
Abstract
Aims/hypothesis
Type 2 diabetes is likely to be an independent risk factor for hippocampal-based memory dysfunction, although this complication has yet to be investigated in detail. As dysregulated glycometabolism in peripheral tissues is a key symptom of type 2 diabetes, it is hypothesised that diabetes-mediated memory dysfunction is also caused by hippocampal glycometabolic dysfunction. If so, such dysfunction should also be ameliorated with moderate exercise by normalising hippocampal glycometabolism, since 4 weeks of moderate exercise enhances memory function and local hippocampal glycogen levels in normal animals.
Methods
The hippocampal glycometabolism in OLETF rats (model of human type 2 diabetes) was assessed and, subsequently, the effects of exercise on memory function and hippocampal glycometabolism were investigated.
Results
OLETF rats, which have memory dysfunction, exhibited higher levels of glycogen in the hippocampus than did control rats, and breakdown of hippocampal glycogen with a single bout of exercise remained unimpaired. However, OLETF rats expressed lower levels of hippocampal monocarboxylate transporter 2 (MCT2, a transporter for lactate to neurons). Four weeks of moderate exercise improved spatial memory accompanied by further increase in hippocampal glycogen levels and restoration of MCT2 expression independent of neurotrophic factor and clinical symptoms in OLETF rats.
Conclusions/interpretation
Our findings are the first to describe detailed profiles of glycometabolism in the type 2 diabetic hippocampus and to show that 4 weeks of moderate exercise improves memory dysfunction in type 2 diabetes via amelioration of dysregulated hippocampal glycometabolism. Dysregulated hippocampal lactate-transport-related glycometabolism is a possible aetiology of type-2-diabetes-mediated memory dysfunction.
Similar content being viewed by others

Introduction
Type 2 diabetes induces several complications, not only in peripheral organs but also in the central nervous system; in particular, hippocampal dysfunction, such as memory dysfunction, is now considered one such complication [1–4]. Hippocampal dysfunction in type 2 diabetes is a serious disability that impairs patient wellbeing and is also associated with the development of dementia, Alzheimer’s disease and depression [5, 6]. Therefore, it is critical to understand its aetiology and to innovate therapeutic strategies for diabetes-induced hippocampal memory dysfunction.
Although there are some factors and/or conditions reported to be involved in diabetes-induced memory dysfunction, such as inflammation, oxidative stress, decreased growth factors (i.e. brain-derived neurotrophic factor [BDNF]) [7, 8], the association of memory dysfunction with hippocampal glycometabolism in type 2 diabetes is yet to be clarified. Indeed, disturbance/alteration of glycometabolism is a common symptom of diabetes complications. Excess glycogen deposition has already been demonstrated in the heart of diabetes mellitus patients as an adaptation process [9, 10], but detailed characterisation of alteration in hippocampal glycogen levels in type 2 diabetes has not yet been explored.
Glucose imported from blood through GLUTs is considered to be a dominant energy source for enhancement of neuronal activity; however, recent studies have shown that lactate is used as an energy substrate [11, 12] and neuromodulator [13] in the hippocampus to enhance memory formation [13–15]. Lactate is taken up from the blood and is also generated from glycogen stored in astrocytes via glycogenolysis and glycolysis [11]. Brain glycogen-derived lactate is transferred to neurons through monocarboxylate transporters (MCTs). Previous studies have demonstrated that MCT2 expressed in neurons plays a role in lactate uptake into neurons [16] and downregulated expression and function of MCT2 induces memory dysfunction [13, 14]. Therefore, memory dysfunction in type 2 diabetes might be caused, at least partially, by diminished utilisation of lactate through downregulated MCT2 expression in the hippocampus.
Because it normalises glycometabolism in peripheral organs [17–21], exercise is useful as a treatment for type 2 diabetes [22]. Exercise enhances memory function in healthy mammals [23–25] and in animal models of type 1 diabetes [26–28]. We demonstrated that hippocampal glycogen increases in non-diabetic rats after 4 weeks of moderate-intensity exercise [29] and that this also enhances memory function [30]. Furthermore, previous studies have shown that exercise improves the downregulated expression of MCT series in peripheral organs in type 2 diabetes [31, 32] and increases the expression of hippocampal MCT series in in type 1 diabetes [33]. Based on these findings, we hypothesised that 4 weeks of moderate exercise would improve memory dysfunction in type 2 diabetes through improvement of hippocampal lactate transport by adaptations in glycogen levels and expression of MCTs.
In the current study, we investigated whether hippocampal lactate-transport-related glycometabolism is impaired in a rat model of type 2 diabetes and, subsequently, whether a regimen of 4 weeks of moderate-intensity exercise improves spatial memory dysfunction associated with adaptation of hippocampal lactate-transport-related glycometabolism.
