNPLA2), CGI-58, G0S2 and PTEN genes were quantified using the housekeeping gene POLR2A as a reference. POLR2A was tested and found equal between interventions. Primers are listed in ESM Methods.
Forearm blood flow
Forearm blood flow (FBF) was measured by venous occlusion plethysmography [17] and was used to assess regional glucose and protein flux in the forearm. Measurements were performed in triplicate towards the end of the basal period (t = 240–260 min) and the insulin treatment period (t = 430–450 min). Means of the three measurements during both periods were used.
Palmitic acid flux
Palmitic acid flux was measured by the isotope dilution technique, using 9,10-[3H]palmitic acid (10.5 Bq/min) (PerkinElmer, Mechelen, Belgium/Department of Clinical Physiology and Nuclear Medicine, Aarhus University Hospital, Denmark) infused from t = 200 min to t = 260 min. Plasma samples, to measure palmitic acid concentration and specific activity (ratio between the isotope and total palmitic acid), were obtained at baseline (t = 0 min) and at t = 240, 250 and 260 min. Palmitic acid concentration and specific activity was measured using [2H31]palmitic acid as an internal standard [11]. A quality control standard was included in every assay.
Glucose kinetics
Forearm glucose disposal was calculated as the difference between arterial and venous plasma glucose concentrations multiplied by the forearm flow as measured by strain-gauge plethysmography [18]. We used a primed continuous infusion of [3H3]glucose (bolus 0.74 MBq; infusion 0.44 MBq/h) (GE Healthcare, Brøndby, Denmark) from t = 20 min to t = 260 min. The glucose rate of appearance (Ragluc) was calculated using the non-steady state equation of Steele [19], where VSteele = 200 ml/kg body weight × 0.65. Ragluc equals EGP (no glucose infused) and Rdgluc equals oxidative glucose disposal (OGD) plus non-oxidative glucose disposal (NOGD). OGD was estimated by indirect calorimetry and NOGD was calculated by subtracting OGD from Rdgluc [20].
Protein kinetics
From t = 20 min to t = 260 min, a primed continuous infusion of [13C]urea was administered (priming 390.6 mg; infusion 42 mg/h). From t = 80 min to t = 260 min, phenylalanine and tyrosine tracers were infused: [15N]phenylalanine (priming 0.75 mg/kg; infusion 0.75 mg kg−1 h−1); l-[ring-2H4]tyrosine (priming 0.5 mg/kg; infusion 0.5 mg kg−1 h−1) and [15N]tyrosine (bolus 0.3 mg/kg). For tracer calculations, arteriovenous plasma samples were drawn in triplicate at t = 240 min, t = 250 min and t = 260 min. GC-MS was used to measure isotopic enrichment of samples.
Whole-body flux of phenylalanine (QPhe) and tyrosine (QTyr) was calculated as described previously [21], and is reported as synthesis, breakdown, and phenylalanine to tyrosine conversion.
Indirect calorimetry
Indirect calorimetry (Deltatrac monitor; Dantes Instrumentarium, Helsinki, Finland) was used from t = 210 min to t = 225 min to measure total energy expenditure and RQ. Averages of all measurements except the first three were used. Urine was collected after both the basal period and the clamp period, for measures of urea excretion rates from which estimates of protein oxidation rates were calculated. Lipid and glucose oxidation rates were calculated as described previously [22].
Biochemical variables
Plasma glucose and lactate were measured using the YSI 2300 STAT plus (YSI Life Sciences, Yellow Springs, OH, USA). Serum insulin was measured by ELISA (Dako, Glostrup, Denmark). Serum C-peptide was measured by ELISA (Alpco, Salem, NH, USA). Plasma glucagon was measured with an RIA kit (EMD Millipore, Darmstadt, Germany). Plasma adrenaline and noradrenaline were measured by electrochemical detection following HPLC [23]. Serum growth hormone was measured using chemiluminescence technology (IDS-iSYS Multi-Discipline Automated Analyzer; Immunodiagnostic Systems Nordic S/A, Copenhagen, Denmark). Serum cortisol was measured by ELISA (DRG Cortisol Enzyme Immunoassay Kit, Marburg, Germany). Serum NEFA was measured with a commercial kit (Wako Chemicals, Neuss, Germany). Whole blood β-Hydroxybutyrate (3-hydroxybutyrate [3-OHB]) was measured by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) [24]. Plasma TNFα, IL-1b, IL-6 and IL-10 were measured using a magnetic beads human chemokine assay (Bio-Rad, Hercules, CA, USA).
