
Abstract
Aims/hypothesis
Tissue-specific amplification of glucocorticoid action through 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) affects the development of the metabolic syndrome. Hexose-6-phosphate dehydrogenase (H6PDH) mediates intracellular NADPH availability for 11β-HSD1 and depends on the glucose-6-phosphate transporter (G6PT). Little is known about the tissue-specific alterations of H6PDH and G6PT and their contributions to local glucocorticoid action in db/db mice.
Methods
We characterised the role of H6PDH and G6PT in pre-receptor metabolism of glucocorticoids by examining the production of the hepatic 11β-HSD1-H6PDH–G6PT system in db/db mice.
Results
We observed that increased production of hepatic H6PDH in db/db mice was paralleled by upregulation of hepatic G6PT production and responded to elevated circulating levels of corticosterone. Treatment of db/db mice with the glucocorticoid antagonist RU486 markedly reduced production of both H6PDH and 11β-HSD1 and improved hyperglycaemia and insulin resistance. The reduction of H6PDH and 11β-HSD1 production by RU486 was accompanied by RU486-induced suppression of hepatic G6pt (also known as Slc37a4) mRNA. Incubation of mouse primary hepatocytes with corticosterone enhanced G6PT and H6PDH production with corresponding activation of 11β-HSD1 and PEPCK: effects that were blocked by RU486. Knockdown of H6pd by small interfering RNA showed effects comparable with those of RU486 for attenuating the corticosterone-induced H6PDH production and 11ß-HSD1 reductase activity in these intact cells. Addition of the G6PT inhibitor chlorogenic acid to primary hepatocytes suppressed H6PDH production.
Conclusions/interpretation
These findings suggest that increased hepatic H6PDH and G6PT production contribute to 11β-HSD1 upregulation of local glucocorticoid action that may be related to the development of type 2 diabetes.
Similar content being viewed by others


Introduction
Excess glucocorticoid (GC) production strongly affects the development of type 2 diabetes and obesity via activation of intracellular GC receptor (GR) [1–5]. In rodents, increased GC production promotes hepatic gluconeogenesis and adipocyte differentiation and induces insulin resistance and obesity in genetically obese db/db and ob/ob mice [5–8]. However, tissue GC action can also be regulated at the pre-receptor level by the 11β-hydroxysteroid dehydrogenases. Metabolic tissues such as liver and adipose tissue abundantly produces 11β-hydroxysteroid dehydrogenase (11β-HSD1), a resident enzyme of the endoplasmic reticulum (ER) lumen that acts in vivo as an NADPH-dependent reductase to generate active cortisol (corticosterone in rodents) from inactive 11-keto cortisone (11-dehydrocorticosterone) [9–11]. 11β-HSD1 plays an important role in the regulation of tissue GC action [12–15]. Indeed, enhanced 11β-HSD1 could result in the production of excess tissue GCs and induction of GC-receptor-mediated local GC action related to glucose homeostasis, insulin action and adiposity, all of which are associated with the development of type 2 diabetes and visceral obesity [16–18]. Pharmacological evidence further validates the role of 11ß-HSD1 in the control of insulin resistance and obesity as 11ß-HSD1 inhibitors reduced hepatic glucose output and improved insulin sensitivity in obese animal models, as well as in patients with type 2 diabetes [19–23]. These studies implicate the importance of 11ß-HSD1 in the pathogenesis of metabolic syndrome and obesity.
The role of 11ß-HSD1 in the pre-receptor activation of GCs is entirely dependent on the production of its cofactor NADPH in the ER lumen [24, 25]. NADPH is regenerated by hexose-6-phosphate dehydrogenase (H6PDH) [26, 27], a microsomal enzyme located in the lumen of the ER and principally produced in hepatocytes and adipocytes, sites of 11β-HSD1 and GR [28, 29]. In these target tissues, H6PDH uses glucose 6-phosphate (G6P) and NADP to produce NADPH. The supply of G6P to H6PDH is ensured by a functional membrane ER protein, the G6P transporter (G6PT), which specifically transports cytosolic G6P into the ER and therefore regulates H6PDH activity via modulation of G6P level within the ER [30, 31]. The generation of NADPH by H6PDH can be used by NADPH-dependent 11ß-HSD1 reductase activity [32, 33]. H6PDH is thus a potential candidate supplying NADPH for 11β-HSD1-induced amplification of tissue GC production [34–36].
In the present study, we characterised the metabolic phenotype of H6PDH and G6PT in relation to pre-receptor metabolism of GCs by examining the effects of the GC antagonist RU486 on the production of the 11β-HSD1–H6PDH–G6PT system in the liver and adipose tissues of db/db mice. We also assessed the direct interaction of G6PT and H6PDH in the control of 11β-HSD1 driving tissue GC action in primary cultures of hepatocytes from db/db mice by using H6pdh small interfering (si)RNA and the G6PT inhibitor chlorogenic acid (CA). Finally, we examined the hormonal regulation of H6PDH and G6PT in these intact cells.
Methods
Animals
Male C57BL/KsJ-obese (db/db) mice and their lean littermates (db/+) were purchased at 10 weeks of age from Taconic Farms (Hudson, NY, USA) and housed in a room illuminated daily from 07:00 to 19:00 hours (12 h light/dark cycle) with free access to water and standard laboratory chow. RU486 (25 mg/kg) or vehicle was given by intraperitoneal injection twice each day (at 07:00 and 19:00 hours) for 3 days or 3 weeks as previously described by Friedman et al. and Liu et al. [5, 15]. Body weight and food intake were recorded daily. All animal experiments were approved by the Charles Drew University Institutional Animal Care and Use Committee.