Methods
Animals
Four-week-old male Otsuka Long–Evans Tokushima Fatty (OLETF) rats (model of human type 2 diabetes) and their genetic control Long–Evans Tokushima (LETO) rats were obtained from Hoshino Laboratory Animals (Ibaraki, Japan). OLETF rats, which were established in 1992 as the animal model for type 2 diabetes, exhibit memory dysfunction and hyperglycaemia after 8 and 18 weeks of age, respectively. Almost all OLETF rats exhibit symptoms of type 2 diabetes, such as hyperinsulinaemia, by 25 weeks of age and display symptoms of type 1 diabetes, such as hypoinsulinaemia, after 40 weeks of age. LETO rats, which were developed from the same colony by selective mating but did not develop diabetes, served as genetic counterpart controls. OLETF and LETO rats were housed and cared for in an animal facility, fed a standard pellet diet (MF; Oriental Yeast, Tokyo, Japan) and given free access to water. The room temperature was maintained at 22–24°C under a 12 h light–dark cycle (light on 07:00–19:00 hours). All experiments were conducted in accordance with the University of Tsukuba Animal Experiment Committee guidelines.
Habituation to treadmill running
At 26 weeks of age, the OLETF and LETO rats were habituated to running on a treadmill (SN-460; Shinano, Tokyo, Japan) using the protocol of previous studies [29, 34]. The running duration was set at 30 min/day and the running speed was gradually increased from 5 to 25 m/min for a total of five sessions over 6 days.
Single bout of exercise
Rats were subjected to exercise for 30 min on a treadmill at moderate-intensity (OLETF rats, 12.5 m/min; LETO rats, 20 m/min) based on their lactate threshold (OLETF rats 22.6 ± 0.3 m/min; LETO rats, 28.3 ± 1.8 m/min) determined according to the method of Beaver et al [35].
Exercise regimen
OLETF and LETO rats were randomly divided into a sedentary group and an exercise group. After exercise habituation, the exercise groups of both breeds were made to run on a treadmill at moderate intensity for 4 weeks (30 min/day, 5 days/week).
Morris water maze test
Hippocampal function in rats was evaluated using the Morris water maze test, which was previously established in our laboratory [36, 37]. The water maze consists of a circular pool, 150 cm in diameter and 45 cm in depth, with an invisible platform (12 cm in diameter; Plexiglas) located in the centre of one quadrant. The platform’s top was 2 cm below the surface of the water. The experimental room contained some extra-maze cues, and their positions were not changed during the testing period. Although all four start points were used during the period of the study, the order in which they were used varied every day. Rats were given a maximum of 60 s to reach the platform and were hand-guided to it if they failed to find it within 60 s. After climbing onto the platform, rats were allowed to remain there for 10 s. During the test period, swimming time (s, escape latency), swim length (cm) and speed (cm/s) were recorded semi-automatically by a video tracking system (35C67-AC5; PANLAB, Barcelona, Spain). Twenty-four hours after the last spatial learning session, the platform was removed from the pool and rats were allowed 60 s probe trial to search for it. The time rats spent in the quadrants that the platform had been placed during spatial learning (s) was measured using the same system.
Blood glucose and HbA1c assays
Glucose levels in blood collected from the jugular vein were measured using an automated glucose–lactate analyser (2300 Stat Plus; YSI, Yellow Springs, OH, USA). We outsourced measurement of HbA1c levels to the Oriental Yeast Co. (Shiga, Japan).
Tissue preparation
Following acute or regular exercise regimen, rats were anaesthetised with isoflurane (30% isoflurane in propylene glycol; Dainippon Sumitomo Pharma Co., Osaka, Japan) in a bell jar and killed using high-power microwave irradiation (10 kW, 1.4 s; NJE-2603; New Japan Radio Co., Tokyo, Japan). The hippocampus was collected using a modified version of Hirano et al’s method [38]. Muscles and liver were also collected. All samples were stored at −80°C for subsequent biochemical analysis.
Glycogen and lactate assays
A modified form of the Kong et al method [39] was used for the measurement of glycogen in the hippocampus, muscles and liver [29, 34] (for details see ESM Methods). Hippocampal lactate levels were detected using ENZYTEC L-Lactic acid (E1254; J. K. International, Tokyo, Japan).
Western blot analysis
Powdered hippocampus was homogenised in LB-TT [38]. Proteins (10 μg) in SDS were loaded on a 5–20% polyacrylamide gradient gel (ATTO Corporation, Tokyo, Japan). The gel was then transferred to polyvinylidene difluoride membranes (RPN2020F; GE Healthcare, Little Chalfont, UK). The membranes were blocked with 5% non-fat skim milk in Tris-buffered saline (154 mmol/l NaCl) containing 0.1% Tween 20 (TBS-T) for 1 h and then incubated for 10 h at 4°C with primary antibodies (see ESM Methods). After washing, the membranes were incubated for 1 h with secondary antibodies (see ESM Methods). Immunocomplexes were made visible using chemiluminescence analysis with the Chemilumi One Super Kit (Nakalai Tesque, Kyoto, Japan) following the manufacturer’s instructions. The signals were digitally scanned and then quantified using image analysis software (GE Healthcare, Bio-Science). Protein levels were normalised to the band intensity of β-actin and phosphorylated protein levels were detected and normalised by total cAMP-response-element-binding protein (CREB), glycogen synthase (GS) and GS kinase-3β (GSK-3β), respectively.