Statistical analyses
Statistical analyses were performed using Stata version 13.0 (StataCorp, College Station, TX, USA) with Student’s paired t test or a mixed linear model when relevant. The mixed model contained visit number, visit order, intervention, time and the interaction between intervention and time as factors. When the t test was used, data were checked for normality by inspection of qq plots by intervention. When the mixed model was used, model validation was performed by inspection of qq plots of the residuals and scatterplots of the predicted values against the fitted values. If data were not normally distributed, logarithmic transformation was performed to obtain normality. Data are presented as crude mean ± SEM or median with range. Comparisons are shown as differences with 95% CIs; p < 0.05 was considered statistically significant.
Ethics statement
This project was approved by the local scientific ethics committee, Viborg, Denmark (1-10-72-247-13), conducted in accordance with the Helsinki Declaration II and volunteers gave their written and oral informed consent. The original protocol also included a separate experiment using injection of growth hormone; data from this arm are under analysis. The study was registered with ClinicalTrials.gov, with identifier NCT02077348.
Results
Participants
Nine volunteers with type 1 diabetes (median [range] age 29 [20–58] years; mean ± SEM BMI 24.2 ± 0.9 kg/m2) completed both study days and were included in the analysis. On the day of insulin deficiency, volunteers experienced mild symptoms of thirst, urinary urgency and nausea and several reported experiencing a ‘feeling of hyperglycaemia’. No serious adverse events occurred.
Hormones, substrates, cytokines and clinical variables
Table 1 shows the clinical and biochemical variables of the participants. Insulin withdrawal decreased insulin levels throughout the basal period in all volunteers (p < 0.001) and increased levels of glucose, NEFA, glucagon, cortisol and growth hormone (p < 0.001), without affecting adrenaline (epinephrine), noradrenaline (norepinephrine) or cytokine levels. Insulin withdrawal also increased levels of 3-OHB (p < 0.001) and decreased HCO3− and pH (p < 0.001).
Energy expenditure and substrate oxidation (basal period)
Insulin withdrawal increased basal energy expenditure by 1207 kJ/day (95% CI 770, 1644 kJ/day; p < 0.001) (Table 2). Heart rate increased from 64 beats/min during the control period to 76 beats/min during insulin withdrawal (Table 1, p < 0.001). Furthermore, insulin withdrawal shifted energy substrate utilisation from glucose to lipids and protein. During insulin withdrawal, glucose oxidation decreased by 941 kJ/day (95% CI −1961, 77 kJ/day; p = 0.07), protein oxidation increased by 582 kJ/day (95% CI −218, 1381 kJ/day; p = 0.13) and lipid oxidation increased by 1613 kJ/day (95% CI 474, 2753 kJ/day; p = 0.01) (Table 2).
Whole-body glucose, protein and lipid metabolism (basal period)
Insulin withdrawal increased EGP by ~70%, from 1.55 ± 0.13 mg kg−1 min−1 under control conditions to 2.70 ± 0.31 mg kg−1 min−1 during insulin withdrawal (p < 0.05, Table 2). Similarly, glucose disposal was increased during insulin withdrawal by ~90%, from 1.81 ± 0.09 mg (kg body weight)−1 min−1 during control conditions to 3.41 ± 0.26 mg (kg body weight)−1 min−1 during insulin withdrawal (p < 0.01, Table 2). Whole-body NOGD, calculated as whole-body glucose disposal minus glucose oxidation, increased by ~20 fold (absolute difference 2.41 mg [kg body weight]−1 min−1 [95% CI 1.77, 3.05 mg (kg body weight)−1 min−1]) during insulin withdrawal (p < 0.001, Table 2).