Insulin tolerance test
For the insulin tolerance test (ITT), animals were fasted for 12 h, and blood samples were drawn at different times following insulin injection (2 U/kg i. p.; Novolin R; Eli Lilly, Indianapolis, IN, USA).
Biochemical assays
Blood glucose levels were determined by the glucose oxidase method [36]. Plasma corticosterone levels were determined by RIA using mouse corticosterone as a standard (ICN Biomedicals, Costa Mesa, CA, USA). Plasma insulin levels were measured by RIA using rat insulin as a standard (Crystal Chemicals, Chicago, IL, USA).
Cell culture and treatment
Primary hepatocytes were isolated from male db/db mice [21] and seeded onto collagen-coated dishes in DMEM/F-12 medium with 10% FBS at 37°C for 4 h. Cells were then washed with PBS, and the medium was changed to DMEM/F-12 medium without FCS. After 12 h, cells were treated with corticosterone \( \left( {1 \times {{10}^{{-7}}}-1 \times {{10}^{{-5}}}{\hbox{mol}}/{\hbox{l}}} \right) \) or insulin (1 × 10−5 mol/l) in the presence or absence of RU486 (1 × 10−6 mol/l) for 48 or 72 h. The G6PT inhibitor CA (0–200 μmol/l) was added to primary cultures of hepatocytes for 48 h. This pharmacological dose of CA is similar to doses previously used in mouse 3T3-L1 pre-adipocytes and glioma cells [37, 38].
siRNA experiments in primary cultures of hepatocytes
Cells were transfected with the H6pd siRNA (SABiosciences siRNA ID KM26265; SABiosciences, Frederick, MD, USA), or negative control siRNA (SABiosciences) using lipofectamine 2000 transfection agent according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). After 4 h, cells were treated with vehicle or corticosterone (1 × 10−6 mol/l) for an additional 48 h.
Microsomal enzymatic activity assays
The liver microsomal pellet was obtained and 11ß-HSD1 activity was evaluated by addition of 0.4–1 mmol/l NADPH and 900 nmol/l unlabelled 11-dehydrocorticosterone (11-DHC) with 100 nmol/l 11-[3H]DHC as tracer to microsomes in Krebs–Ringer buffer solution at 37°C for 10–30 min [36, 39]. The liver microsomes (1 mg/ml protein) were permeabilised with 1% Triton-100 to allow the free access of the cofactor to the intraluminal enzyme. Steroids were separated by thin-layer chromatography (TLC) and analysed by scintillation counting. The percentage of interconversion of [3H]corticosterone and 11-[3H]DHC was calculated from the radioactivity in each fraction. Reductase activity of 11ß-HSD1 was also evaluated by immunoassay of the corticosterone produced from 11-DHC using a sensitive corticosterone ELISA kit.
H6PDH enzyme activity was determined by spectrophotometric measurement of NADPH production in the presence of G6P and NADP using absorbance at 340 nm with a spectrophotometer (Ultrospec 2100, Amersham Biosciences) [36, 40]. Protein, 50 μg, from liver microsomes was incubated with 0.5–5 mmol/l G6P, 1–5 mmol/l NADP and 100 mmol/l glycine buffer solution at 22°C for 0–5 min. Specific activities were calculated and expressed as micromoles NADPH production per minute per milligram protein. The protein concentration was measured by the Bradford assay (Bio-Rad Protein Assay Kit, Bio-Rad, Hercules, CA, USA).
11β-HSD1 and H6PDH activity in primary hepatocytes
Cells were incubated with 2 nmol/l 11-[3H]DHC with 18 nmol/l unlabelled 11-DHC for 10–30 min [41]. Steroids were extracted from 1.0 ml of culture medium with ethyl acetate and separated by TLC. Enzyme activity levels were determined by counting the radioactivity. For the H6PDH activity in vitro, protein extracts, 20 μg, from primary hepatocytes were incubated with 2 mmol/l G6P as substrate in 100 μl total volume of glycine buffer (pH 10.0) with 0.5 mmol/l NADP as a cofactor. The changes in absorbance at 340 nm were measured during 25 min at 5 min intervals [36].
RNA extraction and real-time quantitative PCR analysis
Total RNA was extracted using a single-step extraction method (RNAzol B, Invitrogen). cDNA synthesis from 2.0 μg RNA was performed using high capacity RNA-to-cDNA Kit (Applied Biosystems, Carlsbad, CA, USA). Real-time primers were designed with Primer express software 2.0 (Applied Biosystems) and are listed in the Electronic supplementary material (ESM) Table 1. Amplification of each target cDNA was then performed with SYBR Green I Master Kits in the ABI Prism 7700 Sequence Detection System (Applied Biosystems) according to the protocols recommended by the manufacturer. All reactions were carried out using the following cycling parameters: 55°C for 2 min and 95°C for 10 min, following by 40 cycles of 95°C for 15 s and 60°C for 1 min. Threshold cycle (Ct) readings for each of the unknown samples were then used to calculate the amount of target genes and were normalised to the signal of 18S rRNA. Data analysis is based on the ΔCt method.