Statistical analysis
No data were excluded from the analysis. Data were expressed as mean ± SEM and were analysed using Prism version 5 (MDF, Tokyo, Japan). Comparisons of two groups were performed using an unpaired Student’s t test. Pearson’s correlation analysis was performed on the entire population of experimental rats, both diabetic (OLETF) and control rats (LETO), according to previously published methods [2, 40]. Group comparisons were performed using two-way ANOVA with Bonferroni post hoc tests. Statistical significance was set at p < 0.05.
Results
Physiological and biochemical variables
Physiological and biochemical variables of LETO and OLETF rats are shown in Table 1. OLETF rats exhibited increased body weight (p < 0.01), fat-to-body weight ratio (p < 0.01) and hyperphagia (p < 0.001) compared with LETO rats. In addition, OLETF rats were hyperglycaemic (p < 0.001) and exhibited higher HbA1c levels (p < 0.001).
Memory dysfunction
OLETF rats took five times longer than LETO rats to swim onto the platform (effect of day F (3, 174) = 15.87, p < 0.001; effect of group F (1, 58) = 73.41, p < 0.001; OLETF rats 34.5 ± 3.9 s vs LETO rats 6.6 ± 1.0 s at trial on day 4). The escape path taken by OLETF rats was threefold longer (effect of day F (3, 174) = 21.63, p < 0.001; effect of group F (1, 58) = 11.61, p < 0.001; OLETF rats 538.5 ± 63.4 cm vs LETO rats 166.3 ± 25.0 cm at trial on day 4). The OLETF rats swam significantly slower than LETO rats (effect of day F (3, 174) = 6.52, p < 0.001; effect of group F (1, 58) = 192.1, p < 0.001; OLETF rats 21.1 ± 0.6 cm/s vs LETO rats 27.0 ± 0.6 cm/s at trial on day 4). OLETF rats spent significantly less time than LETO rats in the quadrants in which the platform had been placed during probe trials (OLETF rats 14.5 ± 2.9 s vs LETO rats 20.9 ± 1.0 s, p < 0.05).
Glycogen levels in hippocampus and breakdown of glycogen with a single bout of exercise
OLETF rats exhibited significantly higher glycogen levels (about 30%) in the hippocampus compared with LETO rats (Fig. 1a; p < 0.01). Additionally, hippocampal glycogen levels showed a positive significant correlation with circulatory HbA1c levels (Fig. 1b; r = 0.78, p < 0.001). Hippocampal glycogen levels (based on lactate threshold as an index of moderate-intensity exercise) in both OLETF and LETO rats decreased significantly after a single bout of exercise (Fig. 2a; effect of diabetes F (1, 27) = 42.3, p < 0.001; effect of exercise F (1, 27) = 57.4, p < 0.001; interaction F (1, 27) = 9.35, p < 0.01). Furthermore, the rate of glycogen decrease had a significant positive correlation with the rate of local lactate increase in the hippocampus of exercised rats (Fig. 2b; r = 0.59, p < 0.05).

(a) Hippocampal glycogen levels in LETO and OLETF rats (mean ± SEM). (b) Correlation between hippocampal glycogen levels and circulatory HbA1c levels. (c) Immunoblot analysis of MCT2 expression in hippocampus (β-actin as an internal standard). Data (mean ± SEM) are expressed based on LETO rats as 100%. (d) Correlation between hippocampal glycogen levels and circulatory HbA1c levels. To convert values for HbA1c in % into mmol/mol, subtract 2.15 and multiply by 10.929. White circles, LETO rats (n = 9); black circles, OLETF rats (n = 7). *p < 0.05 and **p < 0.01 vs LETO rats

(a) Pre- and post-exercise (0 and 30 min) hippocampal glycogen levels in LETO and OLETF rats (mean ± SEM). n = 6–9 rats per group. *p < 0.05 and ***p < 0.001 vs LETO rats; † p < 0.01 vs pre-exercise. (b) Correlation between the rate at which glycogen levels decreased and the rate at which local lactate levels increased in hippocampus of exercised rats. White circles, LETO rats; black circles, OLETF rats
Hippocampal MCT2 expression
Hippocampal MCT2 expression decreased significantly (about 20%) in OLETF rats compared with LETO rats (Fig. 1c; p < 0.05). In addition, hippocampal MCT2 expression exhibited a significant inverse correlation with circulatory HbA1c levels (Fig. 1d; r = ˗0.50, p < 0.05).
Effects of exercise regimen on OLETF rats
Physiological and biochemical variables
Symptoms of type 2 diabetes, such as increased body weight and ratio of fat, hyperphagia and hyperglycaemia, were unchanged after 4 weeks of exercise intervention in OLETF rats (Table 2).
Memory dysfunction
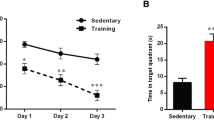
Following exercise, the escape latency (Fig. 3a; effects of exercise F (1, 108) = 12.63 on day 2, F (1, 108) = 9.97 on day 3, F (1, 108) = 21.82 on day 4, p < 0.001; interaction F (1, 108) = 13.94 on day 4, p < 0.001) and the swim path length (Fig. 3b; effect of exercise F (1, 108) = 7.05 on day 2, F (1, 108) = 9.16 on day 3, p < 0.01, F (1, 108) = 19.03 on day 4, p < 0.001; interaction F (1, 108) = 9.00 on day 4, p < 0.01) of OLETF rats were shortened, without alteration of the speed of swimming, at trials on days 2, 3 and 4 (Fig. 3c). The time spent by OLETF rats in the platform area also improved after 4 weeks of exercise (Fig. 3d; effect of exercise F (1, 24) = 4.69, p < 0.05; effect of diabetes F (1, 24) = 6.38, p < 0.05).

