
Abstract
Aims/hypothesis
Proinflammatory and proapoptotic cytokines such as TNF-α are upregulated in human obesity. We evaluated the association between ghrelin isoforms (acylated and desacyl ghrelin) and TNF-α in obesity and obesity-associated type 2 diabetes, as well as the potential role of ghrelin in the control of apoptosis and autophagy in human adipocytes.
Methods
Plasma concentrations of the ghrelin isoforms and TNF-α were measured in 194 participants. Ghrelin and ghrelin O-acyltransferase (GOAT) levels were analysed by western-blot, immunohistochemistry and real-time PCR in 53 biopsies of human omental adipose tissue. We also determined the effect of acylated and desacyl ghrelin (10 to 1,000 pmol/l) on TNF-α-induced apoptosis and autophagy-related molecules in omental adipocytes.
Results
Circulating concentrations of acylated ghrelin and TNF-α were increased, whereas desacyl ghrelin levels were decreased in obesity-associated type 2 diabetes. Ghrelin and GOAT were produced in omental and subcutaneous adipose tissue. Visceral adipose tissue from obese patients with type 2 diabetes showed higher levels of GOAT, increased adipocyte apoptosis and increased expression of the autophagy-related genes ATG5, BECN1 and ATG7. In differentiating human omental adipocytes, incubation with acylated and desacyl ghrelin reduced TNF-α-induced activation of caspase-8 and caspase-3, and cell death. In addition, acylated ghrelin reduced the basal expression of the autophagy-related genes ATG5 and ATG7, while desacyl ghrelin inhibited the TNF-α-induced increase of ATG5, BECN1 and ATG7 expression.
Conclusions/interpretation
Apoptosis and autophagy are upregulated in human visceral adipose tissue of patients with type 2 diabetes. Acylated and desacyl ghrelin reduce TNF-α-induced apoptosis and autophagy in human visceral adipocytes.
Similar content being viewed by others

Introduction
Ghrelin, a 28 amino-acid peptide with an n-octanoyl group at the serine 3 residue, is the endogenous ligand of the growth hormone secretagogue receptor (GHS-R) [1, 2]. Ghrelin is synthesised predominantly in X/A cells of the oxyntic glands in the mucosa layer of the gastric fundus and secreted to the bloodstream in two major forms: acylated (∼5% of total ghrelin) and desacyl ghrelin (the form lacking n-octanoylation and representing ∼95% of total ghrelin) [1, 3, 4]. The porcupine-like enzyme ghrelin O-acyltransferase (GOAT) catalyses the octanoylation of ghrelin in the endoplasmic reticulum (ER) [3, 4] and belongs to the family of membrane-bound O-acyltransferases (MBOAT) [5]. The expression sites of Mboat4, the gene encoding GOAT, in rodents include stomach, intestine, colon and testis, while in humans MBOAT4 is mainly expressed in stomach, intestine and pancreas [3–5]. Acylated and desacyl ghrelin increase adiposity and body weight through central and peripheral mechanisms [6]. On the one hand, ghrelin increases food intake by activating hypothalamic neuropeptide Y/agouti-related peptide neurons, which produce GHS-R type 1a, via the modulation of fatty acid metabolism [7]. On the other hand, adipose tissue is also an important target for the adipogenic actions of ghrelin in rodents and humans [8, 9]. Thus ghrelin (GHRL) gene expression increases during adipogenesis with preproghrelin knockdown reducing insulin-mediated adipogenesis in 3T3-L1 adipocytes [9]. Moreover, acylated and desacyl ghrelin directly stimulate the production of several fat storage-related proteins, including acetyl-CoA carboxylase, fatty acid synthase, lipoprotein lipase and perilipin, in human visceral adipocytes, thereby stimulating intracytoplasmic lipid accumulation [8, 10].
The excessive expansion of adipose tissue during the onset of obesity results in the activation of death receptors and mitochondrial pathways, leading to the activation of effector caspases and adipocyte apoptosis [11, 12]. Other types of cell death, namely autophagy, have been recently shown to be altered in adipose tissue in obesity [13]. Autophagy is a dynamic self-degrading process, characterised by sequestering of cytosolic organelles and proteins in double-membrane vesicles, which are known as autophagosomes and translocate to lysosomes for fusion and content degradation [14]. The proteins encoded by autophagy-related genes (ATG) are required for the formation of autophagic vesicles. Given the anti-apoptotic action of ghrelin in several cell systems, including murine 3T3-L1 adipocytes [15–17], it seems plausible that ghrelin operates as a survival factor that prevents obesity-associated apoptosis and autophagy in human adipocytes. We therefore sought to: (1) characterise the production and regulation of the ghrelin–GOAT system in human adipose tissue; and (2) analyse the effect of acylated and desacyl ghrelin on basal and TNF-α-induced apoptosis, and on autophagy in human visceral adipocytes.
Methods
Patient selection and study design
The study of acylated and desacyl ghrelin plasma levels was performed in a population of 194 participants (111 men, 83 women). BMI was calculated as weight in kilograms divided by the square of height in meters. Obesity was defined as a BMI of ≥30 kg/m2 and normal weight as a BMI of <25 kg/m2. Obese patients were sub-classified into three groups (normoglycaemia, impaired glucose tolerance or type 2 diabetes) following the criteria of the Expert Committee on the Diagnosis and Classification of Diabetes [18]. Participants with type 2 diabetes were not on insulin therapy or medication likely to influence endogenous insulin levels. It should be stressed that the type 2 diabetic patients included in our obese group did not have a long history of diabetes (less than 2 to 3 years, or even de novo diagnosis on the basis of their anamnesis and biochemical determinations). The clinical characteristics of the participants are shown in Table 1.