G6P uptake measurement
Microsomal G6P uptake measurements were performed according to protocols previously published by Hiraiwa et al. [42]. Briefly, 50–100 μg of microsomes were incubated in 50 mmol/l sodium cacodylate buffer containing 250 mmol/l sucrose (pH 6.5) in the presence of 0.2–1 mmol/l G6P plus [U-14C]G6P (American Radiolabeled Chemicals, St Louis, MO, USA) at 22°C for 3 min. The reaction was stopped by filtering through a nitrocellulose filter and samples were quickly washed with an ice-cold buffer containing 50 mmol/l TRIS–HCl, pH 7.4 and 250 mmol/l sucrose. Microsomes were subsequently permeabilised with 0.2% deoxycholate to abolish intravesicular G6P uptake. The radioactivity associated with microsomes retained by filters was measured by liquid-scintillation counting.
Western blot analysis
Protein, 25 μg samples from liver microsomes or 50 μg total cellular proteins from primary hepatocytes, was analysed by SDS-PAGE as described by Liu et al. [36]. Membranes were incubated with a polyclonal anti-11β-HSD1 antibody (1:1,500; Affinity Bioreagents, Rockford, IL, USA) and mouse anti-human H6PDH antibody (1:2,000; Novus Biological, Littleton, CO, USA). The 11β-HSD1 and H6PDH protein signal was quantified with the use of the Eagle Eye II Quantitation System (Stratagene, La Jolla, CA, USA).
Statistical analyses
All data are expressed as the mean ± SEM. The normality of the distribution of data was established using the Wilks–Shapiro test, and outcome measures between groups were compared by Student’s t test. To compare multiple groups, one-way ANOVA used. If ANOVA reveals significant differences, then individual group comparisons were performed by using the Newman–Keul’s post hoc test. The differences among groups were considered significant at p < 0.05.
Results
Characterisation of H6PDH and G6pt levels in db/db mice
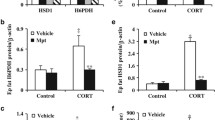
db/db mice had higher body weight and plasma levels of insulin, corticosterone and blood glucose than matched littermates (Table 1). Plasma levels of cholesterol and triacylglycerols in db/db mice were significantly higher than those in lean mice (Table 1). 11β-HSD1 reductase activity in liver microsomes of db/db mice was significantly increased to 47% over that of db/+ controls (p < 0.01; Fig. 1a). Western blot analysis revealed the 11β-HSD1 protein production in the liver of db/db mice was increased 1.7-fold over that of db/+ controls (p < 0.01; Fig. 1b). H6PDH activity and protein production in the liver of db/db mice were increased 1.57- and 1.9-fold, respectively, compared with db/+ control mice (p < 0.001; Fig. 1 c, d). Real-time RT-PCR analysis revealed that hepatic 11ß-Hsd1 (also known as Hsd11b1) mRNA levels were increased 2.8-fold in db/db mice and level was positively correlated with H6pd mRNA levels (p < 0.01), which increased to 1.8-fold in liver over that of db/+ animals (p < 0.01; Fig. 2a). Similarly, the hepatic G6pt (also known as Slc37a4) mRNA levels in db/db mice were significantly increased by 1.9-fold over that of db/+ controls (p < 0.01; Fig. 2a). Moreover, the [U-14C]G6P uptake in hepatic microsomes of db/db mice was higher than that of db/+ controls (p < 0.01; Fig. 2b). The levels of hepatic Pepck and G6Pase mRNA in db/db mice were higher than the respective in db/+ mice (p < 0.01; Fig. 2c). AUC analysis showed that the glucose levels in db/db mice were significantly higher than those in vehicle-treated db/+ animals (Fig. 2d). In addition, subcutaneous and epididymal adipose 11ß-Hsd1 mRNA levels were higher than those of lean controls (Fig. 3a, b). Moreover, H6pd mRNA levels were also markedly increased in subcutaneous fat and in epididymal fat in db/db mice compared with lean levels (p < 0.05), respectively. However, there was no difference in G6pt mRNA levels in subcutaneous and epididymal fat between db/db mice and lean controls (Fig. 3a, b).

