
Abstract
Aims/hypothesis
The aim of this study was to investigate the association of the rs10811661 polymorphism near the CDKN2B/CDKN2A genes with glucose tolerance, insulin sensitivity and insulin release in three samples of white people with European ancestry.
Methods
Sample 1 comprised 845 non-diabetic offspring of type 2 diabetes patients recruited in five European centres participating in the EUGENE2 study. Samples 2 and 3 comprised, respectively, 864 and 524 Italian non-diabetic participants. All individuals underwent an OGTT. Screening for the rs10811661 polymorphism was performed using a TaqMan allelic discrimination assay.
Results
The rs10811661 polymorphism did not show a significant association with age, BMI and insulin sensitivity. Participants carrying the TT genotype showed a significant reduction in insulin release, measured by an OGTT-derived index, compared with carriers of the C allele, in the three samples. When these results were pooled with those of three published studies, and meta-analysed with a random-effects model, the T allele was significantly associated with reduced insulin secretion (−35.09 [95% CI 14.68–55.52], p = 0.0008 for CC+CT vs TT; and −29.45 [95% CI 9.51–49.38], p = 0.0038, for the additive model). In addition, in our three samples, participants carrying the TT genotype exhibited an increased risk for impaired glucose tolerance (IGT) compared with carriers of the C allele (OR 1.55 [95% CI 1.20–1.95] for the meta-analysis of the three samples).
Conclusions/interpretation
Our data, together with the meta-analysis of previously published studies, show that the rs10811661 polymorphism is associated with impaired insulin release and IGT, suggesting that this variant may contribute to type 2 diabetes by affecting beta cell function.
Similar content being viewed by others

Introduction
The pathophysiology of type 2 diabetes is characterised by a combination of impaired insulin action at the level of skeletal muscle, fat and liver, and failure of pancreatic beta cells to compensate for the enhanced insulin demand, ultimately resulting in hyperglycaemia [1, 2]. The pathogenesis of these two components is generally thought to be multifactorial, involving both genetic susceptibility and environmental factors [3]. However, identifying genes that confer susceptibility to type 2 diabetes has proven problematic. The availability of high-density genotyping arrays has enabled a systematic search for type 2 diabetes-associated common variants on a genome-wide scale [4–9]. Results of these studies have led to the identification of novel risk loci for type 2 diabetes. Among these loci, a polymorphism on chromosome 9p (rs10811661), located 125 kb upstream of the CDKN2B and CDKN2A genes (encoding cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) [p15INK4b] and cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4) [p16INK4a], respectively), has been associated with type 2 diabetes in three of the genome-wide association (GWA) studies (OR for pooled studies 1.20 [95% CI 1.14–1.25], p = 5 × 10−15) [6–8]. This association was replicated in several populations including Danish, Norwegian, French, Korean, Japanese and Chinese participants [10–15], but not confirmed in African-Americans and Pima Indians [16, 17]. Furthermore, the polymorphism rs10811661 was not associated with incident diabetes in the Diabetes Prevention Program (DPP) [18].
p15INK4b and p16INK4a, the proteins encoded by the CDKN2B and CDKN2A genes, are tumour suppressors that inhibit cyclin-dependent kinase 6 (CDK6) and CDK4, respectively, two regulators of pancreatic beta cell replication [19–21]. Both CDKN2B and CDKN2A are expressed in pancreatic islets and adipocytes [6–8]. In murine models, it has been demonstrated that increased production of p15INK4b is associated with pancreatic islet hypoplasia and impaired glucose-induced insulin secretion [19]. On the other hand, mice lacking CDK4 exhibit insulin-deficient diabetes due to a reduction in pancreatic beta cells, and mice expressing a mutant CDK4 that cannot bind the cell-cycle inhibitor p16INK4a display pancreatic hyperplasia due to proliferation of beta cells [20, 21]. These data suggest that the rs10811661 polymorphism located upstream of the CDKN2B and CDKN2A genes may confer increased risk for type 2 diabetes by affecting beta cell function. However, studies aimed at evaluating the potential role of the rs10811661 polymorphism in affecting quantitative metabolic traits associated with the risk of diabetes, such as measures of beta cell function and insulin sensitivity, have led to discordant results. Two studies have reported that non-diabetic carriers of the type 2 diabetes risk allele (T), the rs10811661 polymorphism, had lower insulin release during an OGTT [10, 22], whereas other studies carried out in participants of European ancestry and in Pima Indians did not find an association between this polymorphism and insulin secretion [17, 23, 24]. Thus, in order to obtain additional data on the association of the rs10811661 polymorphism with impaired glucose tolerance and pathophysiological quantitative traits, i.e. measures of beta cell function and insulin sensitivity, we studied three well-characterised samples of white Europeans.