The rate of palmitate flux was increased 3.3-fold by insulin withdrawal (95% CI 2.4, 4.4; p < 0.001): during control conditions, the lipolysis rate was 73 (range 39, 104) μmol/min vs 239 (range 151, 474) μmol/min during insulin withdrawal (Table 2). Palmitate concentrations increased from 74 ± 9 nmol/ml during control to 275 ± 37 nmol/ml during insulin withdrawal.
Insulin withdrawal increased whole-body phenylalanine breakdown by ~20%, from 33.0 ± 1.1 μmol kg−1 h−1during control conditions to 39.3 ± 1.3 μmol kg−1 h−1 during insulin withdrawal (p < 0.001) (Table 2). Similarly, phenylalanine synthesis was increased by ~20%, from 30.1 ± 1.0 μmol kg−1 h−1 during control conditions to 36.4 ± 1.2 μmol kg−1 h−1 during insulin withdrawal (p < 0.001, Table 2) and phenylalanine concentrations were increased (Table 1). Conversion of phenylalanine to tyrosine (p = 0.8, Table 2) and urea flux (p = 0.3, Table 2) were not statistically significantly altered.
Regional (forearm) blood flow, glucose disposal and protein synthesis, breakdown and balance during the basal period
Insulin withdrawal increased FBF by ~25% during the basal period (p < 0.01, Fig. 1b). No statistically significant differences in regional glucose disposal were found between interventions at the end of the basal period (p = 0.8; Fig. 1c). Protein breakdown and synthesis in the forearm were not statistically altered during insulin withdrawal (Table 2), although forearm net phenylalanine balance was less negative during control conditions than during insulin withdrawal (p < 0.01; Table 2), indicating an increased net protein loss after insulin withdrawal. Phenylalanine concentrations were comparable between study days; concentrations were increased during insulin withdrawal (36.9 ± 4.5 μmol/l vs control 30.9 ± 3.5 μmol/l) but the difference was not statistically significant (p = 0.06, Table 1).

Measurements of (a) arterial glucose concentrations, (b) FBF, (c) forearm glucose disposal (FA GD) and (d) NEFA concentration throughout the study day; and (e) 3-OHB concentration during the basal period only. Black circles with dashed line, control conditions; white squares with solid line, insulin withdrawal. **p<0.01 and ***p<0.001 vs control. FA, forearm tissue (calculated as ml using venous occlusion plethysmography); GD, glucose disposal; IW, insulin withdrawal. Data are presented as a dot plot for data points shown (b and c) with mean ± SEM or as mean ± SEM, n=9 (a, d, and e)
Signalling and gene expression in adipose tissue
Insulin withdrawal decreased ser473 phosphorylation of Akt compared with control conditions (Fig. 2a, overall p = 0.01). No statistically significant differences were found between interventions with respect to protein kinase A (PKA)-dependent perilipin 1 (PLIN1) phosphorylation, acetyl-CoA carboxylase phosphorylation or hormone-sensitive lipase (HSL) ser552, ser555 or ser660 phosphorylation. Neither did we find any differences in total protein levels of comparative gene identification-58 (CGI-58) or ATGL in adipose tissue samples.

(a) Ratio of phosphorylated ser473 Akt and total Akt, and (c, d) gene expression of G0S2 (c) and CGI-58 (d) in adipose tissue during control conditions and insulin withdrawal at t=0 min and t=270 min (start and end of the basal period). All measurements are expressed as fold change relative to control at t=0. (b) A representative western blot of pAkt ser473 is shown. IW, insulin withdrawal. Horizontal lines show geometric mean (a and c) and mean (d)
Compared with control conditions, insulin withdrawal decreased G0S2 mRNA levels by ~50% (p < 0.01, Fig. 2c) and increased CGI-58 mRNA by more than twofold (p < 0.001, Fig. 2d). No differences were found in ATGL or PTEN mRNA expression (p > 0.05).
Effects of insulin treatment
At the end of the insulin treatment period (clamp), insulin levels were comparable between interventions at ~650 pmol/l (p = 0.32), whereas glucose levels were not (p < 0.001); glucose levels did not reach the goal of 5 mmol/l after insulin withdrawal (Fig. 1a).