In addition, the presence of ghrelin and GOAT, and the effect of the ghrelin isoforms were assessed in paired omental and subcutaneous adipose tissue samples from 53 patients undergoing either laparoscopic Roux-en-Y gastric bypass (n = 43) or Nissen fundoplication (n = 10) at the Clínica Universidad de Navarra. The clinical characteristics of this cohort were essentially similar to those of the cohort in which circulating ghrelin isoforms were studied (Electronic supplementary material [ESM] Table 1). All reported investigations were carried out in accordance with the principles of the Declaration of Helsinki as revised in 2008. The experimental design was approved from an ethical and scientific standpoint by the Research Ethics Committee of the Clínica Universidad de Navarra, which is responsible for research (028/2009); informed consent was obtained from all volunteers. For more details see ESM.
Analytical procedures
Biochemical and hormonal assays performed in the study were done as previously described [8, 10]. Insulin resistance and sensitivity were calculated using the HOMA and QUICKI indices, respectively [19, 20]. Acylated and desacyl ghrelin were determined by commercially available ELISA kits (Linco Research, St Charles, MO, USA) following the manufacturer’s guidelines, as described earlier [8, 10]. TNF-α was measured with a high-sensitivity ELISA (R&D Systems, Minneapolis, MN, USA). For more details see ESM.
RNA extraction and real-time PCR
RNA isolation and purification were performed as described earlier [8, 21]. Transcript levels for ghrelin (GHRL), GOAT (MBOAT4) and the ATGs ATG5, BECN1 and ATG7 were quantified by real-time PCR (7300 Real-Time PCR System; Applied Biosystems, Foster City, CA, USA). Primers and probes (ESM Table 2) were designed using Primer Express 2.0 (Applied Biosystems). For more details see ESM.
Western blot studies
Blots were incubated overnight at 4°C with anti-ghrelin (Alpha Diagnostic, San Antonio, TX, USA), anti-GOAT (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-caspase-3 (Cell Signaling Technology, Danvers, MA, USA), anti-cleaved caspase-3 (Cell Signaling), anti-caspase-8 (Cell Signaling) and anti-cleaved caspase-8 (Cell Signaling) antibodies (diluted 1:1,000 in blocking solution), or with anti-β-actin (Sigma, St Louis, MO, USA) antibody (diluted 1:5,000 in blocking solution). The antigen–antibody complexes were visualised using horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG antibodies (1:5,000), and an enhanced chemiluminescence detection system (ECL Plus; Amersham Biosciences, Little Chalfont, Buckinghamshire, UK). For more details see ESM.
Immunohistochemistry of ghrelin and GOAT
Immunohistochemical detection of ghrelin and GOAT was carried out as described before [8]. Sections were incubated overnight at 4°C with rabbit polyclonal anti-ghrelin (Alpha Diagnostic) or rabbit polyclonal anti-GOAT (Santa Cruz Biotechnology) antibodies diluted 1:50 in TRIS-buffer saline. After three washes with TRIS-buffer saline, slides were incubated for 1 h at room temperature with horseradish peroxidase-conjugated anti-rabbit/mouse (Dako, Glostrup, Denmark). The peroxidase reaction was visualised with a 3,3′-diaminobenzidine (Amersham Biosciences)/H2O2 solution (0.5 mg/ml 3,3′-diaminobenzidine, 0.03% H2O2 diluted in 50 mmol/l TRIS–HCl, pH 7.36) as chromogen and with Harris haematoxylin solution (Sigma) as counterstaining. For more details see ESM.
Confocal immunofluorescence microscopy of ghrelin and GOAT
Differentiated human adipocytes grown on glass coverslips were fixed with 4% paraformaldehyde (wt/vol) (15 min, room temperature), incubated with PBS containing 0.1% saponin (wt/vol) and 1% BSA (wt/vol) (1 h, room temperature), and exposed overnight and at 4°C to rabbit polyclonal anti-ghrelin (Alpha Diagnostic) or anti-GOAT (Santa Cruz) antibodies diluted 1:50 (ghrelin) or 1:250 (GOAT) in PBS containing 0.1% saponin (wt/vol). Thereafter, cells were washed with PBS and incubated at room temperature for 2 h with Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Invitrogen, Paisley, UK) diluted 1:500. After washing, coverslips were mounted on microscope slides and examined under a confocal laser scanning microscope (TCS-SP2-AOBS; Leica, Heidelberg, Germany). For more details see ESM.
Cell cultures
Human stromovascular fraction cells (SVFC) were isolated from omental adipose tissue from obese normoglycaemic participants and differentiated as previously described [8]. Differentiated human omental adipocytes were serum-starved for 24 h and then treated for 24 h with increasing concentrations of insulin (Sigma), leptin (PeproTech, Rocky Hill, NJ, USA), TNF-α (PeproTech), acylated ghrelin (Tocris, Ellisville, MO, USA) or desacyl ghrelin (Tocris). In a subset of experiments quiescent cells were incubated for 24 h with TNF-α (100 ng/ml) in the presence or absence of acylated ghrelin (100 pmol/l) or desacyl ghrelin (100 pmol/l). One sample per experiment was used to obtain control responses in the presence of the solvent. For more details see ESM.
TUNEL assay
The TUNEL assay was performed using an in situ cell death detection kit (POD; Roche, Basel, Switzerland) following the manufacturer’s instructions. TUNEL-positive cells were analysed under the light microscope. For more details see ESM.
Statistical analysis
Data are expressed as mean ± SEM. Statistical differences between mean values were determined using Student’s t test, a χ2 test and one-way ANOVA, followed by Scheffé’s or Dunnet’s tests where appropriate. A value of p < 0.05 was considered statistically significant. Due to their non-normal distribution, plasma concentrations of acylated and desacyl ghrelin, and TNF-α were logarithmically transformed (log10) for statistical analyses. In the analysis of Pearson’s correlation coefficients (r), Bonferroni’s correction was applied when multiple comparisons were performed simultaneously, establishing the significance level at p = 0.003. For more details see ESM.
