
Abstract
Aims/hypothesis
Cytokines stimulate nitric oxide production in pancreatic beta cells, leading to endoplasmic reticulum (ER) stress and apoptosis. Treatment of beta cells with glucose and NEFA induces nitric oxide synthase (NOS) as well as ER stress. However, the role of NO in glucolipotoxicity-induced ER stress in beta cells is not clear.
Methods
We studied the effect of high glucose and palmitate levels on NOS isoform production in rat and Psammomys obesus islets and in insulinoma-1E beta cells. The effects of neuronal NOS (nNOS) inhibition by small interfering RNA or by N ω-nitro-l-arginine methyl ester (l-NAME) on beta cell function, ER stress and apoptosis under conditions of glucolipotoxicity were investigated.
Results
Overnight incubation of rat and P. obesus islets at 22.2 mmol/l glucose with 0.5 mmol/l palmitate induced the production of nNOS but not inducible NOS (iNOS), in contrast with the robust stimulation of iNOS by cytokines. NOS inhibition by l-NAME did not prevent the decrease in glucose-stimulated insulin secretion and proinsulin biosynthesis or the depletion of islet insulin content observed under conditions of glucolipotoxicity. Moreover, treatment of beta cells with palmitate and l-NAME together resulted in marked activation of the IRE1α and PERK pathways of the unfolded protein response. This was associated with increased JNK phosphorylation and apoptosis in islets and beta cells. Moreover, partial nNos knockdown increased JNK phosphorylation and CHOP production, leading to apoptosis.
Conclusions/interpretation
In beta cells subjected to glucolipotoxic conditions, chronic inhibition of NOS exacerbates ER stress and activates JNK. Therefore, induction of nNOS is an adaptive response to glucolipotoxicity that protects beta cells from stress and apoptosis.
Similar content being viewed by others


Introduction
In type 2 diabetes, elevated blood glucose and NEFA cause beta cell dysfunction and apoptosis, leading to exacerbation of diabetes by a process called glucolipotoxicity [1]. Glucolipotoxicity is believed to involve both oxidative and endoplasmic reticulum (ER) stresses [2–4]. The ER stress response, also called the unfolded protein response (UPR), is a complex signalling network that attempts to restore normal ER function by translation attenuation, degradation of misfolded proteins and increased protein folding capacity through augmented transcription and translocation of ER chaperones. UPR is initiated by three ER transmembrane sensor proteins: IRE1 (inositol requiring ER-to-nucleus signal kinase 1), the pancreatic ER kinase PERK (double-stranded RNA-activated protein kinase-like ER-associated kinase) and activating transcription factor 6 [5–7]. Sustained activation of the UPR may lead to cell death through activation of apoptotic signals by c-Jun N-terminal kinase (JNK, downstream to IRE1α) and CCAAT/enhancer binding protein homologous protein (CHOP, downstream to PERK) [3, 8, 9].
NEFAs, mainly palmitate, are potent inducers of ER stress in beta cells [3, 10–12]. We have previously shown that glucose amplifies NEFA-induced ER stress through activation of the mammalian target of rapamycin complex 1 (mTORC1), which leads to apoptosis by activating JNK [13]. However, yet unknown additional mechanisms probably participate in glucolipotoxic ER stress.
Nitric oxide (NO), a mediator of protein nitrosylation and reactive oxygen species, can interfere with protein disulphide bonding and thereby result in protein misfolding and ER stress [14, 15]. Indeed, NO has been suggested to play an important role in stroke and brain ischaemia–reperfusion injury by inducing ER stress [15]. Moreover, NO is an important mediator of beta cell cytokine toxicity in type 1 diabetes, with induction of ER stress and apoptosis [16, 17].
Several studies have suggested that exposure of islets to high glucose or NEFA levels induces production of inducible nitric oxide synthase (iNOS), causing attenuation of the insulin response to glucose [18–22]. Thus, iNOS-dependent NO generation with subsequent induction of ER stress could be the common denominator of beta cell apoptosis induced by inflammatory cytokines and glucolipotoxicity. Nevertheless, it has not yet been shown convincingly that NO production is the cause of beta cell dysfunction and apoptosis during glucolipotoxicity. Moreover, other investigators have failed to find iNos (also known as Nos2) mRNA expression, nitrite production or activation of the nuclear factor κB (NFκB) pathway, an important mediator of cytokine-induced beta cell apoptosis, in rat beta cells treated with NEFA [23–25]. Therefore, the role of NO as a mediator of beta cell glucolipotoxicity remains controversial.