The differences in FBF persisted during insulin treatment, measured 2.5 h after the end of the basal period (Fig. 1b).
At the end of the insulin treatment period, glucose disposal increased ~30-fold (p < 0.001) during control conditions, whereas there were no differences over time during insulin withdrawal (p = 0.7) (Fig. 1c). Comparing control conditions with insulin withdrawal at the end of the insulin treatment period revealed a massive difference of ~70-fold higher glucose uptake during control conditions (p < 0.001) (Fig. 1c).
Discussion
This study was designed to examine the effects of insulin withdrawal per se on the initiating metabolic events leading to DKA in individuals with type 1 diabetes, focusing on the possible role of counter-regulatory hormones and cytokines and on signalling events in adipose tissue. With this controlled experimental approach, we found clear evidence of incipient DKA, in terms of increased levels of 3-OHB and NEFA, and decreased pH and bicarbonate, together with increased energy expenditure and increased fluxes of palmitate, glucose and phenylalanine. Elevated levels of glucagon, cortisol and growth hormone and transcriptional alterations of ATGL modulators could contribute to these catabolic processes, whereas adrenaline and cytokines seem to be of minor importance. In addition, we assessed the metabolic response to insulin treatment and found evidence of severe insulin resistance in skeletal muscle.
It is of interest that our findings, using insulin deprivation alone, to a large degree resemble those recently reported using a combination of LPS-induced acute inflammation and insulin restriction [14, 25]. In both reports, glucagon, cortisol and growth hormone levels rose, 3-OHB concentrations increased to between 1.5 and 2 mmol/l, glucose increased to around 18 mmol/l and palmitate flux tripled. Furthermore, the only detectable signalling events in adipose tissue were increased CGI-58 and decreased G0S2 mRNA contents, suggesting a common pathogenesis, despite insulin concentrations being much lower in the present study (15 vs 30 pmol/l) and cytokine concentrations being higher after LPS administration.
We observed decreased ser473 phosphorylation of Akt and a weakened insulin signal in adipose tissue after insulin withdrawal, which obviously may contribute to the observed increases in palmitate flux and lipolysis [26]. In addition, insulin withdrawal was associated with increased levels of CGI-58 and decreased G0S2 mRNA, suggesting that ATGL activation participates in stimulation of lipolysis [26]. We were not able to detect any differences in pro-lipolytic signalling through the PKA to HSL pathway. This may be due to timing of adipose tissue sampling, as it has been reported that both increased ketone and lactate levels inhibit lipolysis [27,28,29]. Therefore, the pro-lipolytic effects of decreased insulin levels as well as increased growth hormone and cortisol levels could be partly counteracted by the anti-lipolytic effects of increased lactate and ketone levels, causing pro-lipolytic proteins such as PKA and HSL to fluctuate between active and inactive states. We did not find any differences in the total amount of CGI-58 or ATGL, possibly because of increased turnover of these proteins. Overall, it appears that increased ATGL activity plays a role in the increased lipolysis rate during insulin withdrawal, albeit currently there is no reliable measure of ATGL activity to confirm this.
Glucose metabolism was also massively affected by insulin withdrawal. The increased EGP during insulin withdrawal was accompanied by similar increases in glucose disposal, largely due to increased NOGD. The increased EGP may to a large extent be explained by high levels of glucagon and low levels of insulin. A recent study in dogs showed convincingly that insulin suppresses hepatic glucose production both directly through effects in the liver and indirectly by reducing glucagon secretion and lipolysis [30]. The increases in glucose disposal were not explained by increased net skeletal muscle glucose disposal, which was comparable between the two interventions at the end of the basal period (Fig. 1c). Thus, the increased glucose disposal must be explained by increased disposal in non-muscle tissues, such as the liver and the kidneys. It is likely that anaerobic glycolysis may contribute, as suggested by the increased levels of lactate after insulin withdrawal (Table 1). Skeletal muscle glucose disposal was stimulated by insulin during control conditions as expected; interestingly, this effect was completely abolished by prior insulin withdrawal (Fig. 1c) conceivably in part due to the decreasing glucose concentration gradient caused by declining levels of plasma glucose. This indicates that DKA in a clinical setting is accompanied by severe skeletal muscle insulin resistance and the lack of any detectable effect of insulin on net forearm muscle glucose uptake by implication suggests that the inhibition of EGP drives the fall in glucose concentrations. It should however be underlined that during insulin withdrawal circulating glucose concentrations dropped markedly, introducing a non-steady state that may lead to underestimation of the ability of insulin to promote skeletal muscle glucose uptake.
