
Abstract
Aims/hypothesis
Ciliary neurotrophic factor (CNTF) improves metabolic variables of obese animals with characteristics of type 2 diabetes, mainly by reducing insulin resistance. We evaluated whether CNTF was able to improve other metabolic variables in mouse models of type 2 diabetes, such as beta cell mass and insulin clearance, and whether CNTF has any effect on non-obese mice with characteristics of type 2 diabetes.
Methods
Neonatal mice were treated with 0.1 mg/kg CNTF or citrate buffer via intraperitoneal injections, before injection of 250 mg/kg alloxan. HEPG2 cells were cultured for 3 days in the presence of citrate buffer, 1 nmol/l CNTF or 50 mmol/l alloxan or a combination of CNTF and alloxan. Twenty-one days after treatment, we determined body weight, epididymal fat weight, blood glucose, plasma insulin, NEFA, glucose tolerance, insulin resistance, insulin clearance and beta cell mass. Finally, we assessed insulin receptor and protein kinase B phosphorylation in peripheral organs, as well as insulin-degrading enzyme (IDE) protein production and alternative splicing in the liver and HEPG2 cells.
Results
CNTF improved insulin sensitivity and beta cell mass, while reducing glucose-stimulated insulin secretion and insulin clearance in Swiss mice, improving glucose handling in a non-obese type 2 diabetes model. This effect was associated with lower IDE production and activity in liver cells. All these effects were observed even at 21 days after CNTF treatment.
Conclusions/interpretation
CNTF protection against type 2 diabetes is partially independent of the anti-obesity actions of CNTF, requiring a reduction in insulin clearance and increased beta cell mass, besides increased insulin sensitivity. Furthermore, knowledge of the long-term effects of CNTF expands its pharmacological relevance.
Similar content being viewed by others

Introduction
Type 2 diabetes is a complex illness mainly characterised by hyperglycaemia, usually associated with pancreatic islet beta cell dysfunction, reduced insulin action on peripheral organs, and alterations in insulin clearance. Although hyperglycaemia is a defining factor for diagnosis of type 2 diabetes, it is probably not the first change to occur, and the alterations in insulin resistance, secretion and clearance vary according to disease development. At onset of type 2 diabetes, and before hyperglycaemia is established, there is an increase in insulin resistance, coupled with increased pancreatic islet beta cell insulin secretion and reduced insulin clearance, leading to a normoglycaemic/hyperinsulinaemic state.
Insulin resistance is characterised by decreased insulin action on peripheral organs (mainly liver and skeletal muscle) and reduced phosphorylation/activation of the insulin receptor (IR), IRSs and components of the phosphatidylinositol 3-kinase/protein kinase B (AKT) pathway. With time, a loss of beta cell function occurs, as represented by a reduction in glucose-stimulated insulin secretion (GSIS), accompanied by loss of beta cell mass by apoptosis [1], leading to hypoinsulinaemia.
Evidence also suggests that insulin clearance mostly depends on degradation by the liver and plays a role in glycaemic control [2, 3]. In hepatocytes, insulin degradation is mediated primarily by the insulin-degrading enzyme (IDE) [4].
Ciliary neurotrophic factor (CNTF) is a cytokine from the IL-6 family that signals through the GP130 complex, composed, in the case of CNTF, of the CNTF receptor α, leukaemia inhibitory factor receptor and GP130 itself, usually associated with modulation of the JAK/STAT pathway [5, 6]. CNTF administration prevents or ameliorates obesity-induced type 2 diabetes [6–10] due, at least in part, to a reduction in insulin resistance, provoked by obesity, which is either genetic or induced by a high-fat diet [7, 11, 12].
Furthermore, CNTF protects rat pancreatic islet beta cells against apoptosis induced by IL-1β [13] and serum deprivation [14]. Thus it is possible that the anti-diabetogenic effects of CNTF go beyond improvements in insulin sensitivity, since it could contribute to maintenance of beta cell mass. Studies associating CNTF with diabetic variables have been performed in obesity-induced type 2 diabetes models, and have been restricted to investigating metabolic variables of peripheral organs, such as insulin sensitivity, overall metabolic rate or reduction in fat weight [15, 16]. Thus it was not possible to ascertain whether the beneficial effects of CNTF on diabetic metabolic variables were just secondary to the reduction in obesity.
The aim of the present study was to evaluate whether CNTF retains its ability to improve metabolic variables in a well-established model of alloxan-induced non-obese type 2 diabetes, and also to determine whether CNTF controls other physiological processes directly associated with type 2 diabetes besides insulin resistance, namely pancreatic islet beta cell mass and insulin clearance.
Methods
Reagents and solutions
CNTF was kindly provided by A. Negro (Universitá di Padova, Italy). Alloxan tetrahydrate, RT-PCR and western-blot reagents were acquired from Sigma Aldrich, St Louis, MO, USA.
Animals
Newborn (1–2 days old) Swiss mice (Unib:SW strain), acquired from the State University of Campinas, were maintained with their mothers on a 12 h light–dark cycle at 20–21°C with controlled air humidity during the entire experimental period.
Animal experimental design
Type 2 diabetes was induced by intraperitoneal injection of alloxan in 6-day-old mice, as described elsewhere [17]. CNTF-treated mice received six independent 0.1 mg/kg intraperitoneal injections of CNTF before the alloxan injection: the first injection was given to 2-day-old mice, the second was given 30 min later, and the third after 2 h, along with further daily doses until day 6, when mice received CNTF 1 h before a 250 mg/kg dose of alloxan, a total of six doses of CNTF. Experimental procedures and tissue samples were carried out in 26–28-day-old mice. Animals were killed in a CO2-saturated atmosphere immediately followed by decapitation. Animal procedures were performed according to the guidelines of the State University of Campinas Animal Care Ethics Committee.
HOMA-insulin resistance
HOMA-insulin resistance was determined using HOMA calculator software (www.dtu.ox.ac.uk/homacalculator/index.php, accessed 28 December 2011).
