
Abstract
The human genome project and its search for factors underlying human diseases has fostered a major human research effort. Therefore, unsurprisingly, in recent years we have observed an increasing number of studies on human islet cells, including disease approaches focusing on type 1 and type 2 diabetes. Yet, the field of islet and diabetes research relies on the legacy of rodent-based investigations, which have proven difficult to translate to humans, particularly in type 1 diabetes. Whole islet physiology and pathology may differ between rodents and humans, and thus a comprehensive cross-species as well as species-specific view on islet research is much needed. In this review we summarise the current knowledge of interspecies islet cytoarchitecture, and discuss its potential impact on islet function and future perspectives in islet pathophysiology research.
Similar content being viewed by others

It is the pervading law of all things organic and inorganic, of all things physical and metaphysical, of all things human and all things superhuman, of all true manifestations of the head, of the heart, of the soul, that the life is recognizable in its expression, that form ever follows function. This is the law.
Louis H. Sullivan (1896) [1]
Introduction
In his 1869 doctoral thesis, now a medical classic, the 22-year-old German physician Paul Langerhans reported the microscopic appearance of scattered small groups of cells among the acinar glandular cells in the pancreas of the rabbit [2]. In 1893 Edouard Laguesse named these cell clusters the ‘islets of Langerhans’, and first postulated an endocrine role for these clusters in the control of blood glucose levels [3].
Successful extractions of insulin and glucagon and the formal experimental proof that these key hormones regulate glucose levels firmly established the islets of Langerhans as key mini-organs in the diabetes field. Research on the biology and pathophysiology of islet cells burgeoned as a result of the emergence of novel technologies and the application of various diabetes animal models as a surrogate for studies on human islets from healthy and diabetic individuals [4–14].
Preclinical research on rodent models offers several logistical, methodological and financial advantages. But this approach may fall short in terms of reflecting critical aspects of human islet pathophysiology [15, 16]. Because islets of non-human primate (NHP) or human origin are increasingly becoming available (albeit in limited numbers), there is an important and creeping increase in the number of studies on primate islets. Although still in their infancy, such studies have highlighted key structure–function differences between rodents and NHP or human islets, with evidence pointing towards a high degree of similarity between NHP and human islets [8, 14, 17, 18]. Moreover, mouse models may not reflect the complexity of human pathophysiology [19], such as islet amyloid deposition, underscoring the need for a better animal model (i.e. NHP) that truly mirrors human islet biology in health and disease [20].
In this review we will summarise the results on islet architecture and structure–function relationships between rodents, NHPs and humans. In addition, we will briefly highlight the plasticity of islet cell architecture observed under diabetic conditions (reviewed in detail in [21]). As noted earlier, assumptions on human islet function and regulation based on rodent islets are not always true. Therefore, we would like to propose, based on data on interspecies islet cytoarchitecture and functional differences, a paradigm shift whereby investigations of the NHP islet can serve as a successful diabetes translational platform.
Cellular architecture and composition of islets of Langerhans is heterogeneous
Islet morphology has been extensively studied using histology, immunohistology and electron microscopy. In addition, different in vivo and in vitro model systems have been used to address islet cell function [22, 23]. Islets contain a variety of cell types, the most abundant being the insulin-producing beta cells, the glucagon-producing alpha cells and the somatostatin-producing delta cells [24] (Fig. 1). Furthermore, ghrelin-producing epsilon cells and pancreatic polypeptide (PP) cells have been described in the mammalian islet, albeit in smaller numbers [25, 26]. Besides hormone-producing cells, the islet also houses immune cells, autonomic nerve system endings (reviewed in detail in [27]) and is served by an abundant vascular network [8, 28, 29].

The cytoarchitecture of islets of Langerhans of rodents and primates is different. In rodents, islets are composed of beta cells located in the core of the islet, which are surrounded by a mantle of alpha and delta cells. Sympathetic fibres surround the islet, while parasympathetic fibres are distributed throughout the core of the islet, supplying acetylcholine to beta cells. In contrast, NHP and human islets have a heterogeneous distribution of beta, alpha and delta cells. Here, sympathetic fibres target the islet vasculature and parasympathetic fibres are rare, with alpha cells in charge of the acetylcholine supply to beta and delta cells
Together, the different cells are architecturally arranged to allow for a fine-tuned functional interrelationship between all islet cells, possibly orchestrated by autocrine, paracrine and endocrine signalling cascades, like a true mini-organ (Fig. 2). With its unique cell organisation, mammalian islets successfully accomplish their ultimate physiological role, to adapt to constant changes in metabolic demands and to maintain optimal circulating glucose levels throughout life.

Paracrine and autocrine signalling networks in mammalian islet of Langerhans. In response to an increase in blood glucose levels, beta cells secrete insulin. In turn, insulin binds to insulin receptors A and B (IR-A and IR-B) in beta and alpha cells, regulating insulin gene expression and insulin and glucagon exocytosis. ATP is secreted together with insulin and acts on purinergic P2X/Y receptors in beta cells. Beta cells also secrete GABA, which acts on beta cells in an autocrine manner through GABAA and GABAB receptors. When blood glucose levels are low, alpha cells secrete glucagon, glutamate and acetylcholinea. Glucagon binds to glucagon receptors in beta and alpha cells, regulating insulin secretion and glucagon gene expression. Similarly, glutamate binds to AMPA and NMDA receptors in beta and alpha cells, regulating insulin and glucagon exocytosis. Acetylcholine binds to muscarinic receptors in beta and delta cells, regulating insulin and somatostatin secretion. At both high and low blood glucose levels the delta cells secrete somatostatin, which binds to somatostatin receptors (SSTRs) in beta and alpha cells, inhibiting insulin and glucagon secretion. aIn rodent islets, acetylcholine is secreted by parasympathetic fibres that permeate the beta cell core (see Fig. 1)
In the following section we will highlight the differences that separate rodent from primate islet physiology.
Rodents
Small animal research has been the cornerstone of islet research for over a century, pioneered by Langerhans himself in his description of the rabbit islet [2]. Today, mouse and rat islets are widely studied, with consensus that these islets are mainly comprised of an insulin-producing beta cell core surrounded by a mantle of intermingled alpha and delta cells (Fig. 1).