Methods
Three different samples of adult (≥18 years of age) non-diabetic white people of European ancestry were studied.
Sample 1 comprised 845 non-diabetic offspring who had one parent with type 2 diabetes and the other parent with no family history of type 2 diabetes and/or a normal response to an OGTT. The offspring of type 2 diabetic patients were recruited in five different European centres participating in the EUGENE2 project (www.eugene2.com) [25] as follows: Copenhagen, Denmark (n = 268), Kuopio, Finland (n = 217), Tübingen, Germany (n = 152), Catanzaro, Italy (n = 109) and Gothenburg, Sweden (n = 99). Of these, 136 individuals (16.1%) had impaired glucose tolerance (IGT).
All study centres followed the same protocol, as previously reported [25]. Briefly, all participants underwent anthropometrical evaluation, and a 75 g OGTT was performed with 0, 30, 60, 90 and 120 min sampling for plasma glucose and insulin. On the second visit after a 12 h fast, the participants underwent a euglycaemic hyperinsulinaemic clamp study. The rate of total insulin-stimulated glucose disposal (M) was calculated for the last 60 min of the insulin infusion. In the Copenhagen centre, the euglycaemic clamp was not performed. Glucose levels were measured by the glucose oxidase method. Because plasma insulin was measured by different methods (except for the Gothenburg centre, for which insulin was measured in Tübingen), the assay applied in Tübingen (micro-particle enzyme immunoassay; Abbott Laboratories, Tokyo, Japan) was selected as a reference assay. The Catanzaro, Copenhagen and Kuopio centres each sent between 40 and 100 fasting and post-glucose challenge plasma insulin samples for parallel analysis in Tübingen. Plasma insulin levels from these three centres were converted by linear regression analysis to plasma insulin levels corresponding to the Tübingen assay.
Sample 2 comprised 864 unrelated non-diabetic individuals participating in the CAtanzaro MEtabolic RIsk factors Study (CATAMERIS), an observational study focused on assessment of cardio-metabolic risk factors [26]. Of these, 237 individuals (27.5%) had IGT.
Sample 3 comprised 524 unrelated non-diabetic participants consecutively recruited at the Department of Internal Medicine of the University of Rome-Tor Vergata [27]. Of these, 125 individuals (23.9%) had IGT. All participants included in samples 2 and 3 underwent anthropometrical evaluation and a venous blood sample was drawn for laboratory determination. After a 12 h overnight fast, a 75 g OGTT was performed with 0, 30, 60 and 120 min sampling for plasma glucose and insulin. The Matsuda index of insulin sensitivity (ISI) was calculated as described by Matsuda and DeFronzo [28]. Glucose-stimulated insulin secretion was estimated using the Stumvoll index for first-phase insulin secretion and was calculated as: 2,503 + 6.476 × insulin0 (pmol/l) − 126.5 × glucose120 (mmol/l) + 0.954 × insulin120 (pmol/l) − 239.3 × glucose0 (mmol/l), as previously described [29].
The study protocol was approved by appropriate institutional review boards and was in accordance with the Helsinki Declaration II. All study participants gave informed consent.
DNA analysis
DNA was isolated from whole blood using commercial DNA isolation kits. Samples from the EUGENE2 Consortium were genotyped in the Kuopio centre, whereas the two Italian samples were genotyped in the Catanzaro centre. Screening of the rs10811661 polymorphism was performed using a TaqMan allelic discrimination assay (Applied Biosystems, Foster City, CA, USA). The TaqMan genotyping reaction was amplified on a GeneAmp PCR system 2700 (Applied Biosystems) and fluorescence was detected using an ABI Prism 7000 sequence detector (Applied Biosystems). The overall genotyping success rate was 99.5%; genotype distributions obeyed Hardy–Weinberg equilibrium in the three different samples.