Tissue samples
Liver samples from Swiss mice were extracted, snap-frozen in liquid nitrogen, and stored for subsequent protein and mRNA extraction. Pancreatic islets were isolated from 26–28-day-old mice by the collagenase method.
Pancreas and islet morphology, immunohistochemistry and beta cell mass
Methods were used as previously described [18].
Experimental design for cell cultures
HEPG2 cells were cultured for 3 days in RPMI 1640 medium enriched with 1% vol./vol. penicillin, 5% vol./vol. FBS and 10 mmol/l glucose. Cells were divided into four different groups: control (received only citrate buffer); CNTF (received 1 nmol/l CNTF); alloxan (preincubated with citrate buffer for 48 h followed by 24 h with 50 mmol/l alloxan); and CNTF + alloxan (preincubated with 1 nmol/l CNTF for 48 h followed by 24 h incubation with 1 nmol/l CNTF plus 50 mmol/l alloxan).
HEPG2 protein extraction
After 72 h of culture, cells were incubated for 1 h with 10 μIU/ml insulin, then harvested by trypsin/EDTA, washed twice with PBS, homogenised in urea anti-protease/anti-phosphatase buffer, and stored at −80°C.
Western blot
Western blots were carried out as previously described [19].
Real-time RT-PCR
Protein extracts from liver samples of mice or from HEPG2 cells were homogenised in Trizol after phenol/chloroform RNA extraction, according to the manufacturer’s instructions (Gibco-BRL, Gaithersburg, MD, USA). To evaluate mRNA levels and search alternatively spliced transcript variants of different size, RT-PCR was carried out using complementary DNAs as templates with TaqDNA polymerase (Invitrogen/Life Sciences, Grand Island, NY, USA) and corresponding primers as described [20]. Relative quantities of target transcripts were calculated from duplicate samples after normalisation of the data against the endogenous control, β-actin (sense: ′5-AGAGGGAAATCGTGCGTGACA-3′; anti-sense: ′5-CGATAGTGATGACCTGACCGTCA3′).
Pancreatic islet GSIS
Batches of 10 islets each were pre-incubated for 1 h in Krebs–Henseleit buffer solution (KHBS) containing 0.5 g/l BSA and 5.6 mmol/l glucose, and equilibrated with 95% O2 and 5% CO2 at 37°C. The medium was discarded and the islets incubated for a further period of 1 h in 1 ml KHBS now containing 2.8, 11.2 or 22.4 mmol/l glucose; subsequently, the supernatant fraction was collected to evaluate insulin secretion, and the remaining islets were homogenised in alcohol/acid solution for measurement of total insulin content by radioimmunoassay.
Intraperitoneal glucose tolerance test
Swiss mice received an intraperitoneal injection of glucose (1 g/kg in 0.9% NaCl) after 8 h of fasting. Blood samples (75–100 μl) were collected from the tail immediately before injection and after 15, 30, 45 and 120 min to determine glucose and insulin concentrations. Glucose was evaluated by glucose strip on an Accucheck Performa II instrument (Roche, Indianapolis, Indiana, USA), and insulin was measured by radioimmunoassay, as previously described [14].
Intraperitoneal insulin tolerance test
Non-fasted Swiss mice received an intraperitoneal injection of insulin (1 U/kg). Blood glucose was measured using test strips (Accu-check Performa II) at baseline (0 min, before insulin administration) and 5, 10, 15, 20 and 30 min after insulin application. Glucose measurements were then converted into natural logarithm (Ln); the slope was calculated using linear regression (time × Ln[glucose]) and multiplied by 100 to obtain the glucose decay constant rate during the insulin tolerance test (k ITT) per minute (%/min).
In vivo insulin clearance
We evaluated the plasma insulin concentration of Swiss mice submitted to the intraperitoneal insulin tolerance test. Insulin clearance was evaluated as previously described [21]. The constant rate for insulin disappearance (insulin decay) was calculated by first converting insulin measurements into natural logarithm (Ln); the slope was calculated using linear regression (time × Ln[insulin]) and multiplied by 100 to obtain the insulin decay constant rate per minute (%/min).
HEPG2 insulin degradation
After 3 days in culture, as described, HEPG2 cells received a human regular insulin bolus (10 μIU/ml). Samples (200 μl) of the culture medium were collected immediately after insulin had been added (0 min) and subsequently at 15 and 30 min. Insulin in the medium was evaluated by radioimmunoassay, and insulin clearance and decay were calculated as described above.
Statistical analysis
Point-to-point comparisons were made by Student’s t test. Groups were compared by two-way ANOVA using the unpaired Tukey–Kramer post test. Results were considered significantly different if p < 0.05. In RT-PCR experiments, results were considered different only if p < 0.01.
Results
CNTF improves metabolic variables of non-obese type 2 diabetic Swiss mice
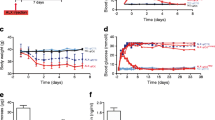
In mice treated with CNTF, fasting and non-fasting plasma insulin levels were lower than those of controls but higher than those of alloxan-treated mice. In addition, fasting and non-fasting blood glucose concentrations in CNTF mice were also lower than those of both control and alloxan-treated mice (Table 1). Alloxan-treated mice had lower fasting insulin, associated with increased fasting plasma glucose, characteristics typically associated with type 2 diabetes. Furthermore, mice treated with CNTF before alloxan partially recovered both blood glycaemia and insulinaemia. Finally, CNTF improved insulin sensitivity and prevented alloxan-induced insulin resistance, as evidenced by the HOMA1− insulin resistance and HOMA2− insulin resistance indexes. None of these changes were accompanied by alterations in total body weight, epididymal fat weight or plasma NEFA (Table 1).