The islet vasculature is responsible for supplying the islet cells not only with oxygen, but also with key metabolic cues (i.e. glucose) and other nutrients, incretin hormones such as glucagon-like peptide-1 (GLP-1) and cytokines [29, 30]. This vast vascular network develops early in islet embryogenesis, providing essential developmental signals [31], and is driven primarily by the secretion of vascular endothelial growth factor-A (VEGF-A) from beta cells [32, 33]. The islet vascular bed consists of both large and small fenestrated capillaries [11, 29]. The islet blood flow arrives from peripheral vascular networks, where the blood flow moves from the beta cell-rich inner core to the outer alpha and delta cells [29, 34]. The islet blood flow pattern and speed has been suggested to be dependent on blood glucose levels, with hyperglycaemia leading to better islet perfusion with faster flow speeds compared with states of hypoglycaemia [7]. Recent evidence suggests a degree of heterogeneity of vascularisation within islets in a single pancreas [35, 36]. Such differences result in distinct islet subpopulations with significant functional implications, with highly perfused islets displaying an increased beta cell proliferation rate and enhanced function [36]. Whether such a phenotype is found in human islets remains unclear. Furthermore, islet vasculature has been suggested to play a crucial role in maintaining islet function in ageing [37].
The location of beta cells within the core of the rodent islet also has important and direct functional consequences. The biological activity between the majority of rodent beta cells within any given islet, commonly measured by changes in intracellular free calcium concentration ([Ca2+]i), is highly synchronised, creating an integrated functional syncytium [38]. This synchronisation is dependent on gap junctions made of Connexin-36 channels between different beta cells [39].
Humans and NHPs
Although human islets have been intensively studied over the last decade, their cellular arrangement is still a matter of debate, largely because of different experimental methodologies. Earlier morphological analysis of different regions of human and NHP pancreases showed that the different islet cell types are intermingled, with beta and alpha cells present in similar numbers with a higher degree of heterotypic islet cell contacts [14, 18]. Thus, human islets have been perceived to be characterised by an intermingled majority of beta cells, followed by alpha and delta cells [14, 18, 24] (Fig. 1). However, other groups have provided evidence of a non-random arrangement of human beta cells within an islet. They have observed that despite the higher percentages of alpha cells within the human islet, beta cells still prefer to remain in contact with one another, in a similar way to rodent beta cells [40–42]. With homotypic cell contacts predominant, paracrine influences within human islets may be limited. In a separate study in which three-dimensional reconstructions of islets were created from serial human pancreatic sections, the islets were reported to consist of a beta cell core with a surrounding mantle of alpha cells and vessels that do not penetrate the islet [43]. However, in the same paper, the authors go on to suggest that, similar to rodents, human islet cells are purposely arranged in a complex trilaminar epithelial plate fold that favours heterologous contacts and paracrine signalling between islet cells [43]. These varied concepts of human beta cell distribution need to be resolved to allow a thorough appreciation of the importance of paracrine interactions within an intact human islet.
Regardless of heterotypic cell arrangements, human islet cell composition is set early in human life, stemming from a high proliferative phase (mainly beta cells) in the first 2 years of life followed by a life-long low-proliferative state [44, 45]. Strikingly, unlike rodents, not much is known about the human (intra)islet vasculature anatomy and flow rate [37, 46], with the exception of early reports that the perfusion of human and NHP islets is similar to that of rodent islets [47, 48]. We have shown that the vasculature of human islets becomes inflamed and fibrotic with ageing, which is also observed in islets from type 2 diabetic patients [37]. In addition, a unique double basement membrane between islet cells and vascular cells has been observed in human islets [49], the function of which is as yet unknown.
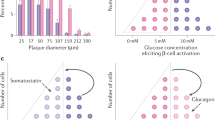
The functional consequences of the unique heterogeneous cellular arrangement of the human islet are even less clear. Some functional differences between rodent, NHP and human islets can be attributed to differences in islet cytoarchitecture [14]. By monitoring changes in islet [Ca2+]i, the authors demonstrate that primate islet cells are activated by changes in low glucose concentrations (from 3 mmol/l to 1 mmol/l), whereas mouse islets are not [14]. Moreover, prolonged exposure of whole mouse islets to 11 mmol/l glucose triggers a synchronised oscillatory [Ca2+]i behaviour, an effect that is absent in primate islets but has been seen in isolated primate beta cells. [14]. However, such [Ca2+]i oscillatory behaviour seen in isolated primate beta cells needs to be verified in an in vivo setting.
Intra-islet signalling is important for human islet function
Rodent islet function relies on homotypic cellular communications between beta cells, configuring a functional syncytium with coordinated islet activity [39, 50]. On the other hand, the cytoarchitecture of the human islet, rich in heterotypic islet cell contacts, suggests that intra-islet paracrine signalling is perhaps less straightforward [14, 24, 43] (Fig. 2). Although reports indicate a rodent-like perfusion pattern of the human islet [47], lending weight to the concept of limited intra-islet paracrine signalling, the trilaminar-fold hypothesis coupled with regulated (and directed) blood flow by vascular sphincters within the human islet suggest that paracrine signalling may yet be important for islet function [8, 51].
Intra-islet communication between different islet cells can be established by the secretion of signalling molecules into islet microvasculature or within the islet’s interstitial space. Once these signalling molecules find their respective receptors on target cells they trigger complex autocrine or paracrine regulatory loops. In addition to hormones, islet cells secrete an array of signalling molecules, including acetylcholine [17, 52], glutamate [53], ATP [54, 55] and γ-aminobutyric acid (GABA) [56–58], all of which are important for islet cell function (Fig. 2).
Acetylcholine
Acetylcholine, the first neurotransmitter to be discovered, is an important modulator of beta cell activity and insulin secretion in rodents [59] as well as in humans [17]. Although acetylcholine has a similar function in the islets of both, the source of acetylcholine is different [17, 59]. In rodents, acetylcholine is delivered through autonomic nervous system varicosities distributed throughout the islet [8], whereas in humans islets it is secreted by alpha cells and influences the activity of neighbouring beta and delta cells [8, 17, 52, 60] (Fig. 2).