Effect of moderate-intensity exercise regimen on spatial memory function. Escape latency (a), swim path length (b) and swim speed (c) during learning test and time spent in platform area during probe test (d) in LETO and OLETF rats (mean ± SEM). Circles, LETO rats; squares, OLETF rats; white symbols and bars, sedentary rats; black symbols and bars, exercised rats, n = 6–8 rats per group. *p < 0.05 and ***p < 0.001 vs LETO rats; † p < 0.05, †† p < 0.01 and ††† p < 0.001 vs sedentary rats
Hippocampal glycogen levels
Elevated hippocampal glycogen levels in OLETF rats increased further (by about 10%) after 4 weeks of exercise (Fig. 4a; effect of exercise F (1, 24) = 9.78, p < 0.01; effect of diabetes F (1, 24) = 146.3, p < 0.001). OLETF rats exhibited much higher total GS levels in the hippocampus compared with LETO rats, although exercise had no effect on total GS level (Fig. 4d; effect of diabetes F (1, 49) = 12.3, p < 0.001; sedentary LETO rats 100%, sedentary OLETF rats 138%, exercised OLETF rats 155%). Phosphorylated GS levels were unaffected by diabetes or exercise (Fig. 4d). On the other hand, glycogen levels in muscle and liver were not altered by the exercise regimen in OLETF rats (Fig. 4b, c).

(a–c) Glycogen levels in hippocampus (a), muscle (b) and liver (c) of LETO and OLETF rats. Data are means ± SEM, n = 6–8 rats for each group. (d) Immunoblot analysis for GS in hippocampus of rats. A representative immunoblot is shown, together with quantification of GS (β-actin as an internal standard) and phosphorylated GS (GS protein as an internal standard) levels. Sed and white bars, sedentary rats; Ex and black bars, exercised rats. Data are means ± SEM, based on sedentary LETO rats as 100%, n = 12–14 rats per group. ***p < 0.001 vs LETO rats, † p < 0.05 and †† p < 0.01 vs sedentary rats
Hippocampal expressions of MCT series
Downregulated MCT2 expression in OLETF rats was restored (almost 100%) following 4 weeks of exercise intervention (Fig. 5a; interaction F (1, 49) = 4.85, p < 0.05). MCT1 and MCT4 were unaffected by type 2 diabetes or 4 weeks of exercise (Fig. 5b, c).

Immunoblot analysis of MCT2 (a), MCT1 (b) and MCT4 expression (c) in the hippocampus of LETO and OLETF rats. Representative blots and quantification are shown, with β-actin as internal standard. Sed and white bars, sedentary rats; Ex and black bars, exercised rats. Data are means ± SEM, based on sedentary LETO rats as 100%, n = 12–14 rats per group. *p < 0.05 vs sedentary LETO rats; † p < 0.05 vs sedentary OLETF rats
Hippocampal expression of GLUT series
Astrocytic GLUT1 expression (as determined by molecular weight of immunoblotting band [41]), which plays a role in astrocytic glucose uptake, was significantly lower in OLETF rats than in LETO rats, but the exercise regimen greatly restored its levels (Fig. 6a; interaction F (1, 49) = 7.83, p < 0.01). Expression of endothelial GLUT1, which plays a role in releasing glucose from blood to extracellular fluid, increased with the exercise regimen in OLETF rats (Fig. 6a; effect of exercise F (1, 49) = 5.26, p < 0.05) irrespective of the unaltered symptom state of OLETF rats compared with that in LETO rats. Expression of GLUT3, which plays a role in neuronal glucose uptake, was unaffected by type 2 diabetes or 4 weeks of exercise (Fig. 6b).

Immunoblot analysis of GLUT1 in astrocytes (45 kDa) and endothelia (55 kDa) (a) and GLUT3 expression (b) in the hippocampus of LETO and OLETF rats. Representative blots and quantification are shown, with β-actin as internal standard. Sed and white bars, sedentary rats; Ex and black bars, exercised rats. Data are means ± SEM, based on sedentary LETO rats as 100%, n = 12–14 rats for each group. *p < 0.05 vs sedentary LETO rats; † p < 0.05 and †† p < 0.01 vs sedentary OLETF rats
Hippocampal levels of protein-related neuroplasticity
OLETF rats exhibited significantly lower levels of BDNF in the hippocampus compared with LETO control rats and the decreased BDNF levels were not improved by 4 weeks of exercise (Fig. 7a; effect of diabetes F (1, 49) = 14.6, p < 0.001; sedentary OLETF rats 82%, exercised OLETF rats 88%, based on sedentary LETO rats as 100%). Levels of tyrosine receptor kinase B (TrkB), phosphorylated CREB and GSK-3β levels in the hippocampus were not altered by type 2 diabetes or exercise (Fig. 7b–d).