CNTF improves glucose tolerance and modifies insulin dynamics in non-obese Swiss mice
We analysed glucose tolerance and insulin dynamics in Swiss mice treated as described. Alloxan led to glucose intolerance (Fig. 1a,c), associated with reduced insulin secretion during the glucose tolerance test (Fig. 1b,d). CNTF improved glucose tolerance (Fig. 1a,c), without concomitant alterations in overall (60 min) insulin secretion during the glucose tolerance test (Fig. 1d). CNTF altered insulin dynamics, presenting a lower insulin secretion peak at 15 min, associated with a higher insulin concentration thereafter (Fig. 1b). Moreover, CNTF pretreatment prevented alloxan-induced deficiencies in glucose tolerance (Fig. 1a,c), insulin secretion (Fig. 1d) and insulin dynamics (Fig. 1b).

CNTF improves glucose tolerance and modifies insulin dynamics in non-obese Swiss mice. (a) Blood glucose and (b) plasma insulin of 8 h fasted 28-day-old Swiss mice at 0, 15, 30, 45 and 60 min after intraperitoneal injection of 1 g/kg glucose. Control (white squares), CNTF (black squares), alloxan (white triangles) and CNTF + alloxan (black triangles). AUC of (c) blood glucose and (d) plasma insulin of non-fasted 28-day-old Swiss mice. Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). n = 4–6. Data are means ± SEM. *p < 0.05 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 vs alloxan
CNTF improved insulin sensitivity and prevented alloxan-induced insulin resistance in non-obese Swiss mice
Alloxan- or CNTF-induced alterations in glucose tolerance and insulin dynamics could be related to various factors, particularly insulin secretion, peripheral insulin sensitivity and insulin degradation. Alloxan induced marked insulin resistance, which was reversed by CNTF in non-obese Swiss mice, as demonstrated by the higher insulin tolerance test (Fig. 2a,b) and lower k ITT (Fig. 2c) of alloxan-treated mice.

CNTF improved insulin sensitivity and prevented alloxan-induced insulin resistance in non-obese Swiss mice. (a) Blood glucose at 0, 5, 10, 15 and 30 min, (b) AUC of blood glucose, and (c) k ITT of 28-day-old Swiss mice after intraperitoneal injection of 1 IU/kg insulin. Control (white squares), CNTF (black squares), alloxan (white triangles) and CNTF + alloxan (black triangles). Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). n = 4–6. Data are means ± SEM. *p < 0.05 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 vs alloxan
CNTF improved insulin signalling (pIR and pAKT) in the liver, skeletal muscle, adipose tissue and hypothalamus of non-obese Swiss mice and in HEPG2 cells
In order to evaluate the effects of CNTF and/or alloxan on insulin signalling, we measured IR and AKT phosphorylation in several insulin-responsive organs of non-fasted Swiss mice and cultured HEPG2 cells, in the presence or absence of CNTF and/or alloxan. Before organ extraction, mice received 1 IU/kg insulin, while HEPG2 cells were exposed to 10 μIU/ml insulin 1 h before harvesting. As expected, alloxan impaired, whereas CNTF increased and reversed alloxan-impaired, IR and AKT phosphorylation in liver (Fig. 3), skeletal muscle, adipose tissue, hypothalamus (electronic supplementary material [ESM] Fig. 1) and HEPG2 cells (Fig. 3).

CNTF improved insulin signalling (pIR and pAKT) in the liver of non-obese Swiss mice (a, b) and in HEPG2 cells (c, d). Phosphorylation of IR (a) and AKT (b) in protein extracts from liver of non-fasted 28-day-old Swiss mice after an intraperitoneal dose of 1 IU/kg insulin. Phosphorylation of IR (c) and AKT (d) of protein extracts from HEPG2 cells after 1 h exposure to 10 μIU/ml insulin. Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). n = 3–4. Data are means ± SEM. *p < 0.05 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 vs alloxan
CNTF partially recovers insulin secretion from pancreatic islets exposed to alloxan
Our results so far have shown that CNTF reversed alloxan-induced glucose intolerance and insulin resistance in mice and that these effects could, at least in part, be explained by a CNTF-induced improvement in hepatocyte cell insulin signalling. However, these effects could not explain the alterations observed in plasma insulin and insulin dynamics. To assess the origin of such alterations, we analysed GSIS in islets isolated from mice treated with alloxan, CNTF or alloxan plus CNTF. First, we found that CNTF, alloxan or a combination of both had no effect on insulin secretion at sub-stimulatory (2.8 mmol/l) glucose concentrations. In addition, both CNTF and alloxan, alone or in combination, impaired insulin secretion at stimulatory (11.2 mmol/l) as well as at super-stimulatory (22.4 mmol/l) glucose concentrations, although alloxan-induced impairment was greater than that observed for CNTF (Fig. 4a). More importantly, CNTF partially recovered the insulin secretion from pancreatic islets of mice exposed to alloxan. Alloxan reduced, whereas CNTF increased and prevented the alloxan-induced reduction in, pancreatic islet insulin content (Fig. 4b).

CNTF partially recovers insulin secretion from islets assessed ex vivo from mice exposed to alloxan. Insulin secretion (a), total insulin content (b) of pancreatic islets isolated from non-fasted 28-day-old Swiss mice and incubated for 1 h in the presence of 2.8, 11.2 or 22.4 mmol/l glucose. Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). The DNA content (nmol/islet) of each group was also evaluated: control 17.5 ± 1.1, CNTF 22.2 ± 1.0*, ALOX 8.3 ± 0.6*† and CNTF + ALOX 18.8 ± 1*‡. n = 8–12. Data are means ± SEM. *p < 0.05 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 vs alloxan
CNTF increases beta cell mass and prevents alloxan-induced loss of beta cell mass
CNTF or alloxan, alone or in combination, did not alter the pancreas weight (data not shown). However, CNTF prevented the alloxan-induced reduction in the number of islets per pancreas (Fig. 5c), but not the alloxan-induced reduction in islet size (Fig. 5d). CNTF increased beta cell area per islet and also prevented the alloxan-induced reduction in beta cell area per islet (Fig. 5e). In addition, alloxan reduced, whereas CNTF increased, beta cell mass, and CNTF prevented the alloxan-induced reduction in beta cell mass. Finally, alloxan increased, whereas CNTF directly reduced and prevented the alloxan-induced increase in DNA fragmentation in pancreatic islets (Fig. 5f).