Nonetheless, in both human and rodent islets acetylcholine secretion is triggered by periods of low circulating levels of glucose, and serves to sensitise beta cells to future rises in blood glucose concentration. The shift from neural to intra-islet acetylcholine delivery by alpha cells indicates that human islets are better able to respond quickly to dynamic shifts in blood glucose levels, bypassing neural networks. This may explain why type 1 diabetic patients can show a reduced dependency on exogenous insulin application as quickly as 20 days after islet transplantation [61], well before complete islet innervation is established.
ATP
ATP is one of the most important signalling molecules in the body [62]. Besides its well-established role in glucose-stimulated insulin secretion [63], ATP may also act as an extracellular signalling molecule within islets. Although ATP is found within insulin granules [9], its secretion seems to precede insulin exocytosis [64, 65].
To date, most of the data available on ATP function in islets is from rodent models. ATP is reported to act via P2Y receptors [66], either stimulating [67] or inhibiting [68] insulin secretion (Fig. 2). Recent studies in human and NHP islets show that ATP similarly may act as an autocrine signalling molecule, taking part in an important positive-feedback loop in active beta cells (Fig. 2). Although ATP is released at low glucose concentrations [54], glucose-stimulated ATP has been suggested to act via ionotropic purinergic P2X3 receptors in the membrane of active beta cells, resulting in P2X3-mediated Na+–Ca2+ influx and further depolarisation of beta cells and enhanced insulin secretion [54]. However, another study produced contradictory data, with P2X isoforms observed to be less relevant than P2Y1 receptors in mediating the positive autocrine role of ATP in human islets [69]. Based on these intriguing findings, further investigations on the role of intra-islet ATP signalling are warranted, especially given the unknown sources of secreted ATP (besides the beta cell) and the conservation of this autocrine mechanism among different islet cells.
GABA
Since its discovery in the 1950s [70], GABA has been characterised as a key inhibitory neurotransmitter in the mammalian central nervous system (CNS), acting via ionotropic GABAA and metabotropic GABAB receptors [71]. In neurons, binding to the various receptor types leads to target cell hyperpolarisation (i.e. electrical inhibition), either through the opening of chloride (Cl−) channels and Cl− influx (GABAA), or by the opening of K+ channels via second-messenger pathways (GABAB) [71]. Again, in islets the majority of data on the function of GABA originates from rodent models. Studies have shown that GABA molecules are found in the cytoplasm of beta cells, both in insulin granules [9] and in small core vesicles [72]. Acting via GABAA receptors, GABA mediates the glucose-dependent inhibition of glucagon secretion in alpha cells [73] and triggers beta cell prolife ration and survival in an autocrine loop [56, 74].
Human islet cells contain high levels of the GABA-synthesising enzyme glutamate decarboxylase 65 (GAD65, also known as GAD2) [75]. Besides inducing beta cell proliferation [56], GABA seems to work both as an inhibitory and as an excitatory molecule. In the presence of physiological glucose concentrations, GABA depolarises beta cells and stimulates glucose-induced insulin secretion [76], whereas the role of GABA in glucagon secretion by alpha cells is controversial [57, 76]. In addition, high levels of GABA can depolarise delta cells, suggesting a role for GABA-mediated regulation of somatostatin release [76].
As all three main islet endocrine cells contain GABA [76], an exciting challenge will be to determine whether GABA secretion from different islet cells is synchronised in vivo, especially given the apparent antagonistic actions of GABA on islet cell function.
Glutamate
Glutamate is one of the key excitatory neurotransmitters in the CNS. Glutamate acts via ionotropic kainite-type α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) or N-methyl-d-aspartate (NMDA) receptors [77]. Similar to acetylcholine, GABA, ATP and glutamate, glutamate-synthesising enzymes and glutamate receptors are found in the three main endocrine cells in rodent islets [78, 79]. According to one school of thought, in NHPs and humans [53], intra-islet glutamate is provided by alpha cells, and glutamate is compartmentalised inside glucagon-containing granules and secreted together with glucagon [80] (Fig. 2). Once secreted, glutamate acts mainly in an autocrine fashion, driving a positive-feedback loop that depolarises the active alpha cell and upregulates glucagon release (Fig. 2). While this autocrine function is conserved in rodents and humans, glutamate action in rodents seems to be mediated by both AMPA and NMDA receptors [78], while in humans only AMPA receptors are involved [53]. This scheme has been challenged by the authors of a recent study using mouse islets who propose a different model [81], in which glutamate is neither secreted together with glucagon nor does it modulate glucagon secretion. Rather, inhibition of glutamate secretion increases cellular pools of both glutamate and α-ketoglutarate, which are suggested to enhance somatostatin secretion release from delta cells via a new AMPA receptor variant [79]. These results highlight the complexity of intra-islet signalling, and underline the need for similar studies to be conducted with human islets.
Insulin
Since the first description of insulin [82], its action and anti-diabetic potential have been studied for almost a century. Insulin is critical for proper control of blood glucose levels and acts in almost all tissues, such as brain, liver, muscle, adipose tissue and pancreatic islets. Insulin action is mediated by two different isoforms of the insulin receptor (namely IR-A and IR-B; Fig. 2). Spatial differences between IR-A and IR-B on cell membranes may explain their differences in intracellular signalling networks [83]. Mice lacking IR expression specifically in beta cells (the βIRKO mouse model) develop a hyperinsulinaemic state and unlike wild-type mice do not show acute glucose-induced insulin secretion [84]. Moreover, insulin released in response to glucose is important for forkhead box protein O1 (FOXO1) transcriptional activity and KATP channel opening in beta cells [85, 86]. In addition, deletion of IR in alpha cells (the αIRKO mouse model) leads to hyperglycaemia, glucose intolerance and hyperglucagonaemia, indicating that insulin acts also at the alpha cell level to suppress glucagon secretion [87].
In humans, the results available on intra-islet insulin action are conflicting. Insulin has been described both as a positive and a negative regulator of islet cell function. Wu et al [88] reported that insulin acts via the phosphoinositide 3-kinase pathway, upregulates pancreatic and duodenal homeobox protein 1 (PDX-1) transcriptional activity and induces insulin gene expression. In contrast, Persaud et al noted a negative effect of insulin on beta cell insulin secretion [89]. Similarly, human pancreases perfused with low glucose and anti-insulin antibodies have impaired glucagon secretion [90]. Given the inconsistency of the experimental data, further studies are necessary to determine the effects of intra-islet insulin.