Results
Plasma levels of acylated and desacyl ghrelin, and TNF-α in obesity and obesity-associated type 2 diabetes
Obese participants showed increased circulating concentrations of acylated ghrelin (obese 5.1 ± 0.3, lean 3.9 ± 0.3 pmol/l [mean ± SEM], p = 0.013) and decreased circulating concentrations of desacyl ghrelin (obese 90.9 ± 4.7, lean 168.1 ± 15.6 pmol/l, p < 0.001) compared with lean individuals (Fig. 1a, b). Acylated ghrelin levels were also higher (p < 0.01) in obese individuals with type 2 diabetes than in obese participants with normoglycaemia or impaired glucose tolerance (Fig. 1a). No effect of glucose intolerance or diabetes was observed on circulating desacyl ghrelin concentrations. Plasma desacyl ghrelin concentrations were increased in women compared with men in the lean group (women 192.5 ± 13.6, men 133.3 ± 13.8 pmol/l, p < 0.001) and in the obese group (women 104.3 ± 12.1, men 88.6 ± 9.4 pmol/l, p < 0.001). This sexual dimorphism was not found in the plasma acylated ghrelin levels of lean participants (women 3.8 ± 0.4, men 4.2 ± 0.5 pmol/l) or in those of the obese patients (women 4.7 ± 0.4, men 5.3 ± 0.3 pmol/l).

(a) Impact of obesity and obesity-associated type 2 diabetes on fasting plasma concentrations of acylated and (b) desacyl ghrelin in lean volunteers (n = 55), obese normoglycaemic (NG) participants (n = 66), obese individuals with impaired glucose tolerance (IGT) (n = 37) and obese patients with type 2 diabetes (T2D) (n = 36). Differences between groups were analysed by one-way ANOVA followed by Scheffé’s test; ***p < 0.001 vs lean NG participants; †† p < 0.01 vs obese NG participants. (c) Correlations between plasma TNF-α and both plasma acylated ghrelin and (d) desacyl ghrelin in lean volunteers (white circles), and obese NG (grey circles), obese IGT (hatched circles) and obese T2D (black circles) patients
Type 2 diabetes was associated with increased circulating levels of well-known inflammatory markers, including TNF-α (Table 1). Interestingly, no correlation was observed between acylated ghrelin and TNF-α (Fig. 1c), whereas desacyl ghrelin showed a negative correlation (r = 0.23, p < 0.05) with this proinflammatory cytokine (Fig. 1d).
Localisation and abundance of ghrelin and GOAT in human adipose tissue
To analyse whether adipose tissue is a production site of ghrelin and its acylating enzyme, GOAT, the presence of both molecules was analysed in biopsies of omental and subcutaneous adipose tissue from lean and obese patients. Analysis was performed by western-blot and real-time PCR. Omental adipose tissue of obese participants showed similar gene expression of ghrelin (p = 0.800), but increased transcript levels of MBOAT4 (p < 0.01) compared with lean individuals (Fig. 2a). Furthermore, transcript levels of GOAT were higher (p < 0.05) in obese individuals with type 2 diabetes than in normoglycaemic patients. No differences in GHRL and MBOAT4 mRNA in subcutaneous fat were found between the lean and obese groups (Fig. 2b). The levels of ghrelin and GOAT in omental and subcutaneous fat depots exhibited a similar pattern to that observed in the gene expression analyses (Fig. 2c, d). A positive correlation between MBOAT4 mRNA levels and serum LDL-cholesterol in omental adipose tissue (r = 0.71, p = 0.002) was found.

Impact of obesity and obesity-associated type 2 diabetes on the expression and abundance of ghrelin and GOAT in omental and subcutaneous adipose tissue. (a, b) The expression of GHRL and MBOAT4 mRNA, and (c, d) the corresponding protein abundance in omental and subcutaneous white adipose tissue (WAT) obtained from lean volunteers (white bars), obese normoglycaemic participants (grey bars), obese individuals with impaired glucose tolerance (hatched bars) and obese patients with type 2 diabetes (black bars); n = 10 per group. Representative blots are shown below the histograms (c, d). Gene expression and protein abundance in lean participants was assumed to be 1. Differences between groups were analysed by one-way ANOVA followed by Scheffé’s test; *p < 0.05 and **p < 0.01 vs lean NG participants; † p < 0.05 vs obese NG participants. (e) Immunohistochemical detection of ghrelin and GOAT in omental and (f) subcutaneous WAT obtained from obese patients. Magnification ×100 for scale bars 80 μm; ×400 for scale bars 20 μm. No immunoreactivity was found without primary antibody (control). (g) Immunocytochemical detection of ghrelin and GOAT proteins in differentiated human adipocytes. Images represent the Z-projection of 20 confocal stacks. Representative images of at least three separate experiments are shown
To identify the cell type responsible for ghrelin and GOAT production, the presence of both molecules in omental adipose tissue was evaluated by immunohistochemistry and real-time PCR. Positive black staining for ghrelin and GOAT was observed in fully mature adipocytes, as well as in the SVFC of omental and subcutaneous fat depots from obese patients (Fig. 2e, f). Adipocytes and SVFC showed similar (p > 0.05) transcript levels for ghrelin and GOAT in omental (GHRL 1.00 ± 0.67 vs 0.38 ± 0.26 arbitrary units [AU]; MBOAT4 1.00 ± 0.40 vs 0.70 ± 0.43 AU) and subcutaneous (GHRL 1.00 ± 0.45 vs 1.52 ± 0.36 AU; MBOAT4 1.00 ± 0.53 vs 0.82 ± 0.33 AU) adipose tissue. To gain more insight into the localisation of ghrelin and GOAT, the subcellular localisation of both proteins was studied in differentiated human adipocytes using confocal immunofluorescence microscopy. As shown in Fig. 2g, ghrelin and GOAT exhibited a cytoplasmic distribution in adipocytes. More specifically, the ghrelin immunosignal surrounded the nucleus and appeared as ring-shaped structures at the periphery of lipid droplets, which is morphologically reminiscent of the appearance of the ER. A similar intracellular distribution was observed for GOAT immunoreactivity (Fig. 2g). No immunoreaction was detected when primary or secondary antibodies were omitted.