This motivated us to study the effects of glucose and palmitate on the production of different NOS isoforms in both rat and Psammomys obesus islets. P. obesus is a gerbil model of type 2 diabetes mellitus characterised by increased susceptibility to beta cell dysfunction and apoptosis in response to high glucose and NEFA levels, as found in islets of patients with type 2 diabetes [26]. We [27] and others [19] have previously shown that NO is a negative regulator of insulin secretion and that short-term NOS inhibition amplifies the response to acute stimulation by glucose and mitochondrial fuels. In the present study we describe the effects of pharmacological and genetic inhibition of neuronal nitric oxide synthase (nNOS) on beta cell function, ER stress and apoptosis under conditions of glucolipotoxicity.
Methods
Islet isolation and beta cell line culture
Diabetes-prone P. obesus (Hebrew University Colony; Harlan, Jerusalem, Israel) aged 2.5–3.5 months were fed a low-energy (9.96 kJ/g) diet (Koffolk, Petach-Tikva, Israel), which maintains normoglycaemia (3–5 mmol/l in this species). Islets were isolated from normoglycaemic P. obesus and Wistar rats (Harlan, Jerusalem, Israel) by collagenase digestion (Collagenase P; Roche Diagnostics, Mannheim, Germany) as described [28], and used after repeated washes with Hanks’ balanced salt solution. Islets were either incubated in suspension for 16 h or cultured in RPMI 1640 medium (Biological Industries, Beit-Haemek, Israel) with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mmol/l l-glutamine (Biological Industries) on extracellular matrix (ECM)-coated plates (Novamed, Jerusalem, Israel) [28] for 5 days at different glucose concentrations with and without 0.5 mmol/l palmitate, as indicated. Insulinoma 1E (INS-1E) beta cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 1 mmol/l sodium pyruvate, 2 mmol/l l-glutamine, 10 mmol/l HEPES, 0.05 mmol/l 2-mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin. Animal use was approved by the Institutional Animal Care and Use Committee of the Hebrew University and the Hadassah Medical Organization.
Experimental methods
INS-1E cells and rat and P. obesus islets were incubated in RPMI medium with 0.55% (wt/vol.) BSA with or without palmitate at various glucose concentrations for different periods of time, as indicated in the figure legends. The palmitate–BSA solution was prepared as described [29]. Briefly, the sodium salt of palmitic acid was dissolved at a concentration of 10 mmol/l in 11% BSA in a shaking water-bath at 37°C for 16 h. The BSA that was used for solubilisation of palmitate was free of fatty acids and endotoxins. The pH of the solution was adjusted to 7.4 with 1 mmol/l NaOH, then filtered through a 0.2 μm filter and stored at −20°C. The palmitate–BSA solution was diluted 1:20 in the incubation medium, the molar ratio of palmitate to BSA being 6:1. The effects of NOS inhibition on beta cell function, stress and apoptosis were studied by treating INS-1E cells and islets with 1 and 10 mmol/l of the NOS inhibitor N ω-nitro-l-arginine methyl ester (l-NAME) for different periods of time or by nNos knockdown. In part of the experiments, a cytokine cocktail (IL-1β 1 × 103 U/μl, TNFα 500 U/μl, IFNγ 1 × 103 U/μl; PeproTech, Rocky Hill, NJ, USA) was used as positive control. All reagents were purchased from Sigma (Rehovot, Israel).
nNos knockdown
nNos knockdown was performed by transient transfection of small interfering RNA oligos for nNos into INS-1E cells, which were plated in 24 well plates and grown overnight to approximately 70% confluence. Transfection was performed in serum-free RPMI medium using Lipofectamine (Invitrogen, Philadelphia, PA, USA) according to the manufacturer’s instructions. The sequence targeted corresponded to bases 3323–3345 of the nNos mRNA (accession number NM_052799). The sense and antisense oligonucleotides were 5′-CGAGGACCUCGUGAAUGCACUCAUU and 5′-AAUGAGUGCAUUCACGAGGUCCUCG, respectively. Six hours after transfection the medium was replaced, and after an additional 24 h the cells were incubated overnight at different glucose concentrations with and without palmitate followed by extraction and analysis for the production of nNOS and different stress markers and for beta cell apoptosis.