11β-HSD1 and H6PDH activity and protein levels in liver microsomes of lean db/+ and obese db/db mice treated with vehicle or RU486. a 11β-HSD1 reductase activity was measured in mouse liver microsomes using 11-DHC as substrate in the presence of NADPH. Production and relative quantification of 11β-HSD1 (b) and H6PDH (c) protein was done relative to the amount of GAPDH. d H6PDH activity was measured in liver microsomes on the basis of NADPH formation using 2 mmol/l G6P as substrate in the presence of NAD. Data are mean ± SEM from seven to eight mice (per group). **p < 0.01, ***p < 0.001 vs db/+ controls; † p < 0.05, †† p < 0.01 vs db/db controls. White bars, vehicle; black bars, RU486

a Quantitative real-time RT-PCR analysis demonstrating the relative alterations of 11β-Hsd1, H6pd and G6pt mRNA expression in the livers of db/+ and db/db mice treated with vehicle or RU486. White bars, db/+; black bars, db/++RU486; hatched bars, db/db; grey bars, db/db+RU486. b Uptake of [U-14C]G6P into the liver microsomes of lean db/+ and db/db mice treated with vehicle or RU486. White squares, db/+; black squares, db/++RU486; white circles, db/db; black circles, db/db+RU486. c Expression levels of Pepck and G6Pase mRNA in the liver of db/+ and db/db mice treated with vehicle or RU486. White bars, db/+; black bars, db/++RU486; hatched bars, db/db; grey bars, db/db+RU486. d The effects of RU486 on AUC of plasma glucose level in ITT. Data are mean ± SEM from seven to eight mice per group. **p < 0.01 vs db/+ controls; †† p < 0.01, ††† p < 0.001 vs db/db controls

Adipose 11β-Hsd1, H6pd and G6pt mRNA expression in (a) subcutaneous and (b) epididymal fat of lean db/+ and obese db/db mice treated with vehicle or RU486. Relative expression of mRNA levels was measured by RT-PCR and normalised to 18S rRNA expression. *p < 0.05, **p < 0.01 vs db/+ control; †† p < 0.01 vs db/db controls. White bars, db/+; hatched bars, db/+ + RU486; black bars, db/db; grey bars, db/db+RU486
The effects of RU486 treatment on the phenotypes of type 2 diabetes in db/db mice
As shown in Table 1, RU486 compared with vehicle treatment for 3 weeks lowered blood glucose levels with no significant changes in body weight and levels of plasma insulin and lipids in db / db mice. However, plasma corticosterone levels were increased in db / db mice after RU486 treatment (p < 0.001). In contrast, hepatic 11β-HSD1 reductase activity was reduced 1.6-fold in RU486-treated db / db mice (Fig. 1a). Western blot analysis revealed that 11β-HSD1 protein production was decreased 2.5-fold in RU486-treated db / db mice (p < 0.01 vs db / db controls; Fig. 1b). Similarly, treatment of db/db mice with RU486 for 3 weeks significantly reduced hepatic H6PDH activity to 45% that of vehicle-treated db / db mice (p < 0.01; Fig. 1d); this reduction in enzyme activity occurred in parallel with decreased hepatic H6PDH protein production in RU486-treated db / db mice (Fig. 1c). Real-time RT-PCR analysis showed that in db / db mice, RU486 reduced hepatic 11β-Hsd1 and H6pd mRNA expression, respectively, to the levels of vehicle-treated db / + mice (Fig. 2a). In parallel with the decrease in H6pd mRNA levels, hepatic G6pt mRNA expression was decreased by twofold in db / db mice after RU486 treatment (Fig. 2a). Consistent with decreased G6pt expression, RU486 reduced [U-14C]G6P uptake in the liver microsomes, although it did not restore G6P uptake levels to those of db/+ controls (Fig. 2b). Moreover, RU486 also significantly reduced hepatic Pepck mRNA levels, but did not exert significant effects on the hepatic G6Pase (also known as G6pc) mRNA expression in db / db mice (Fig. 2c). The AUC glucose levels were reduced in db / db mice after RU486 treatment (Fig. 2d). RU486 treatment for 3 weeks also reduced the G6pt mRNA level with simultaneous reduction in H6PDH and 11β-HSD1 expression in subcutaneous fat of db / db mice as compared with controls (Fig. 3a), but did not alter epididymal fat G6PT, H6PDH and 11β-HSD1 expression (Fig. 3b). However, RU486 did not exert significant effects on 11β-Hsd1, H6pd and G6pt mRNA expression in adipose tissues from db / + mice (Fig. 3a, b). In addition, a shorter treatment of db / db mice with RU486 for 3 days also significantly reduced the expression of 11ß-Hsd1, H6pd and G6pt in the liver and improved glucose levels, but did not change the slope of insulin tolerance curve (data not shown), indicating that blocking the action of GC with RU486 leads to changes in gene expression that are independent of improvement in insulin sensitivity.
H6PDH and G6PT abundance in primary cultures of hepatocytes
To confirm our in vivo observations, we investigated the effects of exogenous corticosterone and insulin on G6PT and H6PDH in primary hepatocytes from db / db mice. Western blot analysis showed that treatment of hepatocytes with increasing doses of corticosterone led to a concentration-dependent induction of H6PDH protein production (Fig. 4a). A concentration of corticosterone (1 × 10−6 mol/l), similar to that occurring in db / db mice in vivo, significantly increased NADPH production in the presence of G6P as a substrate (Fig. 4b). The increase in H6PDH activity was consistent with real-time RT-PCR analyses, which showed that corticosterone increased H6pd mRNA levels 1.6-fold in primary hepatocytes compared with vehicle (p < 0.001; Fig. 4c). In contrast, treatment of hepatocytes with both corticosterone and RU486 (10−6 mol/l) for 48 h failed to increase H6PDH activity (Fig. 4b) or H6pdh mRNA levels (Fig. 4c). Moreover, co-treatment with corticosterone and RU486 also blocked the corticosterone-induced changes in 11ß-Hsd1, Pepck and G6Pase mRNA expression (Fig. 4d) in these intact cells. However, no changes in H6pdh mRNA expression were observed after 48 h in cells exposed to a relatively high concentration of insulin (1 × 10−6 mol/l; Fig. 4c). Extending our findings on H6PDH activity, corticosterone-induced expression of G6pt mRNA in primary hepatocytes was reduced significantly by RU486 (Fig. 5a, b).