We observed a robust increase in NOGD measured by indirect calorimetry during insulin withdrawal. This could relate to increased glycogen synthesis, increased lactate formation/anaerobic glycolysis and increased glycosuria. In this context, it should be stressed that low levels of insulin and high levels of glucagon [31, 32] stimulate gluconeogenesis and fractional extraction of gluconeogenic precursors such as lactate, implying that the 50% increase in lactate concentrations could reflect a high turnover state with increased rates of appearance. In addition, high rates of gluconeogenesis may lead to underestimation of glucose oxidation and overestimation of NOGD due to a low respiratory quotient [22]. Finally, it should be mentioned that 72 h fasting, which in many aspects resembles ketoacidosis metabolically, leads to a 10% increase in human muscle glycogen content [33].
Insulin withdrawal significantly increased whole-body flux of phenylalanine and tyrosine (by ~20%) (Table 2; data for tyrosine flux not shown), in the presence of increased net forearm phenylalanine release and unaltered urea flux, phenylalanine-to-tyrosine conversion rates and protein oxidation, indicating that alterations in protein metabolism are less prominent in the early phase of DKA. Our observations confirm previous findings of increased leucine and phenylalanine flux [11, 34] in studies that included arteriovenous catheterisation techniques and showing that increased protein breakdown occurs in the splanchnic bed and that increased muscle (leg) protein breakdown may contribute, whereas kidney plays a minor role.
It is not certain which mechanisms trigger the release of glucagon, cortisol and growth hormone; we observed no alterations in concentrations of cytokines, so it appears implausible that these substances are involved. Insulin appears to inhibit glucagon secretion directly, implying that the increased levels of glucagon during insulin withdrawal may be caused by hypoinsulinaemia [29, 35]. It has also been reported that glucagon administration, through unknown mechanisms, elicits ACTH, cortisol and growth hormone responses in humans [36], whereas high levels of glucose and lipid intermediates would be expected to inhibit growth hormone secretion [37, 38].
As mentioned above, our forearm data show that insulin withdrawal leads to substantial insulin resistance in muscle, implying that individuals with diabetic ketoacidosis should be treated with relatively high doses of insulin. The peripheral insulin resistance is likely to be caused by a combination of high levels of NEFA, glucose, cortisol and growth hormone. One study of human islet transplantation in individuals with type 1 diabetes has linked improved insulin sensitivity to normalisation of NEFA dynamics [39] and many studies have shown that cortisol and growth hormone induce peripheral insulin resistance after a latency of several hours [38, 40]. When DKA is triggered by systemic inflammation, high levels of cytokines and adrenaline may contribute to muscle insulin resistance [14, 25].
Our study design has limitations. Adipose biopsies were obtained from subcutaneous abdominal depots and the results may have been different if the biopsies had been taken at other time points and/or from other locations. In addition, we only observed modest increments in 3-OHB, implying that our findings only apply to the initial events triggering DKA.
In conclusion, we show that release of counter-regulatory hormones and overall increased catabolism, including lipolysis, are distinct features of evolving ketoacidosis in an insulin withdrawal model. In this model, modulators of ATGL activity (decreased G0S2 and increased CGI-58 mRNA contents in adipose tissue) are transcriptionally involved, whereas cytokines and adrenaline are not detectably altered. The absence of any increase in forearm glucose uptake during insulin therapy indicates severe muscle insulin resistance and is compatible with the notion that insulin primarily lowers blood glucose by inhibition of EGP.
Data availability
The datasets generated and analysed during the current study are available from the corresponding author on request.