Insulin secretion and proinsulin biosynthesis
Groups of 25 islets were cultured on ECM-coated plates for 5 days with treatments as indicated. Insulin secretion of cultured islets was assessed at 24 h and 5 days by static 1 h incubations at 1.7 and 16.7 mmol/l glucose. Medium was collected at the end of the incubations, centrifuged and frozen at −20°C pending insulin assay.
Proinsulin biosynthesis was studied by labelling islets in the centre of the plate in 25 μl fresh KRB HEPES (KRBH)-BSA buffer [28]. The buffer contained different treatments at the same glucose concentration as for the chronic incubation and 0.925 ΜΒq l-[2,3,4,5-3H]leucine (4.44 × 1012 Bq/mmol; ARC, St Louis, MO, USA). After a 15 min pulse at 37°C, leucine incorporation was terminated by adding 1 ml ice-cold glucose-free KRBH-BSA buffer, removal of the islets by scraping and rapid centrifugation. The islet pellet was subjected to immunoprecipitation as described [27].
Insulin assay
Insulin immunoreactivity in P. obesus was determined using a human insulin RIA kit from Linco Research (St Charles, MO, USA) and rat insulin using a rat RIA kit (Linco Research).
Measurement of NOS activity and medium nitrites
NOS activity was determined in rat islets and cerebellum using the NOSdetect assay kit (Stratagene, Cedar Creek, TX, USA) according to the manufacturer’s instructions. The assay is based on the measurement of l-[14C]citrulline, formed by NOS in equimolar concentration to NO. Islets were incubated at different glucose concentrations with and without 0.5 mmol/l palmitate for 16 h, then washed and collected in 300 μl ice-cold homogenisation buffer. The islets were sonicated and stored at −80°C for subsequent measurement of NOS activity. Rat cerebellum homogenate was used as a positive control. Radioactivity was counted in Optiphase HiSafe scintillation cocktail (PerkinElmer, Waltham, MA, USA). Total NOS activity was determined by subtracting the background readings (with l-NAME) from the radioactive counts obtained without l-NAME. Calcium/calmodulin is a cofactor for nNOS activity, whereas iNOS is calcium-independent. iNOS activity was determined by performing the NOS enzymatic reaction in the presence of the calcium chelator EGTA. This was subtracted from total NOS activity to give the nNOS activity [19]. Protein was determined with the Bradford method [30].
Nitrite production was measured in the incubation medium of INS-1E cells under different experimental conditions using the Griess reagent, as described [31]. Palmitate-supplemented medium was used as a blank.
Western blot analysis
Protein production and phosphorylation were studied by western blotting using antibodies against iNOS (Sigma), nNOS, phospho-stress-activated protein kinase (SAPK)/JNK (Thr183/Tyr185), SAPK/JNK, phospho-c-JUN, total IRE1α, total and phospho-eIF2α, phospho-PERK, cleaved and non-cleaved caspase 3 (all from Cell Signaling Technology, Beverley, MA, USA), phospho-IRE1α (kindly provided by F. Urano, University of Massachusetts, Boston, MA, USA), growth arrest- and DNA damage-inducible gene 153 (GADD153)/CHOP and GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Peroxidase-conjugated AffiniPure goat anti-rabbit and anti-mouse IgG from Jackson Immunoresearch Laboratories (West Grove, PA, USA) were used as secondary antibodies. Immunoreactive bands were visualised by chemiluminescence using ECL-Plus (Biological Industries, Rehovot, Israel). We used x-ray film densitometry for quantification (ImageMaster VDS-CL; Amersham Pharmacia Biotech, Uppsala, Sweden). Immunoblots were scanned and signals were quantified using TINA software (University of Manchester, Manchester, UK).
Quantitative real-time RT-PCR
RNA was extracted from INS-1E cells using TRI reagent (Biolab, Jerusalem, Israel) and reverse-transcribed using Moloney murine leukaemia virus reverse transcriptase (Promega, Madison, WI, USA). Quantitative real-time RT-PCR (qPCR) for spliced X-box binding protein-1 (Xbp1) was performed on a Prism 7000 Sequence Detection System using the Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Samples were analysed in triplicate and corrected for GAPDH. The following oligonucleotides were used for the PCR reaction: spliced Xbp1, forward 5′-GAGTCCGCAGCAGGTG-3′, reverse 5′-GAAGAGGCAACAGCGTCAGA-3′; Gadph, forward 5′-AGTTCAACGGCACAGTCAAG-3′, reverse 5′-TACTCAGCACCAGCATCACC-3′.