Statistical analysis
Data analyses were performed using Statistical Package for Social Science (SPSS Inc., Chicago, IL, USA) version 14.0. The results for continuous variables are given as means ± SD. Because of the low number of CC homozygotes in the study samples, the rs10811661 polymorphism was tested using both the additive and the dominant inheritance model. Unpaired Student’s t tests or ANOVAs were used to compare differences among groups for continuous variables, as appropriate, and the χ 2 test for non-continuous variables. A general linear model was used to compare phenotypic differences in the three samples. A multivariable logistic regression analysis, adjusted for age, sex and centre, was used to determine the association between the genotype frequencies and IGT. We report a nominal p value of <0.05 without adjustment for multiple testing given the high prior probabilities for association of the rs10811661 polymorphism with the examined phenotypes. A fixed-effect meta-analysis was performed using a comprehensive meta-analysis programme to assess the global OR for IGT in samples 1–3. To meta-analyse the effect of rs10811661 on insulin secretion in the three samples analysed in this study, together with three previously published studies [10, 22, 23], effect size estimates and standard errors from the six samples were pooled by RGui version 2.10.0 (www.r-project.org) applying the meta package and the combined effect was derived from the random-effects method (weight of studies estimated using the DerSimonian–Laird method [30]).
Results
The clinical characteristics of sample 1 according to the rs10811661 polymorphism are shown in Table 1. The rs10811661 polymorphism did not show any significant association with age, BMI or fasting plasma glucose levels. We also found no association between the rs10811661 polymorphism and whole-body insulin sensitivity as measured by a hyperinsulinaemic, euglycaemic clamp (M value; Table 1). By contrast, the rs10811661 polymorphism was significantly associated with 2 h post-load glucose levels, with carriers of the C allele having significantly lower levels compared with participants carrying the TT genotype (p = 0.03 according to a dominant model; Table 1). In addition, the rs10811661 polymorphism was significantly associated with the OGTT-derived index of insulin secretion, with participants carrying the TT genotype showing a significant reduction in insulin release compared with carriers of the C allele (p = 0.03). In a logistic regression analysis with adjustment for age, sex and centre, participants carrying the TT genotype exhibited an increased risk for IGT compared with carriers of the C allele (OR 1.78 [95% CI 1.12–2.82], p = 0.014).
To replicate these data, two independent samples of non-diabetic Italian individuals were studied. The clinical characteristics of sample 2 according to the rs10811661 polymorphism are shown in Table 2. The rs10811661 polymorphism did not show any significant association with age, BMI, fasting plasma glucose levels or insulin sensitivity as measured by the ISI index. Carriers of the C allele have significantly lower levels of 2 h post-load glucose levels compared with participants carrying the TT genotype (p = 0.04 according to a dominant model; Table 2). Furthermore, the rs10811661 polymorphism was significantly associated with the OGTT-derived index of insulin secretion, with participants carrying the TT genotype showing a significant reduction in insulin release compared with carriers of the C allele (p = 0.05). In a logistic regression analysis with adjustment for age and sex, participants carrying the TT genotype exhibited an increased risk for IGT, compared with carriers of the C allele (OR 1.44 [95% CI 1.02–2.03], p = 0.04).
Clinical characteristics of sample 3 according to the rs10811661 polymorphism are shown in Table 3. The rs10811661 polymorphism was not associated with age, BMI or insulin sensitivity as measured by the ISI index. Carriers of the C allele showed significantly lower fasting and 2 h post-load glucose levels compared with participants carrying the TT genotype (p = 0.01 and p = 0.04 according to the dominant model, for fasting and 2 h post-load glucose, respectively). In addition, the rs10811661 polymorphism was significantly associated with the OGTT-derived index of insulin secretion, with participants carrying the TT genotype showing a significant reduction in insulin release compared with carriers of the C allele (p = 0.03). In a logistic regression analysis with adjustment for age and sex, participants carrying the TT genotype exhibited an increased risk for IGT compared with carriers of the C allele (OR 1.61 [95% CI 1.01–2.59], p = 0.04).
As no genetic heterogeneity across the three samples was evident (p value for heterogeneity = 0.76), data from the three samples were meta-analysed with a fixed-effect model. The results of this meta-analysis showed that participants carrying the TT genotype have an OR for IGT of 1.55 (95% CI 1.20–1.95, p = 0.0001) compared with carriers of the C allele.