CNTF increases beta cell mass and prevents alloxan-induced beta cell mass loss through reduced pancreatic islet DNA fragmentation and apoptosis. (a) Representative image of pancreatic islets obtained by immunohistochemistry from the pancreas of non-fasted 28-day-old Swiss mice. We also evaluated the number of islets per pancreas (b), average size of each individual islet (c), area of the pancreas occupied by beta cells (d), estimated total beta cell mass (e), and DNA fragmentation (f). Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). n = 4–6. Data are means ± SEM. *p < 0.05 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 vs alloxan
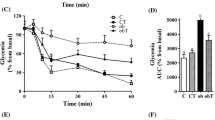
CNTF reduced insulin degradation in the liver of Swiss mice and in HEPG2 cells
In addition, we evaluated the removal of insulin from plasma, or insulin clearance, from non-obese type 2 diabetic mice as well as in vitro insulin degradation by HEPG2 cells. Alloxan had no effect whatsoever on insulin degradation in vivo (Fig. 6a–c) or in vitro (Fig. 6d–f). In vivo, CNTF reduced insulin degradation (Fig. 6a) and insulin decay rate (Fig. 6b), resulting in prolonged exposure of tissues to insulin during the 60 min of measurement (Fig. 6c). As most of the insulin is degraded by the liver, we also evaluated whether CNTF affects the capacity of HEPG2 cells to clear insulin from the culture medium. CNTF reduced in vitro insulin degradation (Fig. 6d) and decay rate (Fig. 6e), resulting in a higher exposure of cells to insulin for 1 h (Fig. 6f).

CNTF reduced insulin degradation in the liver of Swiss mice (a–c) and in HEPG2 cells (d–f). (a) Plasma insulin at 0, 15, 30 and 60 min, (b) insulin decay over 60 min, and (c) AUC of insulin for the entire 60 min experiment after intraperitoneal injection of 1 IU/kg insulin in non-fasted 28-day-old Swiss mice. (d) Plasma insulin at 0, 15, 30 and 60 min, (e) insulin decay over 60 min, and (f) AUC of insulin for the entire 60 min experiment after the addition of 10 μIU/ml insulin to HEPG2 cells cultured for 3 days. n = 4–6. Control (white squares), CNTF (black squares), alloxan (white triangles) and CNTF + alloxan (black triangles). Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). Data are means ± SEM. *p < 0.05 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 vs alloxan
CNTF reduced Ide expression and activity in the liver of Swiss mice and in HEPG2 cells
Finally, we verified how CNTF reduced in vivo and in vitro insulin degradation. We evaluated Ide expression and alternative splicing in liver samples and HEPG2 cells. CNTF reduced IDE protein and Ide mRNA levels in the liver (Fig. 7a,b) and in HEPG2 cells (Fig. 7e,f). Moreover, CNTF reduced both higher-activity (Fig. 7c) and lower-activity (Fig. 7d) IDE isoforms in liver, although it reduced the higher-activity isoform to a greater extent. Similar results were obtained in HEPG2 cells (Fig. 7g,h).

CNTF reduced IDE levels and activity in the liver of Swiss mice (a–d) and in HEPG2 cells (e–h). IDE protein production (a), total Ide mRNA levels (b), high-activity Ide transcript mRNA levels (c), and low-activity Ide transcript mRNA levels (d) from livers of non-fasted 28-day-old Swiss mice. IDE protein production (e), total Ide mRNA levels (f), high-activity Ide transcript mRNA levels (g), and low-activity Ide transcript mRNA levels (h) of HEPG2 cells cultured for 3 days. Control (white bars), CNTF (grey bars), alloxan (hatched bars) and CNTF + alloxan (black bars). n = 4–6 for mice and n = 3–4 for HEPG2 cells. Data are means ± SEM. *p < 0.05 and **p < 0.01 vs controls; †p < 0.05 vs CNTF; ‡p < 0.05 and ‡‡p < 0.01 vs alloxan
Discussion
It is well established that overt type 2 diabetes is characterised by increased insulin resistance, impaired insulin secretion, reduced beta cell mass, and impaired insulin degradation. However, how and when these dysfunctions appear during the development of the illness is still unclear. CNTF improves metabolic profiles and insulin sensitivity in obese type 2 diabetic mice, but whether it affects peripheral insulin sensitivity directly, or indirectly by ameliorating obesity and preventing obesity-induced insulin resistance, is not known. To address this issue, we used a non-obese mouse model that mimics the alterations observed in late type 2 diabetes. In this model, conceived by Kodama et al. [17], alloxan was administered to 6-day-old Wistar rats, which showed higher plasma glucose levels, associated with increased insulin resistance. Also, unlike rats treated with streptozotocin, those treated with alloxan were permanently hyperglycaemic and insulin resistant, since insulin production by pancreatic β cells was not recovered. Thus this is a suitable model of non-obese type 2 diabetes for evaluating CNTF effects. In the present study, the alloxan-treated mice showed glucose intolerance, insulin resistance, hyperglycaemia and hypoinsulinaemia, but without any increase in weight, plasma NEFA levels or adipose tissue (Table 1), justifying its applicability to answer our questions.