Apoptosis ELISA assay
Cells plated in 96-well plates were grown in RPMI 1640 containing 11.1 mmol/l glucose until they reached 70% confluence. They were then treated with 0.55% (wt/vol.) BSA with or without 0.5 mmol/l palmitate at 3.3 and 27.8 mmol/l glucose and l-NAME for 16 h. The cells were lysed and oligonucleosomes in the cytosol, indicative of apoptosis-induced DNA degradation, were quantified using the Cell Death Detection ELISA Plus kit (Roche Diagnostics) according to the manufacturer’s instructions.
Data presentation and statistical analysis
Data shown are mean ± SEM. Statistical significance of differences between groups was determined by one-way ANOVA followed by the Newman–Keuls test using the InStat statistical program (GraphPad Software, San Diego, CA, USA). A paired sample t test was used when the difference between a reference (taken as 100%) and test was analysed. A p value of less than 0.05 was considered significant.
Results
Effects of glucose and palmitate on NOS isoform production and activity in islets
NOS isoforms were studied in P. obesus and rat islet extracts following incubation for 16 h at 22.2 mmol/l glucose with and without 0.5 mmol/l palmitate. Basal (3.3 mmol/l glucose) nNOS production was low in P. obesus islets; it was not increased in response to 22.2 mmol/l glucose either in rat or P. obesus islets. In contrast, 0.5 mmol/l palmitate induced nNOS at 22.2 and 3.3 mmol/l glucose in both species (Fig. 1a–d). Consistent with these findings, the effect of palmitate in INS-1E cells was concentration-dependent (see Electronic supplementary material [ESM] Fig. 1). In P. obesus islets treated with palmitate, nNOS production was higher at a high glucose concentration than at a low glucose concentration (6.5- and 3.4-fold increase in palmitate-treated P. obesus islets at 22.2 and 3.3 mmol/l glucose, respectively, compared with islets at 3.3 mmol/l glucose alone; Fig. 1a, c).

NOS isoform production in rat and P. obesus islets was assessed by western blotting. P. obesus (a, c, e) and rat (b, d, f) islets were incubated at 3.3 (G3.3) and 22.2 (G22.2) mmol/l glucose with and without 0.5 mmol/l palmitate for 16 h. Results are expressed as mean ± SEM of three individual experiments. Extract of INS-1E cells treated with a cocktail of cytokines (1 × 103 U/μl IL-1β, 500 U/μl TNFα and 1 × 103 U/μl IFNγ) at 11.1 mmol/l glucose for 16 h was used as a positive control for iNOS. *p < 0.05, **p < 0.01, ***p < 0.001 for the difference between the indicated groups or between these groups and untreated islets at 3.3 mmol/l glucose
Whereas treatment with cytokines, as expected, resulted in robust induction of iNOS, there was no effect of palmitate on iNOS in rat or P. obesus islets (Fig. 1e, f). iNOS was also undetectable in INS-1E cells treated with palmitate (not shown).
Consistent with the lack of iNOS induction by palmitate, iNOS activity was undetected in islets incubated at 22.2 mmol/l glucose with palmitate (ESM Fig. 2a). Furthermore, total NOS activity of rat islets treated with palmitate was low: islets incubated at high glucose+palmitate concentrations showed <1% NOS activity compared with rat cerebellar tissue. Moreover, there was only a small increase in nitrites in the medium of INS-1E cells treated with palmitate, whereas cytokines markedly increased nitrite production (ESM Fig. 2b).
Collectively, palmitate increased nNOS, but not iNOS, with a small increase in total NOS activity and nitrite production.
Effect of NOS inhibition on beta cell function in islets exposed to glucolipotoxicity
The induction of nNOS by palmitate and high glucose was more pronounced in P. obesus than in rat islets. P. obesus islets are prone to develop beta cell dysfunction in response to high glucose and palmitate [26]. Therefore, to study the role of NOS, P. obesus islets were cultured on ECM-coated plates at 3.3 mmol/l glucose alone or 22.2 mmol/l glucose with and without 0.5 mmol/l palmitate and 10 mmol/l l-NAME for 5 days.