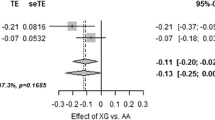
Finally, in order to provide a more confident estimate of the effect of rs10811661 on insulin secretion, the results for the three samples analysed in this study were pooled with those of three available studies on European non-diabetic participants [10, 22, 23]. Borderline significant heterogeneity indicating differential effects between the studies was observed in the meta-analysis (p = 0.068 for the dominant model and p = 0.053 for the additive model). When data for the 13,896 individuals were meta-analysed with a random-effects model, the T allele was significantly associated with reduced insulin secretion (−35.09 [95% CI 14.68–55.52], p = 0.0008 for CC+CT vs TT; and −29.45 [95% CI 9.51–49.38], p = 0.0038, for the additive model; Fig. 1).

Results of the meta-analysis of the association of the rs10811661polymorphism at CDKN2A/B and insulin secretion in 13,986 individuals. a CC+CT vs TT: −35.09 (95% CI 14.68–55.52), p = 0.0008 (diamond) and (b) additive model: −29.45 (95% CI 9.51–49.38), p = 0.0038 (diamond)
Discussion
The present study aimed to elucidate the metabolic effects of the rs10811661 polymorphism, which has been recently identified as a type 2 diabetes susceptibility locus by GWA studies [6–8]. Using three samples of non-diabetic white people of European ancestry, we found that the rs10811661 polymorphism of CDKN2B/CDKN2A was significantly associated with impaired glucose-stimulated insulin release, but not with insulin sensitivity as measured by hyperinsulinaemic, euglycaemic clamp (sample 1) or by an OGTT-derived index of insulin sensitivity (samples 2 and 3). Association of the rs10811661 polymorphism of CDKN2B/CDKN2A with estimates of insulin release derived from an OGTT has been evaluated in previous studies, with divergent results [10, 17, 22–24]. In the Danish population-based Inter99 study, comprising 5,970 middle-aged participants, it was reported that carriers of the C allele had an estimated higher level of insulin release in response to an oral glucose load compared with participants carrying the TT genotype [10]; similar results were observed in a study of 5,327 non-diabetic Finnish men [22]. Interestingly, when we performed a meta-analysis including our samples and data from three previously published studies [10, 22, 23], we observed that the T allele was significantly associated with reduced insulin secretion. Of note, in Pima Indians, the rs10811661 polymorphism was not associated with either type 2 diabetes or insulin release [17]. The reasons for these discrepancies are unclear. Difference in ethnicity is the most obvious explanation for these divergent results. In support of this hypothesis, several type 2 diabetes risk variants found in Europid participants, such as polymorphisms in CDKAL1, SLC30A8, HHEX, EXT2, IGF2BP2 and LOC387761, were not significantly associated with type 2 diabetes in Pima Indians [17]; furthermore it is possible that the lack of data on the causative variant associated with the variant under study may play a role in the discrepancies between the different ethnicities.
We also found that participants carrying the TT genotype have higher 2 h post-load glucose levels and increased risk of IGT (OR 1.55 [95% CI 1.20–1.95] in the pooled data from the three samples) compared with carriers of the C allele. Interestingly, participants carrying the low-risk CC genotype at the CDKN2A/B locus enrolled in the DPP exhibited a greater improvement in beta cell function than those with the high-risk TT genotype after treatment with troglitazone and lifestyle modification for 1 year, suggesting that they may have benefited more from these interventions [18].
Mechanisms underlying the association of the rs10811661 polymorphism with impaired insulin secretion and the risk of IGT and type 2 diabetes are not fully understood. The nearest annotated genes CDKN2B and CDKN2A, encoding p15INK4b and p16INK4a, respectively, have been implicated in pancreatic islet regenerative capacity [19, 31]. p16INK4a has been shown to accumulate in many tissues, including pancreatic islets, as a function of ageing [32, 33]. Transgenic mice overproducing p16INK4a showed decreased islet proliferation with ageing, and aged mice lacking p16INK4a demonstrated enhanced islet proliferation. These observations support the idea that p16INK4a mediates a decline in the replicative capacity of beta cells associated with ageing. Because type 2 diabetes mellitus patients exhibit a decrease in beta cell mass [34], it may be tempting to speculate that increased p16INK4a levels with ageing may contribute to the relative failure of islet proliferation associated with type 2 diabetes mellitus. However, a recent study, evaluating whether genetic variants robustly associated with type 2 diabetes also modulate expression levels of nearby candidate genes, has reported no evidence of an association between the rs10811661 polymorphism and CDKN2B and CDKN2A gene expression in human tissues [35].