Glucagon
Glucagon is the key hormone in mammals that maintains glucose availability during fasting periods. Acting primarily on the liver, glucagon stimulates hepatic glucose production and excursion [91]. The systemic role of glucagon is clearly demonstrated in rodents, where deletion of the glucagon receptor leads to low blood glucose levels, hyperglucagonaemia and improved glucose tolerance [92].
Glucagon signalling within the islet is not well understood, mainly due to a potential crosstalk with the pre-proglucagon–incretin pathway [30, 93]. Glucagon receptors are found in both rodent and human islets and in all three major islet cell types [94, 95]. There is consensus that glucagon causes intracellular cAMP levels to rise, which stimulates beta cell glucose-induced insulin secretion in both rodents and humans [92, 94, 96]. This insulinotropic effect of glucagon is also observed in isolated rodent islets, where deletion or inhibition of the glucagon receptor blocks insulin secretion and impairs glucose oxidation [94, 97]. In perfused human pancreas, antibody neutralisation of glucagon under both low and high glucose conditions increases insulin secretion [90], contradicting the above data from isolated human islets [94] (Fig. 2). Furthermore, glucagon also plays a positive autocrine role by binding to glucagon receptors on alpha cells to enhance its own expression [98].
Somatostatin
The peptide hormone somatostatin is secreted by nerve cells in different regions of the brain, by cells in the digestive system and by pancreatic islet delta cells [99] (Fig. 2). Somatostatin secreted by gastrointestinal cells is the main source of circulating somatostatin [99], whereas delta cell-derived somatostatin plays a key role in intra-islet cell signalling [100] (Fig. 2). In both rodents and humans, somatostatin inhibits alpha and beta cell activity, repressing glucagon and insulin secretion [90, 100]. The actions of somatostatin are mediated by five different isoforms of the somatostatin receptor (SSTR), found throughout the islet [101, 102]. Deletion of the gene encoding somatostatin in mice leads to a hyperinsulinaemic and hyperglucagonaemic state, but, surprisingly, without major whole body metabolic aberrations [100]. Accordingly, somatostatin neutralisation in perfused human pancreas leads to higher glucagon and insulin secretion [90].
Recent data indicate that human delta cells are influenced by alpha and beta cells, thus being an intricate part of a complex paracrine islet cell signalling network [52].
Islet structure–function and diabetes
Structural changes in pancreatic islets have been observed in both type 1 and type 2 diabetes (summarised in Table 1). Most of the studies show that besides the loss in beta cell mass in type 1 diabetes, both rodent and human/NHP islets display an increase in alpha and delta cell mass and alpha cell proliferation [6, 103–105]. Surprisingly, studies have suggested the presence of glucose-responsive beta cells in patients with long standing type 1 diabetes [106, 107].
In rodent models of type 2 diabetes, a large increase in islet volume/mass is commonly observed, largely as a result of increased beta cell proliferation and vascular density [108–113]. This massive increase in beta cell mass is attributed to an intra-islet, rather than systemic, insulin resistance [84, 114]. In type 2 diabetic humans and NHP models, a more heterogeneous scenario is observed. Studies have described a loss in beta cell mass coupled to an increase in amyloid (IAPP) deposition and alpha cell proliferation (Table 1). We refer to a recent review article describing beta cell pathophysiology in type 2 diabetes [115], describing beta cell failure as a central mechanism in both type 1 and type 2 diabetes. Interestingly, in non-diabetic humans, insulin resistance has been reported to contribute to increased beta cell number and islet size [116]. Recently, in a mouse model, beta cell dedifferentiation rather than accelerated death has been suggested as a key driver of beta cell failure [117, 118]. However, at this stage, no direct histopathological evidence has been found in human type 2 diabetes to support this observation besides the well-known imbalance observed in the peripheral blood between alpha and beta cell hormone levels [12].
Nevertheless, to date we are limited to cross-sectional studies on highly selected patient cohorts (Table 1). In view of the fact that the data from the UKPDS indicated a constant loss of beta cell function over time [119], it is imperative to investigate, longitudinally, the natural history of human islet dysfunction from pathological onset to progressive mini-organ failure. Only this approach will allow the dissection the molecular mechanisms responsible for the morphological changes of pancreatic islets in the different metabolic phenotypes [119].
Conclusion
In this review we have summarised key interspecies differences in the islet cytoarchitecture of rodents, NHPs and humans (Fig. 1). Such differences might explain disparities observed in the intra-islet networks regulating islet physiology and pathophysiology between mice and humans (Fig. 2). In view of the evidence provided, we believe that islet and diabetes research should consider differences in islet cytoarchitecture when conducting and interpreting results from one given species, especially rodents. Since human islet composition is heterogeneous and significantly different from that of rodents but similar to that of NHPs, the NHP may serve as a more appropriate animal model for studying human islet pathophysiology from a translational perspective.
The future of islet research to address existing knowledge gaps in islet physiology and pathophysiology
Besides understanding the main differences in microanatomy between the rodent and the human islet, it is essential to study islet physiology and pathophysiology in an in vivo setting. Only by using an in vivo platform can researchers truly appreciate an integrated view of islet biology. The islet micro-environment is highly dynamic, and cross-sectional studies are limited to snapshots of changes in islet cytoarchitecture and function (Fig. 1 and Table 1), ignoring the longitudinal dimension that takes into account the natural history of such changes. Moreover, isolated islets lack functionally important nerve fibres and blood vessels, and are thus not under the influence of crucial peripheral and central regulatory factors. Therefore, as long as the majority of islet research is conducted ex vivo, whether in isolated islets or pancreatic slices, our knowledge of islet pathophysiology will remain incomplete and lacking the translation from bench to bedside. Over the years islet researchers have developed various in vivo islet imaging techniques to provide a glimpse of islet morphology, function, innervation and vascularisation (reviewed in [22, 23]).
The next step in the evolution of these techniques would involve in vivo islet imaging in the NHP, as these islets represent a close surrogate of human islet physiology. An increasing number of publications suggest that islets transplanted into the mouse anterior chamber of the eye (ACE) mirror those of the endogenous pancreas, both structurally and functionally [11, 37, 109, 120, 121]. Whether or not this holds true during diabetes pathogenesis (e.g. in states of glucolipotoxicity and of an immune attack) remains to be elucidated. It is noteworthy that studies on the autologous transplantation of NHP islets following partial pancreatectomy into the ACE are already under way. If successful, these studies may offer a quantum leap in understanding human-like islet plasticity, thus improving translational islet research.