Before starting the long-term experiment with CNTF, we tested its acute in vivo ability to reduce blood glucose of 60-day-old Swiss mice, and found that it was effective under these conditions (ESM Fig. 2). Subsequently, the long-term protocol was initiated and we found that, in an apparent paradox, CNTF reduced glycaemia and insulinaemia in both control and alloxan-treated mice. CNTF does improve insulin sensitivity and overcomes insulin resistance [9, 11, 15] in a multifactorial fashion that seems to involve control of the central nervous system through regulation of hypothalamic AMP-activated protein kinase (AMPK) activation in leptin-resistant obese mice [22], improved skeletal muscle glucose uptake through the phosphatidylinositol 3-kinase pathway in obese mice [23], reversal of obesity-induced skeletal muscle insulin resistance by AMPK activation [12], and overall overcoming of leptin resistance and deficiency [8]. However, there is no information available regarding the ability of CNTF to improve insulin sensitivity when dissociated from obesity and/or leptin resistance, and we found that CNTF also improves insulin sensitivity regardless of its well-known anti-obesity properties [8, 10, 15, 16]. Moreover, CNTF improves insulin signalling in mouse liver and HEPG2 cells (Fig. 3), as well as in skeletal muscle, adipose tissue and hypothalamus of mice (ESM Fig. 1). This is in accordance with previous results showing that CNTF reduces hepatic steatosis in db/db mice [16], and also directly modulates in vivo and in vitro hepatocyte functions [24, 25]. In db/db mice, CNTF reduced plasma insulin, glucose, NEFA, triacylglycerol and cholesterol to levels observed in control (db/?) mice [16], thus reinforcing our initial doubt as to whether the beneficial effects of CNTF treatment occur directly, via effects on insulin secretion and sensitivity, or indirectly, as a result of a reduction in obesity. As CNTF still improved metabolic variables in our non-obese type 2 diabetes mice, we conclude that CNTF directly improves insulin sensitivity, regardless of its anti-obesity properties.
However, improved insulin sensitivity probably does not account for the lower plasma insulin levels in CNTF-treated mice, so we evaluated pancreatic islet insulin secretion and beta cell mass. We found that alloxan disrupts, whereas CNTF impairs, GSIS, in accordance with the literature and with results from our group [13, 14, 26]. The novelty here is that CNTF partially recovers GSIS in pancreatic islets from alloxan-treated mice. These results could, at least in part, explain the lower insulinaemia of CNTF-treated mice, as well as the partial recovery of alloxan-treated mice pretreated with CNTF. We also observed that, as expected, alloxan severely reduced beta cell mass, whereas CNTF increased beta cell mass and protected mice against the loss of beta cell mass induced by alloxan. We previously reported that CNTF protects rat pancreatic islets against apoptosis induced by serum deprivation [14] and IL-1β inflammatory cytokine [13], in vitro; however, these experiments were carried out with isolated islets in a well-controlled environment. Here, we show that CNTF protects pancreatic islets against apoptosis in an in vivo model that closely resembles type 2 diabetes. We have recently found that the apoptosis of pancreatic islets, induced by alloxan as well as IL-1β, required AMPK activation, and that CNTF reduced pancreatic islet apoptosis through downregulation of the AMPK pathway, more precisely via CaMKII/AMPK downregulation [19], indicating a possible mechanism of action of CNTF on beta cell mass. Moreover, given the long-term effects of alterations in CaMKII activity, considered a ‘memory molecule’ [27], we deduce that the downregulation of CaMKII in pancreatic islets by CNTF could, at least in part, explain the prolonged (3 weeks) effects of CNTF.
However, the CNTF-induced reduction of GSIS and increase in beta cell mass does not solely explain the alteration in insulin dynamics observed in CNTF-treated mice: a lower initial insulin burst followed by higher insulin levels. After secretion, insulin is removed from the plasma, mainly by the liver [28], in a process called insulin clearance, which is altered in obesity [29] and type 2 diabetes [2, 30, 31] animal models, and is also commonly associated with obesity [32, 33] and type 2 diabetes in human patients [2, 3, 34]. It is well accepted that, in type 2 diabetes, insulin clearance is usually impaired and associated with insulin resistance and loss of beta cell mass. However, it is not known whether this reduction is an early cause and precedes the other symptoms [2, 30, 35] or whether this is a compensatory mechanism to ameliorate the effects of lower insulin secretion and increased insulin resistance [21, 36, 37].
An additional difficulty to solving this dilemma is that insulin clearance is modulated by NEFA [38, 39] and liver fat content [2, 40], and that most animal models of type 2 diabetes are obesity-induced. We here found that alloxan induced type 2 diabetes by increasing insulin resistance and loss of beta cell mass without alterations in the overall insulin clearance by hepatocytes. However, CNTF reduced insulin clearance by hepatocytes in both control and alloxan-treated mice, in the 60 min acute phase (Fig. 6) and the 24 h extended (ESM Fig. 3) experiments with HEPG2 cells, associated with lower insulin resistance (Figs 2 and 3) and increased beta cell mass (Fig. 5). In addition to the direct protective effects of CNTF on pancreatic islets [13, 14] and increased insulin sensitivity, reduced insulin clearance also helps to overcome insulin resistance, which might attenuate beta cell overload, ultimately increasing their survival. However, at present, we cannot determine exactly how CNTF impairs pancreatic islet GSIS but promotes their survival. It is possible that this apparent controversy is related to increased BCL2 production, as this protein is associated with increased survival as well as reduced insulin secretion in pancreatic islets [14].
Intracellular insulin degradation occurs via the action of IDE [28, 41], a 110 kDa zinc-dependent metalloproteinase present in most insulin-responsive cells, mainly hepatocytes. Given the importance of insulin clearance to type 2 diabetes and the role of IDE in its control, it is not surprising that alterations in IDE levels and activity are closely related to type 2 diabetes onset and development [34, 41]. On the basis of this, we evaluated how CNTF could control hepatic degradation of insulin by measuring Ide expression and alternative splicing in mouse liver and hepatocyte HEPG2 cells. First, IDE production was not altered in control or alloxan-treated mice, in accordance with the fact that alloxan also had no effect on insulin clearance. Second, CNTF reduced IDE production in the liver of control and alloxan-treated mice and also in control and alloxan-treated HEPG2 hepatocytes. Furthermore, CNTF reduced, to a greater extent, the production of high-activity rather than low-activity Ide mRNA, effectively displacing the balance of high/low activity in favour of the low-activity IDE, both in liver and hepatocytes, explaining the reduction in insulin clearance caused by CNTF. As CNTF increases levels of the tissue inhibitor of metalloproteinases-1 in hepatocytes [42], we postulate that this is how CNTF reduces IDE production in liver cells. In addition, CNTF protected mouse liver and HEPG2 cells against alloxan-induced apoptosis, as indicated by lower cleaved caspase 3/pro-caspase 3 ratio, and this effect probably occurs through STAT3 upregulation and STAT1 downregulation (ESM Fig. 4), similarly to that observed in neuronal cells [43, 44] and pancreatic islets [13]. The protective effects of CNTF on mouse liver could also help to, at least in part, explain how CNTF is able to reduce hepatic steatosis and inflammation, but this is a subject yet to be explored.