Effects of corticosterone and insulin on primary cultures of db/db mouse hepatocytes. Levels of H6PDH production (a) and activity (b) in cells treated with corticosterone for 48 h. In (b), white circles, control; black squares, corticosterone; black triangles, corticosterone + RU486. The levels of H6pd, 11β-Hsd1, G6Pase and Pepck mRNA expression (c, d) in cells treated with corticosterone (1 × 10−6 mol/l) or insulin (1 × 10−6 mol/l) in the presence or absence of RU486 (1 × 10−6 mol/l) for 48 h. In (d), white bars, 11β-Hsd1; black bars, G6Pase; grey bars, Pepck. Production and relative quantification of H6PDH protein levels are expressed relative to the amount of GAPDH. Relative H6PDH activity was measured on the basis of NADPH production at 5 min intervals for 20 min. Values are the mean ± SEM from three separate culture preparations. *p < 0.05, **p < 0.01, ***p < 0.001 vs controls; †† p < 0.01, ††† p < 0.001 vs corticosterone-treated hepatocytes. C, control; B, corticosterone; I, insulin; RU, RU486

a Effect of different doses of corticosterone on G6pt mRNA expression in primary cultures of hepatocytes from db/db mice. b Hepatocytes were incubated with corticosterone (10−6 mol/l) or insulin (10−6 mol/l) for 48 h in the absence or present of RU486. Expression and relative quantification of G6pt mRNA levels expressed relative to the amount of 18S in vehicle-treated cells. Values are the mean ± SEM from three separate culture preparations. **p < 0.01, ***p < 0.001 vs vehicle-treated cells. C, control; B, corticosterone; I, insulin; RU, RU486
The effects of H6PDH siRNA and G6PT inhibitor (CA) on H6PDH and 11β-HSD1
As shown in Fig. 6, H6PDH protein and H6pd mRNA levels were markedly reduced in intact hepatocytes transfected with H6pd siRNA in comparison to cells transfected with siRNA control, respectively (p < 0.001, Fig. 6a, b). Similarly, in comparison with control levels, 11β-HSD1 reductase activity in primary hepatocytes after transfection with H6pdh siRNA was decreased by 44% (p < 0.01; Fig. 6c). Furthermore, H6pd siRNA attenuated the corticosterone-mediated increase in H6PDH protein and 11β-HSD1 reductase activity in these intact cells in comparison with control levels (Fig. 6), indicating that suppression of H6PDH by siRNA exerted effects comparable with those of RU486 on reducing the corticosterone-induced H6PDH levels and 11ß-HSD1 activity.

Suppression of H6pd expression by H6pd siRNA decreased 11β-HSD1 reductase activity and attenuated the effects of corticosterone on H6PDH and 11β-HSD1 in primary culture of hepatocytes. Cells were transfected with either H6pd siRNA (si) or negative control siRNA (N) and cultured with or without corticosterone (b; 1 × 10−6 mol/l) for 48 h. a Western blots showing effect of H6pd siRNA inhibition on H6PDH protein in cells. b H6pd and 11β-Hsd1 mRNAs were measured by real-time RT-PCR. White bars, H6pd; black bars, 11β-Hsd1. c 11ß-HSD1 reductase activity was expressed as the percentage of [3H]DHC converted to [3H]corticosterone (b) in medium from hepatocytes. Values are the mean ± SEM from three separate culture preparations. **p < 0.01, ***p < 0.001 vs negative control; ††† p < 0.001 vs corticosterone-treated hepatocytes
The effects of the G6PT inhibitor CA on H6PDH and 11β-HSD1 are shown in Fig. 7. CA significantly reduced H6PDH protein production in primary hepatocytes compared with vehicle treatment for 48 h (Fig. 7a). Consequently, the mRNA levels of H6pd were decreased by 1.6-fold in these intact cells after CA treatment (Fig. 7b). In agreement with reduction of H6PDH, CA treatment also reduced 11β-Hsd1 mRNA expression as compared with that of controls (p < 0.01; Fig. 7b).

The effects of CA on H6PDH and 11β-HSD1 in primary cultures of hepatocytes from db/db mice. a Primary hepatocytes were incubated with increased concentrations of the G6PT inhibitor CA (0–200 μmol/l) for 48 h and H6PDH protein levels were determined by western blot analysis. b Hepatocytes were incubated with CA (150 μmol/l) for 48 h. H6pdh and 11β-Hsd1 mRNA were measured by real-time RT-PCR. Values are the mean ± SEM from three separate culture preparations. **p < 0.01 vs vehicle-treated hepatocytes. White bars, vehicle; black bars, CA
Discussion
We found that the phenotype of type 2 diabetes in db / db mice was associated with the induction of hepatic H6PDH activity and gene expression. H6PDH activity and protein expression were elevated in liver microsomes from db / db mice, and this was accompanied by the induction of hepatic 11ß-HSD1 reductase activity. This elevated production of hepatic H6PDH corresponded with increased hepatic Pepck and G6pase mRNA levels, high blood glucose and insulin resistance, suggesting that elevated hepatic H6PDH levels may contribute to the development of type 2 diabetes in our animal model. Induction of hepatic H6pd expression could increase the generation of the crucial co-factor NADPH and thus enhance 11ß-HSD1 upregulation of local GC production, which would lead to gluconeogenesis linked to the development of hyperglycaemia in diabetic animals [15, 21]. Moreover, H6pd and 11ß-Hsd1 mRNA levels were also increased in the adipose fat of db / db mice. Our present results are consistent with recent reports that adipose H6pd and 11ß-Hsd1 mRNA were increased in patients with type 2 diabetes [43]. To our knowledge, hepatic H6PDH has not been measured in patients with type 2 diabetes. Our findings are also supported by earlier reports that increased hepatic 11ß-HSD1 production may contribute to the pathogenesis of type 2 diabetes in db / db mice [21, 44].