Abbreviations
- ACE:
-
Anterior chamber of the eye
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- [Ca2+]i :
-
Intracellular free calcium
- CNS:
-
Central nervous system
- GABA:
-
γ-Aminobutyric acid
- GLP-1:
-
Glucagon-like peptide-1
- IR:
-
Insulin receptor
- NHP:
-
Non-human primate
- NMDA:
-
N-methyl-d-aspartate
- VEGF-A:
-
Vascular endothelial growth factor-A
References
Sullivan LH (1896) The tall office building artistically considered. Lippincotts Mag 57:403–409
Langerhans P (1869) Beitraege zur mikroskopischen Anatomie der Bauchspeicheldruese. Friedrich-Wilhelm-Universitaet zu Berlin, Inaugural Dissertation (in German)
Laguesse É (1893) Sur la formation des ilots de Langerhans dans le pancreas. Compt Rend Soc Biol 5:819–820 (in French)
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
de Koning EJ, Bodkin NL, Hansen BC, Clark A (1993) Diabetes mellitus in Macaca mulatta monkeys is characterised by islet amyloidosis and reduction in beta-cell population. Diabetologia 36:378–384
Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG (2010) Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia 53:2020–2028
Nyman LR, Ford E, Powers AC, Piston DW (2010) Glucose-dependent blood flow dynamics in murine pancreatic islets in vivo. Am J Physiol Endocrinol Metab 298:E807–E814
Rodriguez-Diaz R, Abdulreda MH, Formoso AL et al (2011) Innervation patterns of autonomic axons in the human endocrine pancreas. Cell Metab 14:45–54
Suckale J, Solimena M (2010) The insulin secretory granule as a signaling hub. Trends Endocrinol Metab 21:599–609
Rutter GA, Hodson DJ (2013) Minireview: intraislet regulation of insulin secretion in humans. Mol Endocrinol 27:1984–1995
Speier S, Nyqvist D, Cabrera O et al (2008) Noninvasive in vivo imaging of pancreatic islet cell biology. Nat Med 14:574–578
Unger RH, Orci L (2010) Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci U S A 107:16009–16012
Fiori JL, Shin YK, Kim W et al (2013) Resveratrol prevents beta-cell dedifferentiation in nonhuman primates given a high-fat/high-sugar diet. Diabetes 62:3500–3513
Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, Caicedo A (2006) The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc Natl Acad Sci U S A 103:2334–2339
Stewart AF (2014) Betatrophin versus bitter-trophin and the elephant in the room: time for a new normal in beta-cell regeneration research. Diabetes 63:1198–1199
Reed JC, Herold KC (2015) Thinking bedside at the bench: the NOD mouse model of T1DM. Nat Rev Endocrinol 11:308–314
Rodriguez-Diaz R, Dando R, Jacques-Silva MC et al (2011) Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans. Nat Med 17:888–892
Kim A, Miller K, Jo J, Kilimnik G, Wojcik P, Hara M (2009) Islet architecture: a comparative study. Islets 1:129–136
Seok J, Warren HS, Cuenca AG et al (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 110:3507–3512
Westermark P, Andersson A, Westermark GT (2011) Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 91:795–826
Mezza T, Kulkarni RN (2014) The regulation of pre- and post-maturational plasticity of mammalian islet cell mass. Diabetologia 57:1291–1303
Wang P, Medarova Z, Moore A (2011) Molecular imaging: a promising tool to monitor islet transplantation. J Transl 2011:202915
Leibiger IB, Caicedo A, Berggren PO (2012) Non-invasive in vivo imaging of pancreatic beta-cell function and survival – a perspective. Acta Physiol 204:178–185
Brissova M, Fowler MJ, Nicholson WE et al (2005) Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem: Off J Histochem Soc 53:1087–1097
Andralojc KM, Mercalli A, Nowak KW et al (2009) Ghrelin-producing epsilon cells in the developing and adult human pancreas. Diabetologia 52:486–493
Wang X, Zielinski MC, Misawa R et al (2013) Quantitative analysis of pancreatic polypeptide cell distribution in the human pancreas. PLoS One 8:e55501
Rodriguez-Diaz R, Caicedo A (2014) Neural control of the endocrine pancreas. Best Pract Res Clin Endocrinol Metab 28:745–756
Westwell-Roper CY, Ehses JA, Verchere CB (2014) Resident macrophages mediate islet amyloid polypeptide-induced islet IL-1β production and β-cell dysfunction. Diabetes 63:1698–1711
Bonner-Weir S, Orci L (1982) New perspectives on the microvasculature of the islets of Langerhans in the rat. Diabetes 31:883–889
Donath MY, Burcelin R (2013) GLP-1 effects on islets: hormonal, neuronal, or paracrine? Diabetes Care 36(Suppl 2):S145–S148
Lammert E, Cleaver O, Melton D (2001) Induction of pancreatic differentiation by signals from blood vessels. Science 294:564–567
Lammert E, Gu G, McLaughlin M et al (2003) Role of VEGF-A in vascularization of pancreatic islets. Curr Biol: CB 13:1070–1074
Gorden DL, Mandriota SJ, Montesano R, Orci L, Pepper MS (1997) Vascular endothelial growth factor is increased in devascularized rat islets of Langerhans in vitro. Transplantation 63:436–443
Samols E, Stagner JI, Ewart RB, Marks V (1988) The order of islet microvascular cellular perfusion is B–A–D in the perfused rat pancreas. J Clin Invest 82:350–353
Olsson R, Carlsson PO (2011) A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes 60:2068–2075
Lau J, Svensson J, Grapensparr L, Johansson A, Carlsson PO (2012) Superior beta cell proliferation, function and gene expression in a subpopulation of rat islets identified by high blood perfusion. Diabetologia 55:1390–1399
Almaca J, Molina J, Arrojo EDR et al (2014) Young capillary vessels rejuvenate aged pancreatic islets. Proc Natl Acad Sci U S A 111:17612–17617
Meissner HP (1976) Electrophysiological evidence for coupling between beta cells of pancreatic islets. Nature 262:502–504
Ravier MA, Guldenagel M, Charollais A et al (2005) Loss of connexin36 channels alters beta-cell coupling, islet synchronization of glucose-induced Ca2+ and insulin oscillations, and basal insulin release. Diabetes 54:1798–1807