This study has some limitations. The present findings obtained in a cross-sectional study of cohorts of Europids are explorative in nature and replication in independent prospective population-based studies with different ethnicities is needed to firmly determine whether the rs10811661 polymorphism affects insulin secretion and whether pancreatic beta cell dysfunction is truly implicated in the pathogenesis of type 2 diabetes.
Abbreviations
- CATAMERIS:
-
CAtanzaro MEtabolic RIsk factors Study
- CDK4:
-
Cyclin-dependent kinase 4
- CDK6:
-
Cyclin-dependent kinase 6
- DPP:
-
Diabetes Prevention Program
- GWA:
-
Genome-wide association
- IGT:
-
Impaired glucose tolerance
- ISI:
-
Matsuda index of insulin sensitivity
- p15INK4a :
-
Cyclin-dependent kinase inhibitor 2A (melanoma, p16, inhibits CDK4)
- p15INK4b :
-
Cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4)
References
Warram JH, Martin BC, Krolewski AS, Soeldner JS, Kahn CR (1990) Slow glucose removal rate and hyperinsulinemia precede the development of type II diabetes in the offspring of diabetic parents. Ann Intern Med 113:909–915
Vauhkonen I, Niskanen L, Vanninen E, Kainulainen S, Uusitupa M, Laakso M (1998) Defects in insulin secretion and insulin action in non-insulin-dependent diabetes mellitus are inherited: metabolic studies on offspring of diabetic probands. J Clin Invest 101:86–96
Permutt MA, Wasson J, Cox N (2005) Genetic epidemiology of diabetes. J Clin Invest 115:1431–1439
Frayling TM, Timpson NJ, Weedon MN et al (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894
Steinthorsdottir V, Thorleifsson G, Reynisdottir I et al (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39:770–775
Zeggini E, Weedon MN, Lindgren CM et al (2007) Replication of genome-wide association signals in U.K. samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Saxena R, Voight BF, Lyssenko V et al (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1341–1345
Scott LJ, Mohlke KL, Bonnycastle LL et al (2007) Genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345
Wellcome TCCC (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678
Grarup N, Rose CS, Andersson EA et al (2007) Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes 56:3105–3111
Hertel JK, Johansson S, Raeder H et al (2008) Genetic analysis of recently identified type 2 diabetes loci in 1,638 unselected patients with type 2 diabetes and 1,858 control participants from a Norwegian population-based cohort (the HUNT Study). Diabetologia 51:971–977
Duesing K, Fatemifar G, Charpentier G et al (2008) Strong association of common variants in the CDKN2A/CDKN2B region with type 2 diabetes in French Europids. Diabetologia 51:821–826
Lee YH, Kang ES, Kim SH et al (2008) Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J Hum Genet 53:991–998
Wu Y, Li H, Loos RJ et al (2008) Common variants in CDKAL1, CDKN2A/B, IGF2BP2, SLC30A8, and HHEX/IDE genes are associated with type 2 diabetes and impaired fasting glucose in a Chinese Han population. Diabetes 57:2834–2842
Tabara Y, Osawa H, Kawamoto R et al (2009) Replication study of candidate genes associated with type 2 diabetes based on genome-wide screening. Diabetes 58:493–498
Lewis JP, Palmer ND, Hicks PJ et al (2008) Association analysis in African Americans of European-derived type 2 diabetes single nucleotide polymorphisms from whole-genome association studies. Diabetes 57:2220–2225
Rong R, Hanson RL, Ortiz D et al (2009) Association analysis of variation in/near FTO, CDKAL1, SLC30A8, HHEX, EXT2, IGF2BP2, LOC387761, and CDKN2B with type 2 diabetes and related quantitative traits in Pima Indians. Diabetes 58:478–488
Moore AF, Jablonski KA, McAteer JB et al (2008) Diabetes Prevention Program Research Group. Extension of type 2 diabetes genome-wide association scan results in the diabetes prevention program. Diabetes 57:2503–2510