Regulation of ghrelin and GOAT in human omental adipocytes
Given the increased abundance of GOAT in omental adipose tissue and the metabolic relevance of this fat depot [22], the subsequent experiments focused on this fat depot. To gain insight into the regulation of ghrelin and GOAT production, the roles of insulin and leptin, two hormones that are tightly associated with obesity and obesity-associated type 2 diabetes and have opposite effects on fat deposition, and the role of the proinflammatory cytokine, TNF-α, were analysed as plausible regulators of the expression of GHRL and MBOAT4 in human omental adipocytes (Fig. 3a–f). GHRL expression was reduced (p < 0.05) by increasing concentrations of insulin and leptin, but augmented (p < 0.05) in response to TNF-α in human visceral fat cells. MBOAT4 transcript levels increased (p < 0.05) in response to insulin, while neither leptin nor TNF-α modified them. The self-regulation of ghrelin in human visceral fat cells was also tested (Fig. 3g–j). GHRL expression was downregulated (p < 0.01) by desacyl ghrelin and upregulated (p = 0.019) by acylated ghrelin. MBOAT4 transcript levels in human visceral adipocytes were significantly increased (p < 0.01) by acylated ghrelin treatment at all the concentrations tested.

Regulation of ghrelin and GOAT production in human omental adipocytes. (a, c, e, g, i) The effect on GHRL and (b, d, f, h, j) MBOAT4 expression of incubating differentiated omental fat cells for 24 h with different concentrations of substances as labelled. Differences between groups were analysed by one-way ANOVA followed by Dunnet’s test; *p < 0.05 and **p < 0.01 vs unstimulated adipocytes
Acylated and desacyl ghrelin prevent TNF-α-induced apoptosis in human visceral adipocytes
TUNEL assays were initially performed to analyse adipocyte apoptosis in human omental adipose tissue from a well-characterised group of lean and obese participants. Adipocyte apoptosis was markedly increased (p < 0.0001) in the visceral adipose tissue of obese participants (Fig. 4a and b), which is in agreement with previous reports [12]. The quantification of TUNEL-positive cells showed that adipocyte apoptosis was further increased (p < 0.05) in adipose tissue of obese type 2 diabetes patients compared with normoglycaemic obese participants.

Effect of acylated and desacyl ghrelin on TNF-α-induced apoptosis. (a) Adipocyte apoptosis in omental white adipose tissue (WAT) obtained from lean volunteers (white bars), obese normoglycaemic (NG) participants (grey bars), obese individuals with impaired glucose tolerance (IGT) (hatched bars) and obese patients with type 2 diabetes (T2D) (black bars); n = 5 per group. Differences between groups were analysed by one-way ANOVA followed by Scheffé’s test; *p < 0.05, **p < 0.01 and ***p < 0.001 vs lean participants; † p < 0.05 vs obese NG. (b) Images on which bar graph (a) is based. (c) The effect of TNF-α (0, 1, 10 and 100 ng/ml) treatment for 24 h on the activation of caspase-8 and (d) caspase-3, and (e) on apoptosis. (f) Effect of co-incubation with acylated ghrelin (100 pmol/l) or desacyl ghrelin (100 pmol/l) on TNF-α (100 ng/ml)-induced activation of caspase-8 and (g) caspase-3, and (h) apoptosis. (c–h) Differences between groups were analysed by one-way ANOVA followed by Dunnet’s test; n = 8 per group; *p < 0.05 and ***p < 0.001 vs unstimulated adipocytes
We evaluated the effect of acylated and desacyl ghrelin on TNF-α-induced caspase activation by examining the appearance of cleaved caspase-8 and caspase-3, while apoptosis was explored by detecting DNA fragmentation with the TUNEL assay. As expected, the incubation of human visceral fat cells with different concentrations of TNF-α for 24 h significantly (p < 0.001) increased the activation of caspase-8 and caspase-3, and apoptotic cell death (Fig. 4c, d and e). The co-incubation with desacyl ghrelin (100 pmol/l) or acylated ghrelin (100 pmol/l) significantly reduced the cleavage of caspase-8 and caspase-3, as well as the apoptotic indices induced by TNF-α (100 ng/ml) (Fig. 4f, g and h).
Acylated and desacyl ghrelin prevent basal autophagy in human visceral adipocytes
To study the regulation of autophagy in obesity, we first examined the expression patterns of several molecular indicators of autophagy (ATG5, ATG7 and Beclin-1/ATG6) in human visceral adipose tissue from lean and obese participants. Obese patients with type 2 diabetes showed significantly (p < 0.05) increased transcript levels of ATG7 and BECN1 in omental adipose tissue compared with lean controls and normoglycaemic obese individuals (Fig. 5b, c). The expression of ATG5 showed a similar trend in obesity-associated type 2 diabetes, but differences did not reach statistical significance (p < 0.10) (Fig. 5a). Adipocytes and SVFC isolated from omental adipose tissue of morbidly obese patients were used to identify which cell type expressed the ATGs in this fat depot. No statistically significant differences were observed in the transcript levels of ATG5, ATG7 and BECN1 in adipocytes and SVFC (Fig. 5d–f).