It is also important to note that all changes promoted by CNTF in insulin dynamics, sensitivity and clearance work in synergy. CNTF reduces mouse insulin secretion and promotes pancreatic islet survival while reducing insulin degradation, which helps to maintain normoglycaemia without overloading pancreatic beta cells and further contributing to functional beta cell mass maintenance. Inhibition of IDE production in hepatocytes increases IR phosphorylation and insulin sensitivity [45]. Reduction of insulin clearance, through downregulation of IDE in mouse hepatocytes, helps to increase insulin sensitivity. Moreover, CNTF protects against alloxan-induced type 2 diabetes without damaging liver cells and without affecting weight loss (Table 1) or weight gain during the whole experiment (ESM Fig. 5).
Last, a clinical trial revealed that daily administration of axokine (a more potent CNTF analogue) for 12 weeks produced weight loss in about 25% of the participants, compared with placebo, only in doses of at least 1 μg/kg. However, generation of CNTF-specific antibodies occurred, reducing the long-term benefits of the therapy [46]. Here we show that CNTF protects non-obese mice against type 2 diabetes with marked differences in treatment: first, no weight loss was observed; second, the mice received only six doses, for 4 days, instead of dozens of doses over 12 weeks; and, third, the CNTF effects in our case remained for at least 3 weeks after treatment. These findings provide evidence for some other potential pharmacological uses of CNTF: for example, pancreatic islets from a donor could be treated with CNTF before transplantation, allowing a better yield of highly viable pancreatic islets, probably prolonging islet survival [47]; CNTF could inhibit or delay rejection of the transplanted islets [48]; and CNTF administration to insulin-resistant patients would maintain their beta cell mass for longer and compensate for the increased insulin resistance, retarding the onset of type 2 diabetes [49, 50].
In summary, our results indicate that CNTF directly increases insulin sensitivity, independently of its anti-obesity effects, and that the effects of CNTF go beyond overcoming insulin resistance: it increases beta cell mass, alters insulin dynamics and lowers insulin secretion coupled to reduced insulin clearance, the latter effect occurring via downregulation of hepatic IDE levels and activity.
Abbreviations
- AKT:
-
Protein kinase B
- AMPK:
-
AMP-activated protein kinase
- CaMKII:
-
Calcium/calmodulin-dependent protein kinase II
- CNTF:
-
Ciliary neurotrophic factor
- GSIS:
-
Glucose-stimulated insulin secretion
- IDE:
-
Insulin-degrading enzyme
- IR:
-
Insulin receptor
- k ITT :
-
Glucose decay constant rate during insulin tolerance test
References
Chandra J, Zhivotovsky B, Zaitsev S, Juntti-Berggren L, Berggren PO, Orrenius S (2001) Role of apoptosis in pancreatic beta-cell death in diabetes. Diabetes 50(Suppl 1):S44–S47
Kotronen A, Juurinen L, Tiikkainen M, Vehkavaara S, Yki-Järvinen H (2008) Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 135:122–130
Anon (1991) Insulin clearance and diabetes. Lancet 338:1270–1271
Valera Mora ME, Scarfone A, Calvani M, Greco AV, Mingrone G (2003) Insulin clearance in obesity. J Am Coll Nutr 22:487–493
Kishimoto T, Akira S, Narazaki M, Taga T (1995) Interleukin-6 family of cytokines and gp130. Blood 86:1243–1254
Febbraio M (2007) gp130 receptor ligands as potential therapeutic targets for obesity. J Clin Invest 117:841–849
Blüher S, Bullen J, Mantzoros CS (2008) Altered levels of adiponectin and adiponectin receptors may underlie the effect of ciliary neurotrophic factor (CNTF) to enhance insulin sensitivity in diet-induced obese mice. Horm Metab Res 40:225–227
Gloaguen I, Costa P, Demartis A et al (1997) Ciliary neurotrophic factor corrects obesity and diabetes associated with leptin deficiency and resistance. Proc Natl Acad Sci U S A 94:6456–6461
Liu QS, Gao M, Zhu SY et al (2007) The novel mechanism of recombinant human ciliary neurotrophic factor on the anti-diabetes activity. Basic Clin Pharmacol Toxicol 101:78–84
Matthews VB, Febbraio MA (2008) CNTF: a target therapeutic for obesity-related metabolic disease? J Mol Med 86:353–361
Ahima R (2006) Overcoming insulin resistance with CNTF. Nat Med 12:511–512
Watt M, Dzamko N, Thomas W et al (2006) CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nat Med 12:541–548
Rezende LF, Vieira AS, Negro A, Langone F, Boschero AC (2009) Ciliary neurotrophic factor (CNTF) signals through STAT3-SOCS3 pathway and protects rat pancreatic islets from cytokine-induced apoptosis. Cytokine 46:65–71
Rezende LF, Stoppiglia LF, Souza KL, Negro A, Langone F, Boschero AC (2007) Ciliary neurotrophic factor promotes survival of neonatal rat islets via the BCL-2 anti-apoptotic pathway. J Endocrinol 195:157–165
Blüher S, Moschos S, Bullen J et al (2004) Ciliary neurotrophic factorAx15 alters energy homeostasis, decreases body weight, and improves metabolic control in diet-induced obese and UCP1-DTA mice. Diabetes 53:2787–2796
Sleeman M, Garcia K, Liu R et al (2003) Ciliary neurotrophic factor improves diabetic parameters and hepatic steatosis and increases basal metabolic rate in db/db mice. Proc Natl Acad Sci U S A 100:14297–14302
Kodama T, Iwase M, Nunoi K, Maki Y, Yoshinari M, Fujishima M (1993) A new diabetes model induced by neonatal alloxan treatment in rats. Diabetes Res Clin Pract 20:183–189
Rafacho A, Cestari TM, Taboga SR, Boschero AC, Bosqueiro JR (2009) High doses of dexamethasone induce increased beta-cell proliferation in pancreatic rat islets. Am J Physiol Endocrinol Metab 296:E681–E689
Santos GJ, Oliveira CA, Boschero AC, Rezende LF (2011) CNTF protects MIN6 cells against apoptosis induced by Alloxan and IL-1β through downregulation of the AMPK pathway. Cell Signal 23:1669–1676