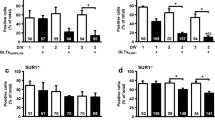
Under these conditions 22.2 mmol glucose did not impair insulin secretion. Both basal and glucose-stimulated insulin secretion and proinsulin biosynthesis were higher at 22.2 than at 3.3 mmol/l glucose (Fig. 2a, b); however, the fold stimulation of insulin secretion was lower in islets cultured at 22.2 mmol/l than at 3.3 mmol/l glucose (2.8- and 5.3-fold stimulation respectively, p < 0.001). Palmitate decreased glucose-stimulated insulin secretion by 37.3% at 24 h (p < 0.01) and 27.5% at 5 days (p < 0.01; Fig. 2a, b). Palmitate induced decreases of 70% in proinsulin biosynthesis and islet insulin content (p < 0.001 for both; Fig. 2c, d). l-NAME did not modify the attenuation of glucose-stimulated insulin secretion and proinsulin biosynthesis, or the depletion of islet insulin content induced by palmitate.

Effect of nNOS inhibition by l-NAME on insulin secretion (a, 24 h; b, 5 days), proinsulin (PI) biosynthesis (c) and insulin content (d) in P. obesus islets. Islets were isolated from normoglycaemic P. obesus and cultured on ECM-coated plates at 3.3 (G3.3) and 22.2 (G22.2) mmol/l glucose with and without 0.5 mmol/l palmitate and 10 mmol/l l-NAME for 5 days. Insulin secretion was analysed by static incubations at 1.7 (white bars) and 16.7 (black bars) mmol/l glucose at 24 h (a) and 5 days (b). Proinsulin biosynthesis was analysed at 5 days after a 15 min pulse with [3H]leucine, as described in the Methods. Results are mean ± SEM of nine individual experiments, each performed on islets pooled from three animals. *p < 0.05, **p < 0.01, ***p < 0.001 for the differences between the indicated groups or between these groups and control islets at 3.3 mmol/l glucose
In summary, NOS inhibition by l-NAME did not prevent the beta cell dysfunction induced by long-term treatment with palmitate and high glucose.
Effect of NOS inhibition on beta cell ER stress response to glucolipotoxicity
Palmitate induces stress in pancreatic beta cells, leading to activation of JNK and consequently apoptosis [13, 32]. We first studied the effects of glucose, palmitate and l-NAME on JNK activation in P. obesus islets (Fig. 3a, b, c). JNK phosphorylation increased in islets treated with palmitate or l-NAME alone, although the effect was relatively small. Addition of both agents stressed the islets synergistically, with major augmentation of JNK phosphorylation at both 3.3 and 22.2 mmol/l glucose. There was a trend for higher JNK phosphorylation in islets treated with palmitate and l-NAME at 22.2 mmol/l glucose relative to 3.3 mmol/l glucose. JNK activation by l-NAME was associated with increased apoptosis (Fig. 3d). Thus, NOS inhibition increased, rather than decreased, JNK activation and apoptosis in islets exposed to glucolipotoxic conditions.

Effect of nNOS inhibition by l-NAME on JNK expression and phosphorylation, and on apoptosis in P. obesus islets. P. obesus islets were incubated at 3.3 and 22.2 mmol/l glucose with and without 0.5 mmol/l palmitate and 10 mmol/l l-NAME for 16 h. a, b, c Phosphorylated and total JNK were assessed by western blotting. d Apoptosis was analysed using an ELISA assay for oligonucleosomes. Results are expressed as mean ± SEM of three individual experiments. *p < 0.05, **p < 0.01, ***p < 0.001 for the difference between the indicated groups or between these groups and control islets at 3.3 mmol/l glucose. AU, arbitrary units
In INS-1E cells, knockdown of nNos induced a moderate (20–40%) decrease in nNOS production (Fig. 4a, ESM Fig. 3a) and nitrite production (ESM Fig. 4). This resulted in increased JNK phosphorylation, CHOP production and augmented beta cell apoptosis (Fig. 4b–e, ESM Fig. 3).