Kilimnik G, Jo J, Periwal V, Zielinski MC, Hara M (2012) Quantification of islet size and architecture. Islets 4:167–172
Wang X, Misawa R, Zielinski MC et al (2013) Regional differences in islet distribution in the human pancreas – preferential beta-cell loss in the head region in patients with type 2 diabetes. PLoS One 8:e67454
Bonner-Weir S, Sullivan BA, Weir GC (2015) Human islet morphology revisited: human and rodent islets are not so different after all. J Histochem Cytochem. doi:10.1369/0022155415570969
Bosco D, Armanet M, Morel P et al (2010) Unique arrangement of alpha- and beta-cells in human islets of Langerhans. Diabetes 59:1202–1210
Meier JJ, Kohler CU, Alkhatib B et al (2010) Beta-cell development and turnover during prenatal life in humans. Eur J Endocrinol Eur Fed Endocr Soc 162:559–568
Gregg BE, Moore PC, Demozay D et al (2012) Formation of a human beta-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab 97:3197–3206
In’t Veld P, Lammert E (2015) The dark side of islet vasculature. Diabetologia 58:4–6
Stagner JI, Samols E (1992) The vascular order of islet cellular perfusion in the human pancreas. Diabetes 41:93–97
Stagner JI, Samols E, Koerker DJ, Goodner CJ (1992) Perfusion with anti-insulin gamma globulin indicates a B to A to D cellular perfusion sequence in the pancreas of the rhesus monkey, Macaca mulatta. Pancreas 7:26–29
Virtanen I, Banerjee M, Palgi J et al (2008) Blood vessels of human islets of Langerhans are surrounded by a double basement membrane. Diabetologia 51:1181–1191
Stozer A, Gosak M, Dolensek J et al (2013) Functional connectivity in islets of Langerhans from mouse pancreas tissue slices. PLoS Comput Biol 9:e1002923
Schaeffer M, Hodson DJ, Lafont C, Mollard P (2011) Endocrine cells and blood vessels work in tandem to generate hormone pulses. J Mol Endocrinol 47:R59–R66
Molina J, Rodriguez-Diaz R, Fachado A, Jacques-Silva MC, Berggren PO, Caicedo A (2014) Control of insulin secretion by cholinergic signaling in the human pancreatic islet. Diabetes 63:2714–2726
Cabrera O, Jacques-Silva MC, Speier S et al (2008) Glutamate is a positive autocrine signal for glucagon release. Cell Metab 7:545–554
Jacques-Silva MC, Correa-Medina M, Cabrera O et al (2010) ATP-gated P2X3 receptors constitute a positive autocrine signal for insulin release in the human pancreatic beta cell. Proc Natl Acad Sci U S A 107:6465–6470
Fernandez-Alvarez J, Hillaire-Buys D, Loubatières-Mariani MM, Gomis R, Petit P (2001) P2 receptor agonists stimulate insulin release from human pancreatic islets. Pancreas 22:69–71
Purwana I, Zheng J, Li X et al (2014) GABA promotes human beta-cell proliferation and modulates glucose homeostasis. Diabetes 63:4197–4205
Taneera J, Jin Z, Jin Y et al (2012) γ-Aminobutyric acid (GABA) signalling in human pancreatic islets is altered in type 2 diabetes. Diabetologia 55:1985–1994
Tian J, Dang H, Chen Z et al (2013) γ-Aminobutyric acid regulates both the survival and replication of human beta-cells. Diabetes 62:3760–3765
Trus MD, Hintz CS, Weinstein JB, Williams AD, Pagliara AS, Matschinsky FM (1979) A comparison of the effects of glucose and acetylcholine on insulin release and intermediary metabolism in rat pancreatic islets. J Biol Chem 254:3921–3929
Renuka TR, Robinson R, Paulose CS (2006) Increased insulin secretion by muscarinic M1 and M3 receptor function from rat pancreatic islets in vitro. Neurochem Res 31:313–320
Markmann JF, Deng S, Huang X et al (2003) Insulin independence following isolated islet transplantation and single islet infusions. Ann Surg 237:741–749, discussion 749-750
Burnstock G, Verkhratsky A (2009) Evolutionary origins of the purinergic signalling system. Acta Physiol 195:415–447
Cook DL, Hales CN (1984) Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature 311:271–273
MacDonald PE, Braun M, Galvanovskis J, Rorsman P (2006) Release of small transmitters through kiss-and-run fusion pores in rat pancreatic beta cells. Cell Metab 4:283–290
Obermuller S, Lindqvist A, Karanauskaite J, Galvanovskis J, Rorsman P, Barg S (2005) Selective nucleotide-release from dense-core granules in insulin-secreting cells. J Cell Sci 118:4271–4282
Leon C, Freund M, Latchoumanin O et al (2005) The P2Y1 receptor is involved in the maintenance of glucose homeostasis and in insulin secretion in mice. Purinergic Signal 1:145–151
Petit P, Manteghetti M, Puech R, Loubatieres-Mariani MM (1987) ATP and phosphate-modified adenine nucleotide analogues. Effects on insulin secretion and calcium uptake. Biochem Pharmacol 36:377–380
Poulsen CR, Bokvist K, Olsen HL et al (1999) Multiple sites of purinergic control of insulin secretion in mouse pancreatic beta-cells. Diabetes 48:2171–2181
Khan S, Yan-Do R, Duong E et al (2014) Autocrine activation of P2Y1 receptors couples Ca2+ influx to Ca2+ release in human pancreatic beta cells. Diabetologia 57:2535–2545
Roberts E, Frankel S (1950) γ-Aminobutyric acid in brain: its formation from glutamic acid. J Biol Chem 187:55–63
Olsen RW, Sieghart W (2009) GABAA receptors: subtypes provide diversity of function and pharmacology. Neuropharmacology 56:141–148
Braun M, Wendt A, Birnir B et al (2004) Regulated exocytosis of GABA-containing synaptic-like microvesicles in pancreatic beta-cells. J Gen Physiol 123:191–204
Rorsman P, Berggren PO, Bokvist K et al (1989) Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 341:233–236
Soltani N, Qiu H, Aleksic M et al (2011) GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc Natl Acad Sci U S A 108:11692–11697
Kim J, Richter W, Aanstoot HJ et al (1993) Differential expression of GAD65 and GAD67 in human, rat, and mouse pancreatic islets. Diabetes 42:1799–1808
Braun M, Ramracheya R, Bengtsson M et al (2010) γ-Aminobutyric acid (GABA) is an autocrine excitatory transmitter in human pancreatic beta-cells. Diabetes 59:1694–1701
Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130:1007S–1015S
Inagaki N, Kuromi H, Gonoi T et al (1995) Expression and role of ionotropic glutamate receptors in pancreatic islet cells. FASEB J: Off Publ Fed Am Soc Exp Biol 9:686–691
Muroyama A, Uehara S, Yatsushiro S et al (2004) A novel variant of ionotropic glutamate receptor regulates somatostatin secretion from delta-cells of islets of Langerhans. Diabetes 53:1743–1753
Hayashi M, Yamada H, Uehara S et al (2003) Secretory granule-mediated co-secretion of l-glutamate and glucagon triggers glutamatergic signal transmission in islets of Langerhans. J Biol Chem 278:1966–1974
Feldmann N, del Rio RM, Gjinovci A, Tamarit-Rodriguez J, Wollheim CB, Wiederkehr A (2011) Reduction of plasma membrane glutamate transport potentiates insulin but not glucagon secretion in pancreatic islet cells. Mol Cell Endocrinol 338:46–57
Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA (1922) Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J 12:141–146
Uhles S, Moede T, Leibiger B, Berggren PO, Leibiger IB (2003) Isoform-specific insulin receptor signaling involves different plasma membrane domains. J Cell Biol 163:1327–1337
Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR (1999) Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339
Martinez SC, Cras-Meneur C, Bernal-Mizrachi E, Permutt MA (2006) Glucose regulates Foxo1 through insulin receptor signaling in the pancreatic islet beta-cell. Diabetes 55:1581–1591
Khan FA, Goforth PB, Zhang M, Satin LS (2001) Insulin activates ATP-sensitive K+ channels in pancreatic beta-cells through a phosphatidylinositol 3-kinase-dependent pathway. Diabetes 50:2192–2198
Kawamori D, Kurpad AJ, Hu J et al (2009) Insulin signaling in alpha cells modulates glucagon secretion in vivo. Cell Metab 9:350–361
Wu H, MacFarlane WM, Tadayyon M, Arch JR, James RF, Docherty K (1999) Insulin stimulates pancreatic-duodenal homoeobox factor-1 (PDX1) DNA-binding activity and insulin promoter activity in pancreatic beta cells. Biochem J 344:813–818
Persaud SJ, Asare-Anane H, Jones PM (2002) Insulin receptor activation inhibits insulin secretion from human islets of Langerhans. FEBS Lett 510:225–228
Brunicardi FC, Kleinman R, Moldovan S et al (2001) Immunoneutralization of somatostatin, insulin, and glucagon causes alterations in islet cell secretion in the isolated perfused human pancreas. Pancreas 23:302–308
Ramnanan CJ, Edgerton DS, Kraft G, Cherrington AD (2011) Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes Metab 13(Suppl 1):118–125
Gelling RW, Du XQ, Dichmann DS et al (2003) Lower blood glucose, hyperglucagonemia, and pancreatic alpha cell hyperplasia in glucagon receptor knockout mice. Proc Natl Acad Sci U S A 100:1438–1443
Moens K, Flamez D, Van Schravendijk C, Ling Z, Pipeleers D, Schuit F (1998) Dual glucagon recognition by pancreatic beta-cells via glucagon and glucagon-like peptide 1 receptors. Diabetes 47:66–72
Huypens P, Ling Z, Pipeleers D, Schuit F (2000) Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia 43:1012–1019
Moens K, Heimberg H, Flamez D et al (1996) Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes 45:257–261
Kieffer TJ, Heller RS, Unson CG, Weir GC, Habener JF (1996) Distribution of glucagon receptors on hormone-specific endocrine cells of rat pancreatic islets. Endocrinology 137:5119–5125
Sorensen H, Winzell MS, Brand CL et al (2006) Glucagon receptor knockout mice display increased insulin sensitivity and impaired beta-cell function. Diabetes 55:3463–3469
Leibiger B, Moede T, Muhandiramlage TP et al (2012) Glucagon regulates its own synthesis by autocrine signaling. Proc Natl Acad Sci U S A 109:20925–20930
Gahete MD, Cordoba-Chacon J, Duran-Prado M et al (2010) Somatostatin and its receptors from fish to mammals. Ann N Y Acad Sci 1200:43–52
Hauge-Evans AC, King AJ, Carmignac D et al (2009) Somatostatin secreted by islet delta-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58:403–411
Kumar U, Sasi R, Suresh S et al (1999) Subtype-selective expression of the five somatostatin receptors (hSSTR1-5) in human pancreatic islet cells: a quantitative double-label immunohistochemical analysis. Diabetes 48:77–85
Wang XP, Yang J, Norman MA, Magnusson J, DeMayo FJ, Brunicardi FC (2005) SSTR5 ablation in islet results in alterations in glucose homeostasis in mice. FEBS Lett 579:3107–3114
Zhang Y, Zhang Y, Bone RN et al (2012) Regeneration of pancreatic non-beta endocrine cells in adult mice following a single diabetes-inducing dose of streptozotocin. PLoS One 7:e36675
Plesner A, Ten Holder JT, Verchere CB (2014) Islet remodeling in female mice with spontaneous autoimmune and streptozotocin-induced diabetes. PLoS One 9:e102843
Dufrane D, Maillart JF, Aouassar N, Goebbels RM, Guiot Y, Gianello P (2009) Native pancreatic alpha-cell adaptation in streptozotocin-induced diabetic primates: importance for pig islet xenotransplantation. Xenotransplantation 16:152–163
Meier JJ, Bhushan A, Butler AE, Rizza RA, Butler PC (2005) Sustained beta cell apoptosis in patients with long-standing type 1 diabetes: indirect evidence for islet regeneration? Diabetologia 48:2221–2228
Walker JN, Johnson PR, Shigeto M, Hughes SJ, Clark A, Rorsman P (2011) Glucose-responsive beta cells in islets isolated from a patient with long-standing type 1 diabetes mellitus. Diabetologia 54:200–202
Carlsson PO, Andersson A, Jansson L (1996) Pancreatic islet blood flow in normal and obese-hyperglycemic (ob/ob) mice. Am J Physiol 271:E990–E995
Ilegems E, Dicker A, Speier S et al (2013) Reporter islets in the eye reveal the plasticity of the endocrine pancreas. Proc Natl Acad Sci U S A 110:20581–20586
Nakamura M, Kitamura H, Konishi S et al (1995) The endocrine pancreas of spontaneously diabetic db/db mice: microangiopathy as revealed by transmission electron microscopy. Diabetes Res Clin Pract 30:89–100