Abbreviations
- 3-OHB:
-
3-hydroxybutyrate (β-hydroxybutyrate)
- ATGL:
-
Adipose triglyceride lipase
- CGI-58:
-
Comparative gene identification-58
- DKA:
-
Diabetic ketoacidosis
- EGP:
-
Endogenous glucose production
- FBF:
-
Forearm blood flow
- HSL:
-
Hormone-sensitive lipase
- LPS:
-
Lipopolysaccharide
- NOGD:
-
Non-oxidative glucose disposal
- OGD:
-
Oxidative glucose disposal
- PKA:
-
Protein kinase A
References
Kitabchi AE, Umpierrez GE, Miles JM, Fisher JN (2009) Hyperglycemic crises in adult patients with diabetes. Diabetes Care 32(7):1335–1343. https://doi.org/10.2337/dc09-9032
Basu A, Close CF, Jenkins D, Krentz AJ, Nattrass M, Wright AD (1993) Persisting mortality in diabetic ketoacidosis. Diabet Med 10(3):282–284. https://doi.org/10.1111/j.1464-5491.1993.tb00060.x
Malone ML, Gennis V, Goodwin JS (1992) Characteristics of diabetic ketoacidosis in older versus younger adults. J Am Geriatr Soc 40(11):1100–1104. https://doi.org/10.1111/j.1532-5415.1992.tb01797.x
Randall L, Begovic J, Hudson M et al (2011) Recurrent diabetic ketoacidosis in inner-city minority patients: behavioral, socioeconomic, and psychosocial factors. Diabetes Care 34(9):1891–1896. https://doi.org/10.2337/dc11-0701
Muyer MT, Buntinx F, Mapatano MA, De Clerck M, Truyers C, Muls E (2010) Mortality of young patients with diabetes in Kinshasa, DR Congo. Diabet Med 27(4):405–411. https://doi.org/10.1111/j.1464-5491.2010.02961.x
Livingstone SJ, Levin D, Looker HC, Scottish Diabetes Research Network epidemiology group; Scottish Renal Registry et al (2015) Estimated life expectancy in a Scottish cohort with type 1 diabetes, 2008-2010. JAMA 313(1):37–44. https://doi.org/10.1001/jama.2014.16425
Umpierrez G, Korytkowski M (2016) Diabetic emergencies - ketoacidosis, hyperglycaemic hyperosmolar state and hypoglycaemia. Nat Rev Endocrinol 12(4):222–232. https://doi.org/10.1038/nrendo.2016.15
Stentz FB, Umpierrez GE, Cuervo R, Kitabchi AE (2004) Proinflammatory cytokines, markers of cardiovascular risks, oxidative stress, and lipid peroxidation in patients with hyperglycemic crises. Diabetes 53(8):2079–2086. https://doi.org/10.2337/diabetes.53.8.2079
Miles JM, Rizza RA, Haymond MW, Gerich JE (1980) Effects of acute insulin deficiency on glucose and ketone body turnover in man: evidence for the primacy of overproduction of glucose and ketone bodies in the genesis of diabetic ketoacidosis. Diabetes 29(11):926–930. https://doi.org/10.2337/diab.29.11.926
Miles JM, Haymond MW, Nissen SL, Gerich JE (1983) Effects of free fatty acid availability, glucagon excess, and insulin deficiency on ketone body production in postabsorptive man. J Clin Invest 71(6):1554–1561. https://doi.org/10.1172/JCI110911
Moller N, Jensen MD, Rizza RA, Andrews JC, Nair KS (2006) Renal amino acid, fat and glucose metabolism in type 1 diabetic and non-diabetic humans: effects of acute insulin withdrawal. Diabetologia 49(8):1901–1908. https://doi.org/10.1007/s00125-006-0287-3
Leslie P, Jung RT, Isles TE, Baty J, Newton RW, Illingworth P (1986) Effect of optimal glycaemic control with continuous subcutaneous insulin infusion on energy expenditure in type I diabetes mellitus. Br Med J (Clin Res Ed) 293(6555):1121–1126. https://doi.org/10.1136/bmj.293.6555.1121
Karakelides H, Asmann YW, Bigelow ML et al (2007) Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 56(11):2683–2689. https://doi.org/10.2337/db07-0378
Svart M, Kampmann U, Voss T et al (2016) Combined insulin deficiency and endotoxin exposure stimulate lipid mobilization and alter adipose tissue signaling in an experimental model of ketoacidosis in subjects with type 1 diabetes: a randomized controlled crossover trial. Diabetes 65(5):1380–1386. https://doi.org/10.2337/db15-1645