Impact of obesity and obesity-associated type 2 diabetes on indicators of autophagy in human omental adipose tissue. Expression of ATG5 (a), ATG7 (b) and BECN1 (c) mRNA in omental white adipose tissue (WAT) obtained from lean volunteers (white bars), obese normoglycaemic participants (grey bars), obese individuals with impaired glucose tolerance (hatched bars) and obese patients with type 2 diabetes (black bars). Gene expression in lean participants was assumed to be 1. Differences between groups were analysed by one-way ANOVA followed by Scheffé’s test; n = 9–13 per group; *p < 0.05 vs lean normoglycaemic participants; † p < 0.05 vs obese normoglycaemic. (d) Comparison of ATG5, ATG7 (e) and BECN1 (f) gene expression in freshly isolated adipocytes and SVFC from omental WAT of obese patients; n = 15. Differences between groups were analysed by two-tailed unpaired Student’s t test
To determine whether acylated and desacyl ghrelin, and TNF-α directly affect autophagy in adipose tissue, differentiated human adipocytes were exposed for 24 h to increasing concentrations of acylated and desacyl ghrelin in the presence or absence of TNF-α and the expression of ATG5, ATG7 and BECN1 was analysed. TNF-α upregulated (p < 0.05) these ATGs at the highest concentration tested (100 ng/ml) (Fig. 6a, e and i). The stimulation of human visceral adipocytes with desacyl ghrelin did not modify the basal expression of ATGs, but acylated ghrelin decreased (p < 0.05) the transcript levels of ATG5 and ATG7 in human visceral adipocytes (Fig. 6c and g). The co-incubation of TNF-α with acylated ghrelin returned the expression of BECN1 to basal levels, while co-incubation with desacyl ghrelin inhibited the TNF-α-induced increase in ATG5, ATG7 and BECN1 (Fig. 6d, h and l).

Regulation of autophagy induced by TNF-α and ghrelin isoforms in human omental adipocytes. The effect of TNF-α (0, 1, 10 and 100 ng/ml) treatment for 24 h on the induction of (a) ATG5, (e) ATG7 and BECN1 (i) mRNA in human omental adipocytes. Effect of desacyl ghrelin on (b) ATG5, (f) ATG7 and BECN1 (j), and acylated ghrelin on (c) ATG5, (g) ATG7 and BECN1 (k) expression. Effect of acylated ghrelin (100 pmol/l) or desacyl ghrelin (100 pmol/l) on TNF-α (100 ng/ml)-induced activation of (d) ATG5, (h) ATG7 and (l) BECN1 expression. Differences between groups were analysed by one-way ANOVA followed by Dunnet’s test; n = 8–10 per group; *p < 0.05 vs unstimulated adipocytes
Discussion
Circulating total ghrelin levels are decreased in obesity, insulin resistance or the metabolic syndrome, all well-known low-grade chronic inflammatory diseases [23–25]. Our findings show that obesity-associated type 2 diabetes is related to low desacyl ghrelin concentrations (the most abundant form of the hormone) and high acylated ghrelin levels, which is in accordance with previously published data [8, 10]. Our study also provides evidence that acylated ghrelin correlates negatively with TNF-α. A similar relationship between ghrelin and TNF-α has been found in other pathological inflammatory conditions, such as juvenile idiopathic arthritis, rheumatoid arthritis or congenital heart disease [25–27]. Since TNF-α and ghrelin exert opposite effects on the immune system and metabolism [28], the present study focused on the interaction of both molecules in human adipose tissue. Despite the negative correlation of TNF-α with plasma desacyl ghrelin, we observed that TNF-α increased GHRL mRNA expression in human visceral adipocytes, suggesting that both molecules interact at the autocrine/paracrine level in fat cells. In this sense, as described below in detail, our data show that acylated and desacyl ghrelin directly act on visceral adipose tissue to regulate TNF-α-induced apoptosis and autophagy in humans.
Human adipose tissue produces all the components of the ghrelin system, namely ghrelin, obestatin, GOAT, and the receptors of ghrelin-related peptides, GHS-R type 1a and G protein-coupled receptor 39 (GPR39) [8, 21, 29, 30]. Our findings show for the first time that ghrelin and GOAT are co-produced in human visceral adipocytes and SVFC. In line with this, we observed a similar intracellular distribution of ghrelin and GOAT immunosignals in human adipocytes, which resembled that observed in the ER. In this sense, it has been suggested that GOAT is located in the membrane of the ER and mediates the translocation of the octanoyl-CoA from the cytosolic side to the ER lumen prior to the transport of acylated ghrelin to the Golgi [31–33]. The subcellular localisation of ghrelin and GOAT found in the present study is similar to that observed in human myometrial smooth muscle cells and in the human embryonic kidney (HEK) 293 cell line [9, 34–36]. Further investigation is warranted to ascertain the mechanisms of ghrelin trafficking in human adipocytes.
Since human obesity and obesity-associated type 2 diabetes are associated with altered levels of circulating acylated and desacyl ghrelin, the potential contribution of human adipose tissue to this dysregulation was analysed. Ghrelin mRNA levels in omental and subcutaneous adipose tissue were similar and comparable within groups, which is in accordance with data published by Knerr and colleagues [29]. To our knowledge, the present study shows for the first time that insulin resistance rather than obesity increases the abundance of GOAT in human visceral adipose tissue, suggesting that this fat depot contributes to the acylation of ghrelin. Moreover, transcript levels of MBOAT4 in omental adipose tissue were positively associated with serum LDL-cholesterol. Interestingly, LDL-cholesterol interacts with acylated ghrelin, and a plausible role of triacylglycerol-rich LDL- and HDL-cholesterol particles as ghrelin transporters in the bloodstream has been suggested [37]. Although correlation does not imply causality, the association between GOAT and LDL-cholesterol may reflect an increased transport of ghrelin into the bloodstream, thereby contributing to the high levels of circulating acylated ghrelin in insulin resistance.