Immunoblot analysis of BDNF (a) and TrkB (b) (β-actin as an internal standard), phosphorylated CREB (c) and GSK-3β (d) (CREB and GSK-3β proteins as internal standard, respectively) in the hippocampus of LETO and OLETF rats. Sed and white bars, sedentary rats; Ex and black bars, exercised rats. Representative blots and quantification are shown. Data are means ± SEM, based on sedentary LETO rats as 100%, n = 12–14 rats per group. *p < 0.05 vs LETO rats; † p < 0.05 vs sedentary rats
Effect of exercise regimen on LETO rats
Food intake and ratio of fat decreased after 4 weeks of exercise in LETO rats (Table 2). Escape latency (Fig. 3a; p < 0.001) and swim path length (Fig. 3b; p < 0.01) shortened without altering swim speed (Fig. 3c) on day 2 after 4 weeks of exercise intervention in LETO rats. In addition, the time spent in the platform area was prolonged in exercised LETO rats compared with non-exercised rats (Fig. 3d; p < 0.05). Concurrently, exercise increased glycogen levels in the hippocampus (Fig. 4a; p < 0.01), muscle (Fig. 4b; p < 0.05) and liver (Fig. 4c; p < 0.05) and increased endothelial GLUT1 expression (Fig. 6a; p < 0.05) in LETO rats, although levels of hippocampal MCT2, MCT1 and MCT4 (Fig. 5a–c) and astrocytic GLUT1 and GLUT3 (Fig. 6a, b) remained unchanged. Furthermore, exercise significantly increased BDNF levels (Fig. 7a; p < 0.05) but not levels of TrkB or phosphorylated CREB and GSK-3β in the hippocampus (Fig. 7b–d).
Discussion
The current findings show that OLETF rats exhibit diminished expression of MCT2 and higher glycogen levels in their hippocampus, together with memory dysfunction, compared with genetic control LETO rats. Interestingly, 4 weeks of moderate exercise normalised MCT2 levels and further enhanced hippocampal glycogen levels and significantly ameliorated spatial memory dysfunction, without any changes in clinical symptoms.
Consistent with earlier reports [42–44], the OLETF rats in this study showed significant dysfunction of spatial memory, thus supporting the appropriateness of their use as a model for spatial memory dysfunction. OLETF rats exhibited increased hippocampal glycogen levels compared with genetic control LETO rats (Fig. 1a). Although tissue-specific alterations in glycogen levels have been demonstrated in other diabetic complications, such as cardiomyopathy [10], the current study is the first to report enhanced glycogen levels in the hippocampus of diabetic rats, with an elevated expression of GS (Fig. 4d). Considering previous findings that epileptic patients with memory dysfunction exhibit elevated hippocampal glycogen and GS content [45], it could be posited that hippocampal hyper-glycogen is a feature of a number of disorders with hippocampal dysfunction; this needs further research. Additionally, hippocampal glycogen levels of rats in the current study had a significant positive correlation with circulatory HbA1c levels (Fig. 1b), suggesting that glycogen levels in the diabetic hippocampus may be related to the circulatory glycaemic state. Our current findings are in contrast to those of a previous study in which Zucker Diabetic Fatty (ZDF) rats (another established model of type 2 diabetes) did not exhibit high glycogen levels in the hippocampus when compared with control rats [46]. However, the difference in data between the two studies is likely due to the methodological difference; the earlier study killed rats using 4 kW microwave irradiation [46] instead of 10 kW, which is ideal for detecting brain glycogen levels correctly [39]. Indeed, we have confirmed that ZDF rats killed by 10 kW microwave irradiation exhibit elevated hippocampal glycogen levels (ZDF 18.2 ± 0.88 μmol/g; control 12.6 ± 0.47 μmol/g; p < 0.001) consistent with OLETF rats.
OLETF rats exhibited downregulated hippocampal MCT2 expression compared with the control rats (Fig. 1c). Further, a significant inverse correlation was noted between low hippocampal MCT2 expression and circulatory HbA1c levels (Fig. 1d). Thus, the circulatory glycaemic state appears to affect the basal MCT2 expression similarly to glycogen levels in the diabetic hippocampus. Importantly, we found that a single bout of exercise enhanced hippocampal glycogen breakdown and lactate production in the diabetic hippocampus (Fig. 2). Based on our findings, elevated hippocampal glycogen in diabetic rats may be an adaptive change to compensate for the decreased lactate and glucose utilisation possibly induced by downregulated MCT2 and astrocytic GLUT1 expression in the hippocampus (Fig. 1c, 5a, 6a). Additionally, hippocampal expression of GLUT3 was unaffected by type 2 diabetes (Fig. 6b). Thus, we suggest that the dysregulated MCT2-mediated neuronal uptake of lactate possibly contributes to the aetiology of memory dysfunction in type 2 diabetes.