Effects of nNos knockdown in INS-1E beta cells on nNOS production, stress markers and apoptosis. INS-1E cells were transfected with RNAi oligos for nNOS (black bars) or with control, scrambled RNA oligos (white bars), as described in the Methods. The cells were then incubated for 16 h at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate (P). a nNOS production analysed by western blotting. b–d Western blot analysis of CHOP production, phosphorylated and total JNK, cleaved caspase 3 and caspase 3. Representative gels and quantification of nNOS production, JNK phosphorylation (normalised to total JNK) and CHOP production (normalised to GAPDH) are shown as fold of G3.3. e Apoptosis was analysed using the apoptosis ELISA assay. Results are expressed as mean ± SEM of at least three individual experiments. *p < 0.05, **p < 0.01, ***p < 0.001 for the difference between similarly treated nNos knockdown and control groups
JNK is a downstream target of the IRE1α branch of the UPR. IRE1α has a dual role during ER stress: (1) it acts as a kinase, phosphorylating apoptosis signalling kinase 1, thereby activating JNK; and (2) it cleaves the mRNA of the transcription factor XBP1. Spliced Xbp1 is an important regulator of ER function [33, 34]. To further characterise the effects of NOS inhibition on the IRE1α pathway, we analysed IRE1α production and phosphorylation and Xbp1 splicing in INS-1E beta cells incubated with palmitate and l-NAME at different glucose concentrations (Fig. 5 and ESM Fig. 5). Palmitate increased IRE1α level and phosphorylation, JNK and levels of c-JUN phosphorylation (Fig. 5a–c) and spliced Xbp1 (Fig. 5d). l-NAME modestly increased the level of ER stress markers at both 3.3 and 27.8 mmol/l glucose. However, treatment with palmitate and l-NAME together markedly increased spliced Xbp1 levels and JNK and c-JUN phosphorylation, indicating further stimulation of the IRE1α pathway.

Effect of nNOS inhibition by l-NAME on IRE1α pathway activation in INS-1E beta cells. INS-1E cells were incubated at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate and 1 or 10 mmol/l l-NAME for 16 h. a Representative gels showing phosphorylated and total IRE1α, phospho-c-JUN, phosphorylated and total JNK, and GAPDH. Quantification of JNK (b) and c-JUN (c) phosphorylation normalised to total JNK, shown as fold of G3.3. d Spliced Xbp1 levels assessed by quantitative RT-PCR. Each experiment was performed in triplicate. Results in b–d are expressed as mean ± SEM of three individual experiments. *p < 0.05, **p < 0.01, ***p < 0.001 for the difference between the indicated groups or between these and untreated cells at the same glucose concentration
We then studied the effects of glucose, palmitate and l-NAME on the PERK-eIF2α-CHOP branch of the UPR (Fig. 6, ESM Fig. 5). Similarly, l-NAME increased PERK, eIF2α phosphorylation and CHOP level and amplified stimulation of eIF2α and CHOP by palmitate.

Effect of nNOS inhibition by l-NAME on PERK pathway activation in INS-1E beta cells. INS-1E cells were incubated at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate and 1 or 10 mmol/l l-NAME for 16 h. a Representative gels showing phosphorylated PERK (p-PERK), phosphorylated and total eIF2α, CHOP and GAPDH levels. Quantification of eIF2α phosphorylation normalised to total eIF2α (b) and of CHOP normalised to GAPDH, (c) shown as fold of G3.3. Results are expressed as mean ± SEM of three individual experiments. *p < 0.05, **p < 0.01, ***p < 0.001 for the difference between the indicated groups or between these and untreated cells at the same glucose concentration
Consistent with our previous report [13], we found that high glucose amplified the activation by palmitate of IRE1α, JNK and c-JUN and the level of CHOP. Moreover, the amplification of palmitate-induced ER stress by l-NAME was more pronounced at 27.8 mmol/l glucose than at 3.3 mmol/l glucose (Figs 5 and 6).
Effect of NOS inhibition on beta cell apoptosis during glucolipotoxicity
The activation of stress markers by palmitate and l-NAME was associated with increased apoptosis in islets and beta cells (Figs 3 and 7a). Treatment of INS-1E cells with palmitate and a low concentration (1 mmol/l) of l-NAME increased beta cell apoptosis compared with cells incubated with palmitate or l-NAME alone. High glucose amplified the effects of palmitate and l-NAME on beta cell apoptosis: the rate of apoptosis induced by addition of both agents was 1.6-fold higher at 27.8 mmol/l than at 3.3 mmol/l glucose.