Several nutritional and hormonal factors are involved in the regulation of the ghrelin/GOAT system [29, 30]. Our data show that insulin and leptin suppressed ghrelin mRNA levels in human visceral fat cells. In this regard, perfusion of the rat stomach with insulin or leptin induced a dose-dependent inhibition of ghrelin release [38] and infusion of insulin at physiological doses throughout prolonged euglycaemic–hyperinsulinaemic clamps significantly decreased desacyl ghrelin in healthy participants and pregnant women with type 2 diabetes [13, 39]. Our results also show that insulin increases GOAT transcript levels in human fat cells. The existing literature on the effect of insulin on GOAT is scarce and controversial, with insulin inhibiting GOAT in INS-1 pancreatic cells, but lacking effect in murine primary pituitary cells [40, 41]. Here, we have also shown that acylated ghrelin promotes ghrelin production and acylation in human adipocytes, a finding in agreement with previously published data on murine primary pituitary cells [41], while desacyl ghrelin acts as a repressor of ghrelin transcription. In this sense, Mboat4 knockout mice, which lack acylated ghrelin, but otherwise exhibit physiological levels of desacyl ghrelin, showed a decreased body weight without changes in fat mass when fed a high-fat diet [42], which may be explained by the negative regulatory effect of desacyl ghrelin on GHRL evidenced in the present study. Taken together, insulin, leptin, TNF-α and ghrelin isoforms themselves act as transcriptional regulators of GHRL and MBOAT4 in human visceral adipocytes.
Acylated and desacyl ghrelin reduce the adipocyte apoptosis induced by TNF-α, a well-known regulator of apoptosis in adipose tissue. Upon binding of TNF-α to its receptor, TNF-α proapoptotic signalling occurs, resulting in caspase-8 cleavage and activation, which further activates caspase-3, leading to adipocyte cell death [43]. Our findings provide evidence that ghrelin isoforms reduce the activation of caspase-8 and caspase-3, and the apoptosis induced by TNF-α. In this regard, ghrelin reportedly prevented apoptosis in murine 3T3-L1 and rat adipocytes [16, 44], as well as in other cell types, such as the murine adult cardiomyocyte cell line HL-1, porcine aortic endothelial cells, rat hypothalamic neurons and cortical oligodendrocytes, rat INS-1E pancreatic beta cells, and the human adrenal gland carcinoma cell line [15, 17, 45–47]. In the present study, we also confirmed increased adipocyte apoptosis in human obesity and obesity-associated type 2 diabetes. Since adipocyte apoptosis constitutes a key initial event that contributes to macrophage infiltration into adipose tissue, insulin resistance and the hepatic steatosis associated with human obesity [12], the inhibition of adipocyte apoptosis by ghrelin isoforms may represent a novel potential therapeutic strategy for the treatment of obesity-associated inflammation (Fig. 7).

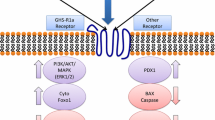
Schematic diagram of the effect of ghrelin isoforms on TNF-α-induced autophagy and apoptosis in human visceral adipocytes. Acylated (pink circles) and desacyl (orange circles) ghrelin inhibit the activation of caspase-3 and caspase-8, and apoptosis induced by TNF-α upon binding to its receptor, TNFR1. Desacyl ghrelin reduces TNF-α-induced transcription of BECN1, an ATG required to initiate the formation of the autophagosome in autophagy, as well as that of ATG7 and ATG5, the corresponding proteins of which participate in the conjugation cascades for autophagosome elongation. Acylated ghrelin reduces the basal expression of the ATGs ATG5 and ATG7. The imbalance of ghrelin isoforms and TNF-α in states of hyperglycaemia may contribute to the altered apoptosis and autophagy observed in patients with type 2 diabetes
Autophagy plays an important role in the regulation of adipose mass and differentiation [14, 48, 49], since ATG5 and ATG7 knockdown in 3T3-L1 adipocytes inhibits lipid accumulation and decreases the abundance of the adipogenic transcription factors, peroxisome proliferator-activated receptor γ and CCAAT/enhancer binding protein α (C/EBPα)[49]. Human adipose tissue contains autophagosomes and obesity is associated with altered levels of the autophagy-related molecules microtubule-associated protein 1 light chain 3 I (LC3-I) and II (LC3-II) and ATG5 [13]. Our data provide evidence that ATG5, beclin-1/ATG6 and ATG7 are present in human adipocytes and SVFC, and that hyperglycaemia, rather than obesity, is associated with altered expression of BECN1 and ATG7 in adipose tissue of obese patients with type 2 diabetes. In this sense, insulin constitutes a major inhibitor of autophagy, with insulin resistance being a potential activator of this process, since patients with type 2 diabetes show elevated formation of autophagosomes in subcutaneous adipose tissue [50].
The activation of autophagy in type 2 diabetes may reflect an underlying cell death of hypertrophied adipocytes [13]. In this regard, Beclin-1/ATG6 has been proposed as a potential link between apoptosis and autophagy [51]. Moreover, TNF-α reportedly induces the production of Beclin-1/ATG6 in vascular smooth muscle cells in atherosclerotic plaques, and Beclin-1/ATG6 is associated with cell death in the plaque [52]. In this context, we analysed the role of the pro-apoptotic TNF-α and the anti-apoptotic ghrelin in the regulation of ATGs. TNF-α increased the transcript levels of ATG5, BECN1 and ATG7, while acylated ghrelin reduced ATG5 and ATG7 expression in human visceral adipocytes. In addition, desacyl ghrelin reduced the TNF-α-induced expression of ATG5, ATG7 and BECN1. Taken together, we have thus shown for the first time that acylated ghrelin and TNF-α exert opposite effects on the regulation of autophagy in human visceral adipocytes (Fig. 7). Furthermore, the imbalance of both molecules in hyperglycaemic states may contribute to the altered autophagy observed in patients with type 2 diabetes.
In summary, the main findings of the present study are that: (1) GOAT abundance, but not that of ghrelin, is increased in human visceral adipose tissue in patients with type 2 diabetes; (2) insulin, leptin, TNF-α and ghrelin itself constitute transcriptional regulators of GHRL and MBOAT4 in human visceral adipocytes; and (3) acylated and desacyl ghrelin reduce TNF-α-induced apoptosis and basal autophagy in human visceral adipocytes. These results broaden the current understanding of the defective mechanisms underlying type 2 diabetes.