A notable finding of the current study is the improvement of spatial memory dysfunction in diabetic rats after 4 weeks of moderate-intensity exercise (Fig. 3), with no change in clinical circulatory profiles (Table 2). The lack of effect on clinical variables may be due to the relatively short duration of the exercise regimen since earlier animal studies with longer exercise interventions reported improvement [47, 48]. This exercise regimen could be utilised as a basis in developing new treatment for cerebral complications in type 2 diabetes. In contrast, a previous study demonstrated that long-term exercise intervention (brisk walking, 175 min weekly for 6 months) did not alter cognitive function in patients with type 2 diabetes [49]. Note that this study in humans examined cognitive function, such as executive function and verbal memory, but not spatial memory.
Hippocampal glycogen levels in rats with type 2 diabetes further increased with 4 weeks of exercise (Fig. 4a). We interpret these data as an adaptation by which to enhance lactate production. Although exercise-induced enhancement of hippocampal glycogen levels would be partially due to enhancement of glucose uptake in astrocytes via exercise-mediated GLUT1 upregulation (Fig. 6a), the functional relevance needs to be investigated in depth. Further, in the same experimental animals, muscular and hepatic glycogen levels were unaltered after exercise (Fig. 4b, c). Thus, these findings lead us to postulate that the brain in type 2 diabetes has a higher sensitivity to exercise than do peripheral tissues in terms of glycometabolism.
Notably, while the expression of MCT1 and MCT4 was unaltered (Fig. 5b, c), the exercise regimen normalised MCT2 levels in the diabetic hippocampus (Fig. 5a). Since an increase in MCT2 expression enhances lactate uptake into neurons [33, 50], it is conceivable that the exercise regimen improved MCT2-mediated lactate uptake in diabetic neurons. While MCT2 expression was restored, hippocampal protein levels related to neuronal glucose uptake (GLUT3) and neuroplasticity (BDNF, TrkB, CREB, GSK-3β) were unaltered in OLETF rats by exercise intervention. Although hippocampal BDNF levels are decreased in type 2 diabetes, as revealed by the current findings and in agreement with other published articles [8], the short duration of the exercise regimen may explain the unaltered levels of BDNF since longer exercise intervention has been shown to improve its levels [51]. Further, we did not observe exercise-induced upregulation of phosphorylated CREB, although lactate was shown to increase phosphorylated CREB in a previous study [13]. These data led us to speculate that the CREB phosphorylation site (Ser-133) may not be related to, or that other phosphorylation sites of CREB may be related to, type-2-diabetes-mediated memory dysfunction, warranting in-depth future studies. Based on our findings, we suggest that enhancing the supply of glycogen-derived lactate to neurons via increase in glycogen levels and MCT2 expression may contribute to exercise-induced improvement of memory dysfunction in type 2 diabetes.
Consistent with other reports [29, 36], we also confirmed that the exercise regimen increases hippocampal glycogen and BDNF levels and, concurrently, enhances spatial memory in control LETO rats. In contrast, the molecular alteration in lactate transporters was only observed in the OLETF rat hippocampus after exercise (Fig. 5a) and not in LETO rats. Thus, the present findings imply that the molecular pathways underlying memory improvement after 4 weeks of moderate exercise are different in control condition and type 2 diabetes. Collectively, exercise has effects on the hippocampus that are specific to type 2 diabetes; this reinforces the notion of profound brain dysregulation in this disease.
The current study has several limitations. First, the in vivo nature of this study poses technical difficulties; it was not feasible to fully delineate the temporal profiles of glycogen and associated molecules such as MCT2 before and after exercise treatment. Second, we did not investigate the effects of the exercise regimen used in the current study on other factors reported to be involved in diabetes-induced memory dysfunction, such as inflammation or oxidative stress. Third, we did not distinguish the spatial memory dysfunction reported here from other forms of memory dysfunction or determine how these may relate to exercise in type-2-diabetes-induced cognitive dysfunction. Indeed, the effect of exercise on memory is a new entity that has only just begun to be appreciated and warrants further investigation. Finally, due to the current experimental design and settings, it was not possible to completely clarify the link between the effects of diabetes and exercise on memory based solely on the results presented in this article, and further studies are warranted in this regard.
Here, we found that 4 weeks of moderate-intensity exercise improves memory dysfunction in a rat model of type 2 diabetes, accompanied by increased glycogen levels and restoration of MCT2 expression in the hippocampus. Our findings suggest that dysregulation of MCT2-mediated neuronal uptake of lactate in the diabetic hippocampus may contribute to the aetiology of spatial memory dysfunction and that moderate exercise could be used as a treatment for type-2-diabetes-induced memory dysfunction.