Pivovarova O, Gögebakan O, Pfeiffer AF, Rudovich N (2009) Glucose inhibits the insulin-induced activation of the insulin-degrading enzyme in HepG2 cells. Diabetologia 52:1656–1664
Ahrén B, Thomaseth K, Pacini G (2005) Reduced insulin clearance contributes to the increased insulin levels after administration of glucagon-like peptide 1 in mice. Diabetologia 48:2140–2146
Steinberg G, Watt M, Fam B et al (2006) Ciliary neurotrophic factor suppresses hypothalamic AMP-kinase signaling in leptin-resistant obese mice. Endocrinology 147:3906–3914
Steinberg G, Watt M, Ernst M, Birnbaum M, Kemp B, Jørgensen S (2009) Ciliary neurotrophic factor stimulates muscle glucose uptake by a PI3-kinase-dependent pathway that is impaired with obesity. Diabetes 58:829–839
Hu X, Zhao Y, He X et al (2008) Ciliary neurotrophic factor receptor alpha subunit-modulated multiple downstream signaling pathways in hepatic cancer cell lines and their biological implications. Hepatology 47:1298–1308
Zvonic S, Cornelius P, Stewart WC, Mynatt RL, Stephens JM (2003) The regulation and activation of ciliary neurotrophic factor signaling proteins in adipocytes. J Biol Chem 278:2228–2235
Wadt KA, Larsen CM, Andersen HU, Nielsen K, Karlsen AE, Mandrup-Poulsen T (1998) Ciliary neurotrophic factor potentiates the beta-cell inhibitory effect of IL-1beta in rat pancreatic islets associated with increased nitric oxide synthesis and increased expression of inducible nitric oxide synthase. Diabetes 47:1602–1608
Griffith LC (2004) Calcium/calmodulin-dependent protein kinase II: an unforgettable kinase. J Neurosci 24:8391–8393
Duckworth WC, Bennett RG, Hamel FG (1998) Insulin degradation: progress and potential. Endocr Rev 19:608–624
Strömblad G, Björntorp P (1986) Reduced hepatic insulin clearance in rats with dietary-induced obesity. Metabolism 35:323–327
Hansen BC, Striffler JS, Bodkin NL (1993) Decreased hepatic insulin extraction precedes overt noninsulin dependent (Type II) diabetes in obese monkeys. Obes Res 1:252–260
Rabkin R, Reaven GM, Mondon CE (1986) Insulin metabolism by liver, muscle, and kidneys from spontaneously diabetic rats. Am J Physiol 250:E530–E537
Polonsky KS, Given BD, Hirsch L et al (1988) Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 81:435–441
Polonsky KS, Gumbiner B, Ostrega D, Griver K, Tager H, Henry RR (1994) Alterations in immunoreactive proinsulin and insulin clearance induced by weight loss in NIDDM. Diabetes 43:871–877
Rudovich N, Pivovarova O, Fisher E et al (2009) Polymorphisms within insulin-degrading enzyme (IDE) gene determine insulin metabolism and risk of type 2 diabetes. J Mol Med 87:1145–1151
Jones CN, Pei D, Staris P, Polonsky KS, Chen YD, Reaven GM (1997) Alterations in the glucose-stimulated insulin secretory dose–response curve and in insulin clearance in nondiabetic insulin-resistant individuals. J Clin Endocrinol Metab 82:1834–1838
Mittelman SD, van Citters GW, Kim SP et al (2000) Longitudinal compensation for fat-induced insulin resistance includes reduced insulin clearance and enhanced beta-cell response. Diabetes 49:2116–2125
Nestler JE, Clore JN, Blackard WG (1988) Reduced insulin clearance in normal subjects due to extreme hyperinsulinemia. Am J Med Sci 295:15–22
Yoshii H, Lam TK, Gupta N et al (2006) Effects of portal free fatty acid elevation on insulin clearance and hepatic glucose flux. Am J Physiol Endocrinol Metab 290:E1089–E1097
Wiesenthal SR, Sandhu H, McCall RH et al (1999) Free fatty acids impair hepatic insulin extraction in vivo. Diabetes 48:766–774
Kotronen A, Vehkavaara S, Seppälä-Lindroos A, Bergholm R, Yki-Järvinen H (2007) Effect of liver fat on insulin clearance. Am J Physiol Endocrinol Metab 293:E1709–E1715
Fernández-Gamba A, Leal MC, Morelli L, Castaño EM (2009) Insulin-degrading enzyme: structure–function relationship and its possible roles in health and disease. Curr Pharm Des 15:3644–3655
Roeb E, Graeve L, Hoffmann R, Decker K, Edwards DR, Heinrich PC (1993) Regulation of tissue inhibitor of metalloproteinases-1 gene expression by cytokines and dexamethasone in rat hepatocyte primary cultures. Hepatology 18:1437–1442
Rezende AC, Vieira AS, Rogério F et al (2008) Effects of systemic administration of ciliary neurotrophic factor on Bax and Bcl-2 proteins in the lumbar spinal cord of neonatal rats after sciatic nerve transection. Braz J Med Biol Res 41:1024–1028
Vieira AS, Rezende AC, Grigoletto J et al (2009) Ciliary neurotrophic factor infused intracerebroventricularly shows reduced catabolic effects when linked to the TAT protein transduction domain. J Neurochem 110:1557–1566
Leissring MA, Malito E, Hedouin S et al (2010) Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One 5:e10504
Ettinger MP, Littlejohn TW, Schwartz SL et al (2003) Recombinant variant of ciliary neurotrophic factor for weight loss in obese adults: a randomized, dose-ranging study. JAMA 289:1826–1832
van Belle TL, Coppieters KT, von Herrath MG (2011) Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev 91:79–118
Rønn SG, Börjesson A, Bruun C et al (2008) Suppressor of cytokine signalling-3 expression inhibits cytokine-mediated destruction of primary mouse and rat pancreatic islets and delays allograft rejection. Diabetologia 51:1873–1882
Del Prato S, Wishner WJ, Gromada J, Schluchter BJ (2004) Beta-cell mass plasticity in type 2 diabetes. Diabetes Obes Metab 6:319–331
Rhodes CJ (2005) Type 2 diabetes—a matter of beta-cell life and death? Science 307:380–384
Acknowledgements
We thank S. M. Ferreira and C. A. M. Oliveira for scientific help, and M. M. C. Brunelli for technical assistance (all from the Department of Structural and Functional Biology, Institute of Biology, State University of Campinas [UNICAMP], Brazil), and N. Conran (Hemocentro, State University of Campinas [UNICAMP], Brazil) for English revision.