Effect of pharmacological inhibition of nNOS and JNK on glucose- and palmitate-induced beta cell apoptosis. a INS-1E cells were incubated for 16 h at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate and 1 mmol/l l-NAME. Apoptosis was assessed using the Cell Death Detection ELISA Plus kit. b INS-1E cells were incubated for 16 h at 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate, 1 mmol/l l-NAME and 20 nmol/l of the JNK inhibitor SP600125. The effect of JNK inhibition on apoptosis was analysed using the apoptosis ELISA assay. Quantification of three individual experiments, each performed in triplicate, is shown. *p < 0.05, **p < 0.01, ***p < 0.001 for the difference between the indicated groups or between these and untreated cells at the same glucose concentration. AU, arbitrary units
JNK inhibition reduces beta cell apoptosis under conditions of ER stress [13]. Indeed, we found that the JNK inhibitor SP600125 completely prevented palmitate-induced beta cell apoptosis and decreased apoptosis in response to palmitate+l-NAME (Fig. 7b).
Thus, nNOS inhibition in beta cells augments glucolipotoxicity-induced ER stress and apoptosis, at least in part through activation of JNK.
Discussion
The aim of this study was to investigate the potential role of NO as a mediator of beta cell ER stress and apoptosis under conditions of glucolipotoxicity. We found that palmitate induced the production of nNOS in INS-1E beta cells and rat and P. obesus islets; in contrast, there was no induction of iNOS even under maximally stressful conditions generated by high glucose and palmitate. The lack of iNOS production in P. obesus islets was unexpected, since they are highly susceptible to glucose and NEFA toxicity [26]. Moreover, total islet NOS activity and nitrite production of beta cells incubated with high glucose and palmitate were low. This was in marked contrast with the robust activation of iNOS by cytokines.
These results are in contrast with previous suggestions that both glucose and palmitate induce iNOS in beta cell lines and rat islets [19–22]. Moreover, it was recently shown that induction of iNOS and consequently excessive generation of NO is responsible for the suppression of glucose-stimulated insulin secretion by palmitate; this was corrected by thiazolidinediones, probably through inhibition of G-protein-coupled receptor 40 (GPR40) [18]. However, other investigators, like ourselves, found that culture of rat islets or beta cells with fatty acids did not increase iNos gene expression [24], intracellular NO or peroxide levels [35]. Similarly, treatment of human islets with NEFA did not affect iNOS expression [36]. Inhibition of NOS by l-NAME failed to restore beta cell function under conditions of glucolipotoxicity (Fig. 2) [35]. Thus, we do not believe that iNOS plays a significant role in glucolipotoxicity-induced beta cell dysfunction.
In contrast with the above, we found that palmitate induced nNOS production; therefore, we studied the role of NOS in the regulation of glucolipotoxicity-induced ER stress and apoptosis. Surprisingly, pharmacological or genetic inhibition of NOS by itself induced beta cell stress and apoptosis and amplified the stress response to palmitate, the effect being more pronounced at a high glucose concentration. Moreover, a modest degree of inhibition of nNos by small interfering RNA was sufficient to worsen beta cell stress and apoptosis. Similar results were obtained using two different small interfering RNA sequences (Fig. 4, ESM Fig. 3), suggesting that the pro-apoptotic effect of nNos knockdown results from nNOS inhibition rather than being an off-target effect. nNos knockdown induced beta cell stress and apoptosis even in the presence of a small decrease in nitrite production. NO measurements probably underestimate the effect of NOS inhibition on NO production due to its trapping in the cell by S-nitrosylation [37], as supported by the observation that the potent NOS inhibitor l-NAME also had a small effect on nitrite production. It is also possible that a minute decrease in NO, although not detected by the nitrite assay, is detrimental to the beta cell. Nonetheless, our findings indicate that nNOS activity is essential for beta cell survival, and its upregulation in islets exposed to glucolipotoxicity is probably an important adaptive mechanism in the defence of beta cells against metabolic stress. Notably, treatment of cardiomyocytes with palmitate increased NO production via constitutive NOS, and its inhibition increased palmitate toxicity [38], consistent with the hypothesis that endogenous production of NO during metabolic stress is protective rather than noxious.