Abbreviations
- BDNF:
-
Brain-derived neurotrophic factor
- CREB:
-
cAMP-response-element-binding protein
- GS:
-
Glycogen synthase
- GSK-3β:
-
GS kinase-3β
- LETO rat:
-
Long–Evans Tokushima Otsuka rat
- MCT:
-
Monocarboxylate transporter
- OLETF rat:
-
Otsuka Long–Evans Tokushima fatty rat
- TrkB:
-
Tyrosine receptor kinase B
- ZDF rat:
-
Zucker Diabetic Fatty rat
References
Cukierman T, Gerstein HC, Williamson JD (2005) Cognitive decline and dementia in diabetes--systematic overview of prospective observational studies. Diabetologia 48:2460–2469
Gold SM, Dziobek I, Sweat V et al (2007) Hippocampal damage and memory impairments as possible early brain complications of type 2 diabetes. Diabetologia 50:711–719
McCrimmon RJ, Ryan CM, Frier BM (2012) Diabetes and cognitive dysfunction. Lancet 379:2291–2299
Umegaki H, Hayashi T, Nomura H et al (2013) Cognitive dysfunction: an emerging concept of a new diabetic complication in the elderly. Geriatr Gerontol Int 13:28–34
Pouwer F, Beekman ATF, Nijpels G et al (2003) Rates and risks for co-morbid depression in patients with type 2 diabetes mellitus: results from a community-based study. Diabetologia 46:892–898
Ahtiluoto S, Polvikoski T, Peltonen M et al (2010) Diabetes, Alzheimer disease, and vascular dementia: a population-based neuropathologic study. Neurology 75:1195–1202
Whitmer RA (2007) Type 2 diabetes and risk of cognitive impairment and dementia. Curr Neurol Neurosci Rep 7:373–380
Stranahan AM (2015) Models and mechanisms for hippocampal dysfunction in obesity and diabetes. Neuroscience 309:125–139
Bhattacharjee M, Venugopal B, Wong KT et al (2006) Mitochondrial disorder, diabetes mellitus, and findings in three muscles, including the heart. Ultrastruct Pathol 30:481–487
Shearer J, Ross KD, Hughey CC et al (2011) Exercise training does not correct abnormal cardiac glycogen accumulation in the db/db mouse model of type 2 diabetes. Am J Physiol Endocrinol Metab 301:E31–E39
Benarroch EE (2010) Glycogen metabolism: metabolic coupling between astrocytes and neurons. Neurology 74:919–923
Tsacopoulos M, Magistretti PJ (1996) Metabolic coupling between glia and neurons. J Neurosci 16:877–885
Suzuki A, Stern SA, Bozdagi O et al (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144:810–823
Newman LA, Korol DL, Gold PE (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One 6:e28427
Duran J, Saez I, Gruart A et al (2013) Impairment in long-term memory formation and learning-dependent synaptic plasticity in mice lacking glycogen synthase in the brain. J Cereb Blood Flow Metab 33:550–556
Pierre K, Pellerin L (2005) Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem 94:1–14
Sigal RJ, Kenny GP, Boulé NG et al (2007) Effects of aerobic training, resistance training, or both on glycemic control in type 2 diabetes: a randomized trial. Ann Intern Med 147:357–369
Holloszy JO (2005) Exercise-induced increase in muscle insulin sensitivity. J Appl Physiol 99:338–343
O’Gorman DJ, Karlsson HKR, McQuaid S et al (2006) Exercise training increases insulin-stimulated glucose disposal and GLUT4 (SLC2A4) protein content in patients with type 2 diabetes. Diabetologia 49:2983–2992
Mu J, Brozinick JT, Valladares O et al (2001) A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell 7:1085–1094
Sriwijitkamol A, Coletta DK, Wajcberg E et al (2007) Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes 56:836–848
Balducci S, Iacobellis G, Parisi L et al (2006) Exercise training can modify the natural history of diabetic peripheral neuropathy. J Diabet Complicat 20:216–223
van Praag H, Christie BR, Sejnowski TJ, Gage FH (1999) Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc Natl Acad Sci U S A 96:13427–13431
Pereira AC, Huddleston DE, Brickman AM et al (2007) An in vivo correlate of exercise-induced neurogenesis in the adult dentate gyrus. Proc Natl Acad Sci U S A 104:5638–5643
Alomari MA, Khabour OF, Alzoubi KH, Alzubi MA (2013) Forced and voluntary exercises equally improve spatial learning and memory and hippocampal BDNF levels. Behav Brain Res 247:34–39
de Senna PN, Ilha J, Baptista PPA et al (2011) Effects of physical exercise on spatial memory and astroglial alterations in the hippocampus of diabetic rats. Metab Brain Dis 26:269–279
Reisi P, Alaei H, Babri S et al (2009) Effects of treadmill running on spatial learning and memory in streptozotocin-induced diabetic rats. Neurosci Lett 455:79–83
de Senna PN, Xavier LL, Bagatini PB et al (2015) Physical training improves non-spatial memory, locomotor skills and the blood brain barrier in diabetic rats. Brain Res 1618:75–82
Matsui T, Ishikawa T, Ito H et al (2012) Brain glycogen supercompensation following exhaustive exercise. J Physiol 590:607–616
Liu YF, Chen H, Yu L et al (2008) Upregulation of hippocampal TrkB and synaptotagmin is involved in treadmill exercise-enhanced aversive memory in mice. Neurobiol Learn Mem 90:81–89
Juel C, Holten MK, Dela F (2004) Effects of strength training on muscle lactate release and MCT1 and MCT4 content in healthy and type 2 diabetic humans. J Physiol 556:297–304