In db / db mice, excess corticosterone production has been shown to be crucial for the development of hyperglycaemia, insulin resistance and obesity, and adrenalectomy reverses these changes [6–8]. In the present study, we observed that the induction of hepatic H6PDH activity and H6pd gene expression was associated with the elevated circulating levels of corticosterone. This was validated using isolated hepatocytes from db / db mice in which corticosterone increased H6PDH activity at the transcriptional level. Increased H6PDH activity could then provide the cofactor NADPH to 11ß-HSD1, suggesting that GCs could positively promote their own pre-receptor metabolism through activation of NADPH availability to 11β-HSD1 reductase activity. Our results are consistent with those of earlier reports that cortisone increased H6PDH activity leading to 11ß-HSD1 amplifying cortisol production in rat liver microsomal fractions [34]. These data support our suggestion that the induction of hepatic H6PDH production may, in part, result from excess circulating corticosterone action.
It is well known that pharmacological blockade of GR by RU486 antagonises hypercortisolaemia-related hyperglycaemia and insulin resistance in db / db mice as well as in patients with Cushing’s syndrome [5, 8]. In the present study, we found that treatment of db / db mice with RU486 markedly reduced production of hepatic H6PDH and 11β-HSD1 and improved hyperglycaemia and insulin resistance. Moreover, we observed that RU486 treatment reversed corticosterone-induced production of H6PDH activity and NADPH and prevented activation of 11β-HSD1 and PEPCK production, consistent with a recent study reporting that the dexamethasone-induced upregulation of H6pd mRNA can be blocked by RU486 in mouse 3T3-L1 adipocytes [45]. Moreover, we also observed that suppression of H6pd by siRNA decreased 11β-HSD1 reductase activity and showed effects comparable with those of RU486 on blocking the corticosterone-induced H6PDH and 11ß-HSD1 in intact hepatocytes. Reduction of hepatic H6PDH expression by RU486 could decrease the ability of H6PDH utilising NADP to regenerate NADPH, thereby limiting the intraluminal NADPH exposure to 11β-HSD1 linked to the reduction of 11β-HSD1 and the improvement of glycaemic variables. This is in agreement with a recent study reporting that the role of RU486 in glycaemic homeostasis is associated with reduction of hepatic 11β-HSD1 levels in type 2 diabetic mice [46].
In addition, the current study also observed a positive relationship between hepatic H6PDH activity and G6PT abundance in db / db mice and db / + controls. Indeed, earlier studies showed that H6PDH requires G6P transport to maintain its metabolic substrate G6P availability within the ER lumen [30, 47, 48]. We observed that the induction of H6PDH production is associated with an elevation of G6pt mRNA levels with increased hepatic intraluminal G6P uptake. In contrast, decreased hepatic G6PT abundance after RU486 treatment reduced the intraluminal hepatic G6P uptake with corresponding suppression of H6PDH activity. In agreement with the reduction of H6PDH levels, the current study also showed that the G6PT inhibitor CA, at doses used in vitro [37, 38], markedly reduced H6PDH levels and led to the suppression of 11β-HSD1 in intact mouse hepatocytes, indicating that G6PT is required to maintain H6PDH levels. Our findings are consistent with recent studies reporting that G6P availability in the ER lumen was reduced, leading to decreased H6PDH and 11β-HSD1 activity in mice with global deletion of G6PT [48]. This is also indirectly supported by a recent study demonstrating that the G6PT inhibitor S3483 (derived from CA) decreased G6P-induced H6PDH levels and resulted in the inhibition of reductase activity of 11β-HSD1 in rat liver microsomes [33, 49]. These data support our hypothesis that suppression of G6PT production in hepatocytes may be an additional mechanism of the RU486-mediated reduction of H6PDH production. Moreover, we also observed that RU486 prevented corticosterone-mediated induction of G6PT production in mouse hepatocytes, suggesting that endogenous GCs could exert positive effects on G6PT production in liver that is likely to occur through a GR-mediated mechanism. These findings are supported by a recent study which showed that there are three GC response elements present in the promoter region of the G6pt gene [50]. This is also in agreement with previous studies showing that the GR signalling pathway was required for the regulation of 11β-HSD1 and PEPCK production by GCs [15, 51].
In summary, we showed that increased hepatic H6PDH activity and G6PT expression may contribute to 11β-HSD1 upregulation of local GC action linked to the development of type 2 diabetes. We also found that the induction of hepatic G6PT and H6PDH production may be mediated, at least in part, through the action of elevated circulating GCs. The benefits of RU486 in db / db mice may be associated with the endogenous inactivation of 11β-HSD1 amplifying tissue GC action through reducing production of H6PDH and G6PT. These findings raise the possibility that tissue-specific modulation of H6PDH and G6PT production may be a new strategy to be coupled with the investigation of 11β-HSD1 as a therapeutic target in the metabolic syndrome.