Dai C, Brissova M, Reinert RB et al (2013) Pancreatic islet vasculature adapts to insulin resistance through dilation and not angiogenesis. Diabetes 62:4144–4153
Paulsen SJ, Jelsing J, Madsen AN et al (2010) Characterization of beta-cell mass and insulin resistance in diet-induced obese and diet-resistant rats. Obesity (Silver Spring) 18:266–273
Ribeiro RA, Santos-Silva JC, Vettorazzi JF et al (2012) Taurine supplementation prevents morpho-physiological alterations in high-fat diet mice pancreatic beta-cells. Amino Acids 43:1791–1801
Okada T, Liew CW, Hu J et al (2007) Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc Natl Acad Sci U S A 104:8977–8982
Halban PA, Polonsky KS, Bowden DW et al (2014) β-Cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment. Diabetes Care 37:1751–1758
Mezza T, Muscogiuri G, Sorice GP et al (2014) Insulin resistance alters islet morphology in nondiabetic humans. Diabetes 63:994–1007
Talchai C, Xuan S, Lin HV, Sussel L, Accili D (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150:1223–1234
Weir GC, Bonner-Weir S (2004) Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 5(Suppl 3):S16–S21
UKPDS (1995) U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 44:1249–1258
Abdulreda MH, Faleo G, Molano RD et al (2011) High-resolution, noninvasive longitudinal live imaging of immune responses. Proc Natl Acad Sci U S A 108:12863–12868
Rodriguez-Diaz R, Speier S, Molano RD et al (2012) Noninvasive in vivo model demonstrating the effects of autonomic innervation on pancreatic islet function. Proc Natl Acad Sci U S A 109:21456–21461
Savinov AY, Wong FS, Stonebraker AC, Chervonsky AV (2003) Presentation of antigen by endothelial cells and chemoattraction are required for homing of insulin-specific CD8+ T cells. J Exp Med 197:643–656
Akirav EM, Baquero MT, Opare-Addo LW et al (2011) Glucose and inflammation control islet vascular density and beta-cell function in NOD mice: control of islet vasculature and vascular endothelial growth factor by glucose. Diabetes 60:876–883
Reddy S, Pathipati P, Bai Y, Robinson E, Ross JM (2005) Histopathological changes in insulin, glucagon and somatostatin cells in the islets of NOD mice during cyclophosphamide-accelerated diabetes: a combined immunohistochemical and histochemical study. J Mol Histol 36:289–300
Esterhazy D, Stutzer I, Wang H et al (2011) Bace2 is a beta cell-enriched protease that regulates pancreatic beta cell function and mass. Cell Metab 14:365–377
Devedjian JC, George M, Casellas A et al (2000) Transgenic mice overexpressing insulin-like growth factor-II in beta cells develop type 2 diabetes. J Clin Invest 105:731–740
Guillam MT, Hummler E, Schaerer E et al (1997) Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat Genet 17:327–330
Höög A, Sandberg-Nordqvist AC, Abdel-Halim SM et al (1996) Increased amounts of a high molecular weight insulin-like growth factor II (IGF-II) peptide and IGF-II messenger ribonucleic acid in pancreatic islets of diabetic Goto-Kakizaki rats. Endocrinology 137:2415–2423
Agudo J, Ayuso E, Jimenez V et al (2012) Vascular endothelial growth factor-mediated islet hypervascularization and inflammation contribute to progressive reduction of beta-cell mass. Diabetes 61:2851–2861
Guardado-Mendoza R, Davalli AM, Chavez AO et al (2009) Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc Natl Acad Sci U S A 106:13992–13997
Guardado-Mendoza R, Jimenez-Ceja L, Majluf-Cruz A et al (2013) Impact of obesity severity and duration on pancreatic beta- and alpha-cell dynamics in normoglycemic non-human primates. Int J Obes (Lond) 37:1071–1078
Jurgens CA, Toukatly MN, Fligner CL et al (2011) β-Cell loss and β-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 178:2632–2640
Henquin JC, Rahier J (2011) Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia 54:1720–1725
Tomita T (2012) Islet amyloid polypeptide in pancreatic islets from type 2 diabetic subjects. Islets 4:223–232
Yoneda S, Uno S, Iwahashi H et al (2013) Predominance of beta-cell neogenesis rather than replication in humans with an impaired glucose tolerance and newly diagnosed diabetes. J Clin Endocrinol Metab 98:2053–2061
Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC (2008) Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10(Suppl 4):32–42
Marselli L, Suleiman M, Masini M et al (2014) Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia 57:362–365
Butcher MJ, Hallinger D, Garcia E et al (2014) Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 57:491–501
Acknowledgements
This work was supported by grants from the Singapore Ministry of Education, Academic Research Fund Tier 1(2014-T1-001-149 to Y.A.) and by Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore (NTU) start-up grants (separately for Y.A., P.O.B. and B.O.B.). The Lee Kong Chian School of Medicine is a partnership between Nanyang Technological University, Singapore (NTU) and Imperial College London. Due to space constraints we have not been able to provide a comprehensive presentation of the theme and have thereby disregarded many important contributions to the field of our colleagues.
Duality of interest
POB is co-founder and CEO of Biocrine AB, a diabetes-related biotech company.
Contribution statement
All authors were responsible for drafting the article and revising it critically. All authors approved the version to be published.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Rafael Arrojo e Drigo and Yusuf Ali contributed equally to this work.
Rights and permissions
About this article

Cite this article
Arrojo e Drigo, R., Ali, Y., Diez, J. et al. New insights into the architecture of the islet of Langerhans: a focused cross-species assessment. Diabetologia 58, 2218–2228 (2015). https://doi.org/10.1007/s00125-015-3699-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-015-3699-0