Nikooie R, Rajabi H, Gharakhanlu R et al (2013) Exercise-induced changes of MCT1 in cardiac and skeletal muscles of diabetic rats induced by high-fat diet and STZ. J Physiol Biochem 69:865–877
Aveseh M, Nikooie R, Sheibani V, Esmaeili-Mahani S (2014) Endurance training increases brain lactate uptake during hypoglycemia by up regulation of brain lactate transporters. Mol Cell Endocrinol 394:29–36
Matsui T, Soya S, Okamoto M et al (2011) Brain glycogen decreases during prolonged exercise. J Physiol 589:3383–3393
Beaver WL, Wasserman K, Whipp BJ (1985) Improved detection of lactate threshold during exercise using a log-log transformation. J Appl Physiol 59:1936–1940
Inoue K, Hanaoka Y, Nishijima T et al (2015) Long-term mild exercise training enhances hippocampus-dependent memory in rats. Int J Sports Med 36:280–285
Lee MC, Okamoto M, Liu YF et al (2012) Voluntary resistance running with short distance enhances spatial memory related to hippocampal BDNF signaling. J Appl Physiol 113:1260–1266
Hirano M, Rakwal R, Shibato J et al (2006) New protein extraction/solubilization protocol for gel-based proteomics of rat (female) whole brain and brain regions. Mol Cells 22:119–125
Kong J, Shepel PN, Holden CP et al (2002) Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci 22:5581–5587
Rodríguez-Mañas L, Angulo J, Vallejo S et al (2003) Early and intermediate Amadori glycosylation adducts, oxidative stress, and endothelial dysfunction in the streptozotocin-induced diabetic rats vasculature. Diabetologia 46:556–566
Vannucci SJ (1994) Developmental expression of GLUT1 and GLUT3 glucose transporters in rat brain. J Neurochem 62:240–6
Matsushita H, Akiyoshi J, Kai K et al (2003) Spatial memory impairment in OLETF rats without cholecystokinin - a receptor. Neuropeptides 37:271–276
Li XL, Aou S, Hori T, Oomura Y (2002) Spatial memory deficit and emotional abnormality in OLETF rats. Physiol Behav 75:15–23
Kwon KJ, Lee EJ, Kim MK et al (2015) Diabetes augments cognitive dysfunction in chronic cerebral hypoperfusion by increasing neuronal cell death: implication of cilostazol for diabetes mellitus-induced dementia. Neurobiol Dis 73:12–23
Dalsgaard MK, Madsen FF, Secher NH et al (2007) High glycogen levels in the hippocampus of patients with epilepsy. J Cereb Blood Flow Metab 27:1137–1141
Sickmann HM, Waagepetersen HS, Schousboe A et al (2010) Obesity and type 2 diabetes in rats are associated with altered brain glycogen and amino-acid homeostasis. J Cereb Blood Flow Metab 30:1527–1537
Lee H, Chang H, Park J et al (2011) Exercise training improves basal blood glucose metabolism with no changes of cytosolic inhibitor B kinase or c-Jun N-terminal kinase activation in skeletal muscle of Otsuka Long-Evans Tokushima fatty rats. Exp Physiol 96:689–698
Jenkins NT, Padilla J, Arce-Esquivel AA et al (2012) Effects of endurance exercise training, metformin, and their combination on adipose tissue leptin and IL-10 secretion in OLETF rats. J Appl Physiol 113:1873–1883
Espeland MA, Rapp SR, Bray GA et al (2014) Long-term impact of behavioral weight loss intervention on cognitive function. J Gerontol A Biol Sci Med Sci 69:1101–1108
Bliss TM, Ip M, Cheng E et al (2004) Dual-gene, dual-cell type therapy against an excitotoxic insult by bolstering neuroenergetics. J Neurosci 24:6202–6208
Stranahan AM, Lee K, Martin B et al (2009) Voluntary exercise and caloric restriction enhance hippocampal dendritic spine density and BDNF levels in diabetic mice. Hippocampus 19:951–961
Acknowledgements
The authors are grateful to R. Rakwal, M. Chul Lee, N. Omori, K. Saitoh, H. Omuro, K. Takahashi and M. Hamasaki (University of Tsukuba, Japan) for technical support and discussion and to M. Noguchi (ELCS English Language Consultation, Japan) for help with the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was supported in part by the ‘Global Initiative for Sports Neuroscience (GISN): For Development of Exercise Prescription Enhancing Cognitive Functions’, by special funds for Education and Research of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) granted to the ‘Body and Mind Integrated Sports Sciences (BAMIS) Project’ and to the ‘Human High Performance (HHP) Research Project’ and by the Japan Society for the Promotion of Science (Grants-in-aid for Scientific Research A, No. 15H01828; Challenging Exploratory Research, No. 23650384).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
TS, TM and HS designed the research. TS, TM, MO, MS, Y-FL and KI contributed to the acquisition of the data. TS, TM, SJ, MO, BSM, IT-A and HS analysed and interpreted the data. TS, SJ, TM, MO, MS, Y-FL, KI and HS wrote the manuscript. BSM and IT-A reviewed and edited the manuscript. All authors approved the final version of the manuscript. HS is responsible for the integrity of the work as a whole.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Methods
(PDF 117 kb)
Rights and permissions
About this article

Cite this article
Shima, T., Matsui, T., Jesmin, S. et al. Moderate exercise ameliorates dysregulated hippocampal glycometabolism and memory function in a rat model of type 2 diabetes. Diabetologia 60, 597–606 (2017). https://doi.org/10.1007/s00125-016-4164-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-016-4164-4