Abbreviations
- 11β-HSD1:
-
11β-Hydroxysteroid dehydrogenase type 1
- CA:
-
Chlorogenic acid
- 11-DHC:
-
11-Dehydrocorticosterone
- ER:
-
Endoplasmic reticulum
- GC:
-
Glucocorticoid
- G6P:
-
Glucose 6-phosphate
- G6Pase:
-
Glucose-6-phosphatase
- G6PT:
-
Glucose-6-phosphate transporter
- GR:
-
Glucocorticoid receptor
- H6PDH:
-
Hexose-6-phosphate dehydrogenase
- ITT:
-
Insulin tolerance test
- TLC:
-
Thin layer chromatography
References
Rizza RA, Mandarino LJ, Gerich JE (1982) Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 54:131–138
Bjorntorp P, Rosmond R (2000) Obesity and cortisol. Nutrition 16:924–936
Kahn BB, Flier JS (2000) Obesity and insulin resistance. J Clin Invest 106:473–481
Havel PJ, Busch BL, Curry DL et al (1996) Predominately glucocorticoid agonist actions of RU-486 in young specific-pathogen-free Zucker rats. Am J Physiol 271:R710–R717
Friedman JE, Sun Y, Yun JS (1997) Phosphoenolpyruvate carboxykinase (GTP) gene transcription and hyperglycemia are regulated by glucocorticoids in genetically obese db/db mice transgenic mice. J Biol Chem 272:31475–31481
Bamberger CM, Schulte HM, Chrousos GP (1996) Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev 17:245–261
Shimomura Y, Bray GA, Lee M (1987) Adrenalectomy and steroid treatment in obese (ob/ob) and diabetic (db/db) mice. Horm Metab Res 19:295–299
Nieman LK, Chrousos GP, Kellner C et al (1985) Successful treatment of Cushing’s syndrome with the glucocorticoid antagonist RU486. J Clin Endocrinol Metab 61:536–540
Seckl JR, Walker BR (2001) Minireview: 11 beta-hydroxysteroid dehydrogenase type: a tissue-specific amplifier of glucocorticoid action. Endocrinology 142:1371–1376
Jamieson PM, Chapman KE, Edwards CR et al (1995) 11 beta-hydroxysteroid dehydrogenase is an exclusive 11 beta-reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology 136:4754–4761
Bujalska IJ, Kumar S, Stewart PM (1997) Does central obesity reflect Cushing’s disease of the omentum? Lancet 349:1210–1213
Stewart PM, Boulton A, Kumar S, Clark PM, Shackleton CH (1999) Cortisol metabolism in human obesity: impaired cortisone→cortisol conversion in subjects with central adiposity. J Clin Endocrinol Metab 84:1022–1027
Napolitano A, Voice MW, Chapman KE et al (1998) 11β-hydroxysteroid dehydrogenase 1 in adipocytes: expression is differentiation-dependent and hormonally regulated. J Steroid Biochem Mol Biol l64:251–260
Rajan V, Edwards CR, Seckl JR (1996) 11β-Hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. J Neurosci 16:65–70
Liu Y, Nakagawa Y, Friedman TC et al (2005) Increased glucocorticoid receptor and 11β-hydroxysteroid dehydrogenase type 1 expression in hepatocytes may contribute to the phenotype of type 2 diabetes in db/db mice. Diabetes 54:32–40
Tiosano D, Eisentein I, Hochberg Z et al (2003) 11beta-Hydroxysteroid dehydrogenase activity in hypothalamic obesity. J Clin Endocrinol Metab 88:379–384
Masuzaki H, Paterson J, Flier JS et al (2001) A transgenic model of visceral obesity and the metabolic syndrome. Science 294:2166–2170
Paterson JM, Morton NM, Fievet C et al (2010) Metabolic syndrome without obesity: hepatic overexpression of 11β-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA 101:7088–7093
Kotelevtsev Y, Holmes MC, Burchell A et al (1997) 11beta-Hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid-inducible responses and resist hyperglycemia on obesity or stress. Proc Natl Acad Sci USA 94:14924–14929
Alberts P, Engblom L, Edling N et al (2002) Selective inhibition of 1beta-hydroxysteroid dehydrogenase type 1 decrease blood glucose concentrations in hyperglycaemic mice. Diabetologia 45:1528–1532
Liu Y, Yan C, Wang Y et al (2006) Liver X receptor agonist T0901317 inhibition of glucocorticoid receptor expression in hepatocytes may contribute to the amelioration of diabetic syndrome in db/db mice. Endocrinology 147:5061–5068
Wang SJ, Birtles S, de Schoolmeeter J et al (2006) Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 reduces food intake and weight gain but maintains energy expenditure in diet-induced obese mice. Diabetologia 49:1333–1337
Andrews RC, Rooyackers O, Walker BR (2003) Effects of the 11beta-hydroxysteroid dehydrogenase inhibitor carbenoxolone on insulin sensitivity in men with type 2 diabetes. J Clin Endocrinol Metab 88:285–291
Odermatt A, Arnold P, Stauffer A et al (1999) The N-terminal anchor sequences of 11beta-hydroxysteroid dehydrogenases determine their orientation in the endoplasmic reticulum membrane. J Biol Chem 274:28762–28770
Mziaut H, Korza G, Hand AR et al (1999) Targeting proteins to the lumen of endoplasmic reticulum using N-terminal domains of 11beta-hydroxysteroid dehydrogenase and the 50-kDa esterase. J Biol Chem 274:14122–14129
Mason PJ, Stevens D, Diez A et al (1999) Human hexose-6-phosphate dehydrogenase (glucose 1-dehydrogenase) encoded at 1p36: coding sequence and expression. Blood Cells Mol Dis 25:30–37
Ozols J (1993) Isolation and the complete amino acid sequence of lumenal endoplasmic reticulum glucose-6-phosphate dehydrogenase. Proc Natl Acad Sci USA 90:5302–5306
Hewitt KN, Walker EA, Stewart PM (2005) Minireview: hexose-6-phosphate dehydrogenase and redox control of 11beta-hydroxysteroid dehydrogenase type 1 activity. Endocrinology 146:2539–2543