Abbreviations
- ATG:
-
Autophagy-related genes
- AU:
-
Arbitrary units
- ER:
-
Endoplasmic reticulum
- GHS-R:
-
Growth hormone secretagogue receptor
- GOAT:
-
Ghrelin O-acyltransferase
- MBOAT:
-
Membrane-bound O-acyltransferases
- SVFC:
-
Stromovascular fraction cells
References
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth-hormone releasing acylated peptide from stomach. Nature 402:656–660
Frühbeck G, Gómez-Ambrosi J (2003) Control of body weight: a physiologic and transgenic perspective. Diabetologia 46:143–172
Frühbeck G, Díez Caballero A, Gil MJ (2004) Fundus functionality and ghrelin concentrations after bariatric surgery. N Engl J Med 350:308–309
Hosoda H, Kojima M, Matsuo H, Kangawa K (2000) Ghrelin and des-acyl ghrelin: two major forms of rat ghrelin peptide in gastrointestinal tissue. Biochem Biophys Res Commun 279:909–913
Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL (2008) Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132:387–396
Chen CY, Asakawa A, Fujimiya M, Lee SD, Inui A (2009) Ghrelin gene products and the regulation of food intake and gut motility. Pharmacol Rev 61:430–481
López M, Lage R, Saha AK et al (2008) Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7:389–399
Rodríguez A, Gómez-Ambrosi J, Catalán V et al (2009) Acylated and desacyl ghrelin stimulate lipid accumulation in human visceral adipocytes. Int J Obes (Lond) 33:541–552
Gurriarán-Rodríguez U, Al-Massadi O, Crujeiras AB et al (2011) Preproghrelin expression is a key target for insulin action on adipogenesis. J Endocrinol 210:R1–R7
Rodríguez A, Gómez-Ambrosi J, Catalán V et al (2010) Association of plasma acylated ghrelin with blood pressure and left ventricular mass in patients with metabolic syndrome. J Hypertens 28:560–567
Sorisky A, Magun R, Gagnon AM (2000) Adipose cell apoptosis: death in the energy depot. Int J Obes Relat Metab Disord 24(Suppl 4):S3–S7
Alkhouri N, Gornicka A, Berk MP et al (2010) Adipocyte apoptosis, a link between obesity, insulin resistance, and hepatic steatosis. J Biol Chem 285:3428–3438
Kovsan J, Bluher M, Tarnovscki T et al (2011) Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab 96:E268–E277
Singh R, Cuervo AM (2011) Autophagy in the cellular energetic balance. Cell Metab 13:495–504
Baldanzi G, Filigheddu N, Cutrupi S et al (2002) Ghrelin and des-acyl ghrelin inhibit cell death in cardiomyocytes and endothelial cells through ERK1/2 and PI 3-kinase/AKT. J Cell Biol 159:1029–1037
Kim MS, Yoon CY, Jang PG et al (2004) The mitogenic and antiapoptotic actions of ghrelin in 3T3-L1 adipocytes. Mol Endocrinol 18:2291–2301
Granata R, Settanni F, Biancone L et al (2007) Acylated and unacylated ghrelin promote proliferation and inhibit apoptosis of pancreatic beta-cells and human islets: involvement of 3′,5′-cyclic adenosine monophosphate/protein kinase A, extracellular signal-regulated kinase 1/2, and phosphatidyl inositol 3-Kinase/Akt signaling. Endocrinology 148:512–529
Genuth S, Alberti KG, Bennett P et al (2003) Follow-up report on the diagnosis of diabetes mellitus. Diabetes Care 26:3160–3167
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC (1985) Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 28:412–419
Katz A, Nambi SS, Mather K et al (2000) Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab 85:2402–2410
Catalán V, Gómez-Ambrosi J, Rotellar F et al (2007) The obestatin receptor (GPR39) is expressed in human adipose tissue and is down-regulated in obesity-associated type 2 diabetes mellitus. Clin Endocrinol (Oxf) 66:598–601
Rodríguez A, Catalán V, Gómez-Ambrosi J, Frühbeck G (2007) Visceral and subcutaneous adiposity: are both potential therapeutic targets for tackling the metabolic syndrome? Curr Pharm Des 13:2169–2175
Tschöp M, Weyer C, Tataranni A, Devanarayan V, Ravussin E, Heiman ML (2001) Circulating ghrelin levels are decreased in human obesity. Diabetes 50:707–709
Poykko SM, Ukkola O, Kauma H, Kellokoski E, Horkko S, Kesaniemi YA (2005) The negative association between plasma ghrelin and IGF-I is modified by obesity, insulin resistance and type 2 diabetes. Diabetologia 48:309–316
Otero M, Nogueiras R, Lago F, Dieguez C, Gomez-Reino JJ, Gualillo O (2004) Chronic inflammation modulates ghrelin levels in humans and rats. Rheumatol (Oxford) 43:306–310
Yilmaz E, Ustundag B, Sen Y, Akarsu S, Kurt AN, Dogan Y (2007) The levels of ghrelin, TNF-α, and IL-6 in children with cyanotic and acyanotic congenital heart disease. Mediat Inflamm 2007:32403
Karagiozoglou-Lampoudi T, Trachana M, Agakidis C et al (2011) Ghrelin levels in patients with juvenile idiopathic arthritis: relation to anti-tumor necrosis factor treatment and disease activity. Metabolism 60:1359–1362
Himmerich H, Sheldrick AJ (2010) TNF-α and ghrelin: opposite effects on immune system, metabolism and mental health. Protein Pept Lett 17:186–196
Knerr I, Herzog D, Rauh M, Rascher W, Horbach T (2006) Leptin and ghrelin expression in adipose tissues and serum levels in gastric banding patients. Eur J Clin Invest 36:389–394
Lim CT, Kola B, Grossman A, Korbonits M (2011) The expression of ghrelin O-acyltransferase (GOAT) in human tissues. Endocr J 58:707–710