Funding
FAPESP, CNPq and INOD provided financial support.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
LFR and GJS were responsible for the conception of the study, data analysis and drafting the article. JCS-S, EMC and ACB contributed to data analysis and interpretation, and revised the manuscript. All authors approved the final version.
Author information
Authors and Affiliations
Corresponding author
Additional information
L. F. Rezende and G. J. Santos contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Figure 1
CNTF reduced blood glucose acutely in fasted non-obese Swiss mice. Blood glucose (a) and AUC (b) of 4 h fasted non-obese 60 days old Swiss mice at 0 min (before CNTF or Citrate Buffer intraperitoneal injection) and 5, 15, 30 60 and 120 min thereafter. Control (black square), CNTF (white circle). Control (white bars), CNTF (grey bars). N = 3–4. Data means ± SEM. *Significantly different from Control (PDF 98 kb)
ESM Figure 2
CNTF improved insulin signaling (pIR and pAKT) in the skeletal muscle, adipose tissue and hypothalamus of non-obese Swiss mice. Phosphorylation of Insulin Receptor and AKT in protein extracts from skeletal muscle (a/b), adipose tissue (c/d) and hypothalamus (e/f) of non-fasted 28-day old Swiss mice after receiving an intraperitoneal dose of 1 IU/kg of insulin. Control (white bars), CNTF (grey bars), Alloxan (hatched bars) and CNTF + Alloxan (black bars). N = 3–4. Data are means ± SEM. *Significantly different from Control. †Significantly different from CNTF. #Significantly different from Alloxan (PDF 91 kb)
ESM Figure 3
CNTF reduced insulin degradation by HEPG2 cells in a 24 h follow-up experiment. (a) Serum insulin at 0, 5, 15, 30, 60 and 120 min, 6 h and 24 h and AUC of insulin for the entire 24 h experiment after adding 10 μIU/ml insulin to HEPG2 cells cultured for 3 days. Control (white square), CNTF (black square), Alloxan (white triangle) and CNTF + Alloxan (black triangle). Control (white bars), CNTF (grey bars), Alloxan (hatched bars) and CNTF + Alloxan (black bars). N = 3–5 for HEPG2 cells Data are means ± SEM. *Significantly different from Control. †Significantly different from CNTF. #Significantly different from Alloxan (PDF 129 kb)
ESM Figure 4
CNTF protects liver from Swiss mice and HEPG2 cells from Alloxan induced apoptosis via increased STAT3 and Reduced STAT1 phosphorylation. (a) Caspase-3 Cleavage, (b) STAT3 phosphorylation and (c) STAT1 phosphorylation in protein extracts from the liver of non-fasted 28-day old Swiss mice after receiving an intraperitoneal dose of 1 IU/kg of Insulin. (d) Caspase-3 Cleavage, (e) STAT3 phosphorylation and (f) STAT1 phosphorylation on protein extracts from HEPG2 cells after 1 h exposure to 10 μIU/ml insulin. Control (white bars), CNTF (grey bars), Alloxan (hatched bars) and CNTF + Alloxan (black bars). N = 3–4. Data are means ± SEM. *Significantly different from Control. †Significantly different from CNTF. #Significantly different from Alloxan (PDF 185 kb)
ESM Figure 5
CNTF had no effect on weight or weight gain of Swiss mice. (a) Weight of Swiss mice (g) at days 0 (before treatment), 7, 14, 21 and 28. Control (white square), CNTF (black square), Alloxan (white triangle) and CNTF + Alloxan (black triangle). Control (white bars), CNTF (grey bars), Alloxan (hatched bars) and CNTF + Alloxan (black bars). (b) Weight gain evaluated as an average AUC of each individual mouse’ weight during the entire experiment. Control (white bars), CNTF (grey bars), Alloxan (hatched bars) and CNTF + Alloxan (black bars). N = 3–4. Data are means ± SEM. *Significantly different from Control. †Significantly different from CNTF. #Significantly different from Alloxan (PDF 119 kb)
Table 1
PDF 62 kb
Rights and permissions
About this article
Cite this article
Rezende, L.F., Santos, G.J., Santos-Silva, J.C. et al. Ciliary neurotrophic factor (CNTF) protects non-obese Swiss mice against type 2 diabetes by increasing beta cell mass and reducing insulin clearance. Diabetologia 55, 1495–1504 (2012). https://doi.org/10.1007/s00125-012-2493-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-012-2493-5