McCormick KL, Wang X, Mick GJ (2006) Evidence that the 11 beta-hydroxysteroid dehydrogenase (11beta-HSD1) is regulated by pentose pathway flux. Studies in rat adipocytes and microsomes. J Biol Chem 281:341–347
van Schaftingen E, Gerin I (2002) The glucose-6-phosphatase system. Biochem J 362:513–532
Chou JY, Matern D, Mansfield BC et al (2002) Type I glycogen storage diseases: disorders of the glucose-6-phosphatase complex. Curr Mol Med 2:121–143
Atanasov AG, Nashev LG, Schweizer RA et al (2004) Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett 571:129–133
Banhegyi G, Benedetti A, Fulceri R et al (2004) Cooperativity between 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase in the lumen of the endoplasmic reticulum. J Biol Chem 279:27017–27021
Draper N, Walker EA, Bujalska IJ et al (2003) Mutations in the genes encoding 11beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet 34:434–439
Lavery GG, Walker EA, Araper N et al (2006) Hexose-6-phosphate dehydrogenase knock-out mice lack 11beta-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J Biol Chem 281:6546–6551
Liu YJ, Wang Y, Nakagawa Y et al (2008) Inhibition of hepatic glucocorticoid receptor and hexose-6-phosphate dehydrogenase expression ameliorates diet-induced insulin resistance and obesity in mice. J Mol Endocrinol 41:53–64
Hsu CL, Huang SL, Yen GC (2006) Inhibitory effect of phenolic acids on the proliferation of 3T3-L1 preadipocytes in relation to their antioxidant activity. J Agric Food Chem 54:4191–4197
Belkaid A, Currie JC, Desganes J et al (2006) The chemopreventive properties of chlorogenic acid reveal a potential new role for the microsomal glucose-6-phosphate translocase in brain tumor progression. Cancer Cell Int 6:7
Marcolongo P, Senesi S, Gava B et al (2008) Metyrapone prevents cortisone-induced preadipocyte differentiation by depleting luminal NADPH of the endoplasmic reticulum. Biochem Pharmacol 76:382–390
Nammi S, Dembele K, Nyomba BLG (2007) Increased 11beta-hydroxysteroid dehydrogenase type-1 and hexose-6-phosphate dehydrogenase in liver and adipose tissue of rat offspring exposed to alcohol in utero. Am J Physiol Regul Integr Comp Physiol 292:R1101–R1109
Liu Y, Nakagawa Y, Wang Y et al (2003) Leptin activation of corticosterone production in hepatocytes may contribute to the reversal of obesity and hyperglycemia in leptin-deficient ob/ob mice. Diabetes 52:1409–1416
Hiraiwa H, Pan CJ, Lin B et al (1999) Inactivation of the glucose 6 phosphate transporter causes glycogen storage disease type 1b. J Biol Chem 274:5532–5536
Uckaya G, Karadurmus N, Kutlu O et al (2008) Adipose tissue 11-beta-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase gene expressions are increased in patients with type 2 diabetes mellitus. Diabetes Res Clin Pract 2:S135–140
Nakano S, Inada Y, Masuzaki H et al (2007) Bezafibrate regulates the expression and enzyme activity of 11beta-hydroxysteroid dehydrogenase type 1 in murine adipose tissue and 3T3-L1 adipocytes. Am J Physiol Endocrinol Metab 292:E1213–1222
Balachandran A, Guan H, Sellan M et al (2008) Insulin and dexamethasone dynamically regulate adipocyte 11beta-hydroxysteroid dehydrogenase type 1. Endocrinology 149:4069–4407
Taylor AI, Frizzell N, McKillop AM et al (2009) Effect of RU486 on hepatic and adipocyte gene expression improves diabetes control in obesity-type 2 diabetes. Horm Metab Res 41:899–904
Piccirella S, Czegle I, Lizak B et al (2006) Uncoupled redox systems in the lumen of the endoplasmic reticulum. Pyridine nucleotides stay reduced in an oxidative environment. J Biol Chem 281:4671–4677
Warlker EA, Ahmed A, Lavery GG et al (2007) 11beta-Hydroxysteroid dehydrogenase type 1 regulation by intracellular glucose 6-phosphate provides evidence for a novel link between glucose metabolism and hypothalamo-pituitary–adrenal axis function. J Biol Chem 282:27030–270306
Marcolongo P, Piccirella S, Schesi S et al (2007) The glucose-6-phosphate transporter-hexose-6-phosphate dehydrogenase-11beta-hydroxysteroid dehydrogenase type 1 system of the adipose tissue. Endocrinology 148:2487–2495
Hiraiwa H, Chou YJ (2001) Glucocorticoids activate transcription of the gene for the glucose-6-phosphate transporter, deficient in glycogen storage disease type 1b. DNA Cell Biol 20:447–453
Opherk C, Tronche F, Kellendonk C et al (2004) Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol 18:1346–1353
Acknowledgements
Y. Liu is supported by NIH grants KO1 DK073272 and SC1DK087655. T. C. Friedman is supported by NIH grants R01 DA14659, K21 DA00276 and an endowment grant S21MD000103-NIH/NCMHD.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Y. Nakagawa and Y. Wang contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
Oligonucleotide sequences of primers used for real-time PCR (PDF 61 kb)
Rights and permissions
About this article
Cite this article
Wang, Y., Nakagawa, Y., Liu, L. et al. Tissue-specific dysregulation of hexose-6-phosphate dehydrogenase and glucose-6-phosphate transporter production in db/db mice as a model of type 2 diabetes. Diabetologia 54, 440–450 (2011). https://doi.org/10.1007/s00125-010-1956-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1956-9