Romero A, Kirchner H, Heppner K, Pfluger PT, Tschöp MH, Nogueiras R (2010) GOAT: the master switch for the ghrelin system? Eur J Endocrinol 163:1–8
Yang J, Zhao TJ, Goldstein JL, Brown MS (2008) Inhibition of ghrelin O-acyltransferase (GOAT) by octanoylated pentapeptides. Proc Natl Acad Sci U S A 105:10750–10755
Al Massadi O, Tschöp MH, Tong J (2011) Ghrelin acylation and metabolic control. Peptides 32:2301–2308
O’Brien M, Earley P, Morrison JJ, Smith TJ (2010) Ghrelin in the human myometrium. Reprod Biol Endocrinol 8:55
Camiña JP, Carreira MC, El Messari S, Llorens-Cortes C, Smith RG, Casanueva FF (2004) Desensitization and endocytosis mechanisms of ghrelin-activated growth hormone secretagogue receptor 1a. Endocrinology 145:930–940
Casanueva FF, Camina JP, Carreira MC, Pazos Y, Varga JL, Schally AV (2008) Growth hormone-releasing hormone as an agonist of the ghrelin receptor GHS-R1a. Proc Natl Acad Sci U S A 105:20452–20457
De Vriese C, Hacquebard M, Gregoire F, Carpentier Y, Delporte C (2007) Ghrelin interacts with human plasma lipoproteins. Endocrinology 148:2355–2362
Kamegai J, Tamura H, Shimizu T, Ishii S, Sugihara H, Oikawa S (2004) Effects of insulin, leptin, and glucagon on ghrelin secretion from isolated perfused rat stomach. Regul Pept 119:77–81
Gibson W, Liu J, Gaylinn B et al (2010) Effects of glucose and insulin on acyl ghrelin and desacyl ghrelin, leptin, and adiponectin in pregnant women with diabetes. Metabolism 59:841–847
An W, Li Y, Xu G et al (2010) Modulation of ghrelin O-acyltransferase expression in pancreatic islets. Cell Physiol Biochem 26:707–716
Gahete MD, Cordoba-Chacon J, Salvatori R, Castano JP, Kineman RD, Luque RM (2010) Metabolic regulation of ghrelin O-acyl transferase (GOAT) expression in the mouse hypothalamus, pituitary, and stomach. Mol Cell Endocrinol 317:154–160
Kirchner H, Gutierrez JA, Solenberg PJ et al (2009) GOAT links dietary lipids with the endocrine control of energy balance. Nat Med 15:741–745
Prins JB, Niesler CU, Winterford CM et al (1997) Tumor necrosis factor-α induces apoptosis of human adipose cells. Diabetes 46:1939–1944
Baragli A, Ghe C, Arnoletti E, Granata R, Ghigo E, Muccioli G (2011) Acylated and unacylated ghrelin attenuate isoproterenol-induced lipolysis in isolated rat visceral adipocytes through activation of phosphoinositide 3-kinase gamma and phosphodiesterase 3B. Biochim Biophys Acta 1811:386–396
Iglesias MJ, Pineiro R, Blanco M et al (2004) Growth hormone releasing peptide (ghrelin) is synthesized and secreted by cardiomyocytes. Cardiovasc Res 62:481–488
Lee JY, Oh TH, Yune TY (2011) Ghrelin inhibits hydrogen peroxide-induced apoptotic cell death of oligodendrocytes via ERK and p38MAPK signaling. Endocrinology 152:2377–2386
Delhanty PJ, van Koetsveld PM, Gauna C et al (2007) Ghrelin and its unacylated isoform stimulate the growth of adrenocortical tumor cells via an anti-apoptotic pathway. Am J Physiol Endocrinol Metab 293:E302–E309
Shibata M, Yoshimura K, Furuya N et al (2009) The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 382:419–423
Singh R, Xiang Y, Wang Y et al (2009) Autophagy regulates adipose mass and differentiation in mice. J Clin Invest 119:3329–3339
Ost A, Svensson K, Ruishalme I et al (2010) Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol Med 16:235–246
Maiuri MC, Criollo A, Kroemer G (2010) Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J 29:515–516
Jia G, Cheng G, Gangahar DM, Agrawal DK (2006) Insulin-like growth factor-1 and TNF-α regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 84:448–454
Acknowledgements
We gratefully acknowledge the valuable collaboration of all members of the Department of Surgery and the Nutrition Unit of the Clínica Universidad de Navarra (Pamplona, Spain) for their technical support.
Funding
This work was funded by the Instituto de Salud Carlos III (FIS PS09/02330 and PI10/01677), by grants from the Department of Health (4/2006) of the Gobierno de Navarra, Spain, by the Plan de Investigación de la Universidad de Navarra (PIUNA) (2011-2012) and by the Ministerio de Economía y Competitividad (BFU2010-17116). CIBER de Fisiopatología de la Obesidad y Nutrición (CIBERobn) is an initiative of the Instituto de Salud Carlos III, Spain.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
AR, JG-A, VC, FR, VV, CS, CM, MRP, RV, JS, MMM, IC and GF researched the data. AR, JG-A, VC, MMM and GF contributed to the discussion. AR and GF wrote the manuscript. All authors reviewed/edited the manuscript and approved the final version. GF is the guarantor of this work, had full access to all the data and takes full responsibility for the integrity of data and the accuracy of data analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 40 kb)
ESM Table 1
(PDF 26 kb)
ESM Table 2
(PDF 14 kb)
Rights and permissions
About this article
Cite this article
Rodríguez, A., Gómez-Ambrosi, J., Catalán, V. et al. The ghrelin O-acyltransferase–ghrelin system reduces TNF-α-induced apoptosis and autophagy in human visceral adipocytes. Diabetologia 55, 3038–3050 (2012). https://doi.org/10.1007/s00125-012-2671-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2671-5