Moritani M, Yamasaki S, Kagami M et al (2005) Hypoplasia of endocrine and exocrine pancreas in homozygous transgenic TGF-beta1. Mol Cell Endocrinol 229:175–184
Rane SG, Dubus P, Mettus RV et al (1999) Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet 22:44–52
Tsutsui T, Hesabi B, Moons DS et al (1999) Targeted disruption of CDK4 delays cell cycle entry with enhanced p27 (Kip1) activity. Mol Cell Biol 19:7011–7019
Stancáková A, Kuulasmaa T, Paananen J et al (2009) Association of 18 confirmed susceptibility loci for type 2 diabetes with indices of insulin release, proinsulin conversion, and insulin sensitivity in 5,327 nondiabetic Finnish men. Diabetes 58:2129–2136
Pascoe L, Tura A, Patel SK et al (2007) Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic beta-cell function. Diabetes 56:3101–3104
Groenewoud MJ, Dekker JM, Fritsche A et al (2008) Variants of CDKAL1 and IGF2BP2 affect first-phase insulin secretion during hyperglycaemic clamps. Diabetologia 51:1659–1663
Laakso M, Zilinskaite J, Hansen T et al (2008) Insulin sensitivity, insulin release and GLP-1 levels in subjects with IFG and/or IGT in the EUGENE2 Study. Diabetologia 51:502–511
Succurro E, Marini MA, Arturi F et al (2009) Elevated one-hour post-load plasma glucose levels identifies subjects with normal glucose tolerance but early carotid atherosclerosis. Atherosclerosis 207:245–249
Sesti G, Cardellini M, Marini MA et al (2003) A common polymorphism in the promoter of UCP2 contributes to the variation in insulin secretion in glucose-tolerant subjects. Diabetes 52:1280–1283
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diab Care 9:1462–1470
Stumvoll M, van Haeften T, Fritsche A, Gerich J (2001) Oral glucose tolerance test indexes for insulin sensitivity and secretion based on various availabilities of sampling times. Diab Care 24:796–797
Der Simonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7:177–188
Krishnamurthy J, Ramsey MR, Ligon KL et al (2006) p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 443:453–457
Krishnamurthy J, Torrice C, Ramsey MR et al (2004) Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114:1299–1307
Nielsen GP, Stemmer-Rachamimov AO, Shaw J, Roy JE, Koh J, Louis DN (1999) Immunohistochemical survey of p16INK4A expression in normal human adult and infant tissues. Lab Invest 79:1137–1143
Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52:102–110
Cotsapas C, Prokunina-Olsson L, Welch C et al (2010) Expression analysis of loci associated with type 2 diabetes in human tissues. Diabetologia 53:2334–2339
Acknowledgements
This study was supported by a grant from the European Community’s FP6 EUGENE2 no. LSHM-CT-2004-512013. The EUGENE2 Consortium (www.eugene2.com) consists of the laboratories of: U. Smith (Sweden), M. Laakso (Finland), H. Haring (Germany), G. Sesti (Italy), O. Pedersen (Denmark), J. Zierath (Sweden), H.-G. Joost (Germany), F. Beguinot (Italy), E. van Obberghen (France), J. Auwerx (France), F. Bosch (Spain), P. Lind (Sweden).
Duality of interest
N. Grarup, T. Hansen and O. Pedersen hold personal shares in Novo Nordisk. M. L. Hribal, I. Presta, T. Procopio, M. A. Marini, A. Stančáková, J. Kuusisto, F. Andreozzi, A. Hammarstedt, P-A Jansson, M. Walker, N. Stefan, A. Fritsche, H. U. Häring, U. Smith, M. Laakso and G. Sesti declare that they have no conflict of interest associated with this manuscript.
Author information
Authors and Affiliations
Consortia
Corresponding author
Rights and permissions
About this article
Cite this article
Hribal, M.L., Presta, I., Procopio, T. et al. Glucose tolerance, insulin sensitivity and insulin release in European non-diabetic carriers of a polymorphism upstream of CDKN2A and CDKN2B . Diabetologia 54, 795–802 (2011). https://doi.org/10.1007/s00125-010-2038-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-2038-8