Nielsen TS, Vendelbo MH, Jessen N et al (2011) Fasting, but not exercise, increases adipose triglyceride lipase (ATGL) protein and reduces G(0)/G(1) switch gene 2 (G0S2) protein and mRNA content in human adipose tissue. J Clin Endocrinol Metab 96(8):E1293–E1297. https://doi.org/10.1210/jc.2011-0149
Gilda JE, Gomes AV (2013) Stain-free total protein staining is a superior loading control to beta-actin for Western blots. Anal Biochem 440(2):186–188. https://doi.org/10.1016/j.ab.2013.05.027
Cooper KE, Edholm OG, Mottram RF (1955) The blood flow in skin and muscle of the human forearm. J Physiol 128(2):258–267. https://doi.org/10.1113/jphysiol.1955.sp005304
Whitney RJ (1953) The measurement of volume changes in human limbs. J Physiol 121(1):1–27. https://doi.org/10.1113/jphysiol.1953.sp004926
Steele R (1959) Influences of glucose loading and of injected insulin on hepatic glucose output. Ann N Y Acad Sci 82:420–430
Moller N, Jorgensen JO, Alberti KG, Flyvbjerg A, Schmitz O (1990) Short-term effects of growth hormone on fuel oxidation and regional substrate metabolism in normal man. J Clin Endocrinol Metab 70(4):1179–1186. https://doi.org/10.1210/jcem-70-4-1179
Thompson GN, Pacy PJ, Merritt H et al (1989) Rapid measurement of whole body and forearm protein turnover using a [2H5]phenylalanine model. Am J Phys 256:E631–E639
Ferrannini E (1988) The theoretical bases of indirect calorimetry: a review. Metabolism 37(3):287–301. https://doi.org/10.1016/0026-0495(88)90110-2
Orskov L, Bak JF, Abildgard N et al (1996) Inhibition of muscle glycogen synthase activity and non-oxidative glucose disposal during hypoglycaemia in normal man. Diabetologia 39(2):226–234. https://doi.org/10.1007/BF00403967
Sorensen LK, Rittig NF, Holmquist EF et al (2013) Simultaneous determination of β-hydroxybutyrate and β-hydroxy-β-methylbutyrate in human whole blood using hydrophilic interaction liquid chromatography electrospray tandem mass spectrometry. Clin Biochem 46(18):1877–1883. https://doi.org/10.1016/j.clinbiochem.2013.08.011
Svart MV, Rittig N, Kampmann U, Voss TS, Møller N, Jessen N (2017) Metabolic effects of insulin in a human model of ketoacidosis combining exposure to lipopolysaccharide and insulin deficiency: a randomised, controlled, crossoverstudy in individuals with type 1 diabetes. Diabetologia 60(7):1197–1206. https://doi.org/10.1007/s00125-017-4271-x
Nielsen TS, Jessen N, Jorgensen JO, Moller N, Lund S (2014) Dissecting adipose tissue lipolysis: molecular regulation and implications for metabolic disease. J Mol Endocrinol 52(3):R199–R222. https://doi.org/10.1530/JME-13-0277
Senior B, Loridan L (1968) Direct regulatory effect of ketones on lipolysis and on glucose concentrations in man. Nature 219(5149):83–84. https://doi.org/10.1038/219083a0
Liu C, Wu J, Zhu J et al (2009) Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 284(5):2811–2822. https://doi.org/10.1074/jbc.M806409200
Moller N, Jorgensen JO, Moller J et al (1990) Substrate metabolism during modest hyperinsulinemia in response to isolated hyperketonemia in insulin-dependent diabetic subjects. Metabolism 39(12):1309–1313. https://doi.org/10.1016/0026-0495(90)90189-J
Edgerton DS, Kraft G, Smith M et al (2017) Insulin’s direct hepatic effect explains the inhibition of glucose production caused by insulin secretion. JCI Insight 2:e91863
Edgerton DS, Ramnanan CJ, Grueter CA et al (2009) Effects of insulin on the metabolic control of hepatic gluconeogenesis in vivo. Diabetes 58(12):2766–2775. https://doi.org/10.2337/db09-0328
Stevenson RW, Steiner KE, Davis MA et al (1987) Similar dose responsiveness of hepatic glycogenolysis and gluconeogenesis to glucagon in vivo. Diabetes 36(3):382–389. https://doi.org/10.2337/diab.36.3.382
Vendelbo MH, Clasen BF, Treebak JT et al (2012) Insulin resistance after a 72-h fast is associated with impaired AS160 phosphorylation and accumulation of lipid and glycogen in human skeletal muscle. Am J Physiol Endocrinol Metab 302(2):E190–E200. https://doi.org/10.1152/ajpendo.00207.2011
Nair KS, Ford GC, Ekberg K et al (1995) Protein dynamics in whole body and in splanchnic and leg tissues in type I diabetic patients. J Clin Invest 95(6):2926–2937. https://doi.org/10.1172/JCI118000
Unger RH (1985) Glucagon physiology and pathophysiology in the light of new advances. Diabetologia 28(8):574–578. https://doi.org/10.1007/BF00281991
Arvat E, Maccagno B, Ramunni J et al (2000) Interaction between glucagon and human corticotropin-releasing hormone or vasopressin on ACTH and cortisol secretion in humans. Eur J Endocrinol 143(1):99–104. https://doi.org/10.1530/eje.0.1430099
Lim CT, Khoo B (2000) Normal physiology of ACTH and GH release in the hypothalamus and anterior pituitary in man. In: De Groot LJ, Chrousos G, Dungan K et al (eds) Endotext. MDText.com, Inc., South Dartmouth
Moller N, Jorgensen JO (2009) Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev 30(2):152–177. https://doi.org/10.1210/er.2008-0027
Rickels MR, Naji A, Teff KL (2006) Insulin sensitivity, glucose effectiveness, and free fatty acid dynamics after human islet transplantation for type 1 diabetes. J Clin Endocrinol Metab 91(6):2138–2144. https://doi.org/10.1210/jc.2005-2519
Christiansen JJ, Djurhuus CB, Gravholt CH et al (2007) Effects of cortisol on carbohydrate, lipid, and protein metabolism: studies of acute cortisol withdrawal in adrenocortical failure. J Clin Endocrinol Metab 92(9):3553–3559. https://doi.org/10.1210/jc.2007-0445
Acknowledgements
The authors thank the following people at Aarhus University Hospital, Denmark for their excellent technical assistance: A. Mengel, K. Nyborg Rasmussen, E. Søgaard Hornemann and K. Mathiassen, Medical Research Laboratory; H. Zibrandtsen, Research Laboratory for Biochemical Pathology; L. Pedersen, Department of Endocrinology and Internal Medicine.
Contribution statement
TSV recruited the participants, conducted the trial and performed the statistical analyses. TSV, MHV, UK, NJ and NM contributed to conception and design of the study. NM, NJ, MVS, MJ, SBP, TSN and TSV collected and interpreted the data. TSV and NM drafted the manuscript and all authors revised it critically and approved the final version to be published. NM is the guarantor of this work and, as such, takes responsibility for the integrity of the data and the accuracy of the data analyses.
Funding
This study was supported by Aarhus University and the KETO Study Group/Danish Agency for Science Technology and Innovation (grant no. 0603-00479 [to NM]).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that there is no duality of interest associated with this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM Methods
(PDF 119 kb)
Rights and permissions
About this article

Cite this article
Voss, T.S., Vendelbo, M.H., Kampmann, U. et al. Substrate metabolism, hormone and cytokine levels and adipose tissue signalling in individuals with type 1 diabetes after insulin withdrawal and subsequent insulin therapy to model the initiating steps of ketoacidosis. Diabetologia 62, 494–503 (2019). https://doi.org/10.1007/s00125-018-4785-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-018-4785-x