Nitric oxide may thus have a dual, opposing effect on beta cell survival depending on the spatial and temporal production of different NOS isoforms, the NO level achieved and the duration of exposure. NO can be scavenged by a rapid reaction with superoxide (O −2 ) to generate peroxynitrite (ONOO−), which is a potent oxidant and the primary component of nitro-oxidative stress. At high concentrations, ONOO− can undergo cleavage to produce additional highly reactive oxidative species. This initiates a cascade of redox reactions which can trigger apoptosis [39]. Induction of iNOS by cytokines increases NO production massively, thereby leading to nitro-oxidative stress, which contributes to beta cell apoptosis in type 1 diabetes [40]. On the other hand, low levels of NO or of other nNOS product(s) may serve as antioxidant buffers, which dissipate reactive oxygen species. Notably, NOS activation may increase the production of the rate-limiting enzyme for glutathione synthesis and of genes involved in antioxidant defence [41]. Similarly, NO has a dual role in the regulation of neuronal cell survival, being neuroprotective through S-nitrosylation of NMDA receptors, and yet neurodestructive by the formation of peroxynitrite [42]. The dual role of NO in the regulation of beta cell survival is also demonstrated by its opposite effects on ER stress. While nNOS-derived NO alleviates the ER stress response to glucose and NEFA (Figs 4, 5, 6), excessive NO production by cytokines may indeed induce ER stress in beta cells [17]. The contribution of ER stress to cytokine-induced beta cell apoptosis is controversial [43]; nevertheless, nitrosylation of ER-associated calcium channels, such as sarcoendoplasmic reticulum Ca2+-ATPase (SERCA), and consequently calcium depletion in the ER and inactivation of protein disulfide isomerase may lead to protein misfolding [44–46], thereby contributing to beta cell dysfunction and apoptosis [16, 17].
Our data suggest a novel mechanism by which NOS protects the beta cell during glucolipotoxicity by attenuating the ER stress response. Indeed, NOS inhibition by l-NAME amplified palmitate-stimulated ER stress in a concentration-dependent manner, as shown by its activation of the IRE1α and PERK–eIF2α–CHOP pathways of the UPR. Inducers of ER stress cause apoptosis through activation of JNK and CHOP. We found that l-NAME and nNos knockdown increased JNK/c-JUN phosphorylation and CHOP production. Consistent with our previous report [13], treatment with a pharmacological inhibitor of JNK prevented beta cell apoptosis under conditions of glucolipotoxicity and alleviated the deleterious effect of NOS inhibition under these conditions.
The recent report that incubation of INS-1 beta cells with a low concentration of diethylenetriamine nitric oxide adduct (DETA)/NO decreased apoptosis induced by the ER stressor thapsigargin, whereas a high concentration of this compound induced beta cell apoptosis [47], supports our conclusion that NO has a dual role in the regulation of beta cell survival and that low concentrations of NO are protective in the context of ER stress.
The precise mechanism by which NO regulates ER stress is unknown. Pharmacological induction of ER stress has been shown to increase endogenous NO production in the mouse insulinoma 6 (MIN6) beta cell line. This in turn augmented the expression of antioxidant genes, including endoplasmic reticulum oxidoreductin 1 (Ero1) and protein disulphide isomerase (Pdi), which regulate oxidative protein folding in the ER, thereby reducing cellular stress [41].
Based on our results, we suggest the following paradigm for the role of nNOS in beta cell glucolipotoxicity: NEFAs stimulate the production of nNOS, but not iNOS, in islets. Although iNOS induction and subsequent excessive NO production may lead to ER stress and apoptosis, the small amounts of NO (and/or other byproducts) generated by nNOS during glucolipotoxicity attenuate the ER stress response in beta cells. Thus, induction of nNOS by palmitate is an important adaptive response to metabolic stress, protecting the beta cells from apoptosis.
Abbreviations
- CHOP:
-
CCAAT/enhancer binding protein homologous protein
- ECM:
-
Extracellular matrix
- ER:
-
Endoplasmic reticulum
- iNOS:
-
Inducible nitric oxide synthase
- INS-1E:
-
Insulinoma 1E
- IRE1:
-
Inositol requiring ER-to-nucleus signal kinase 1
- JNK:
-
c-Jun N-terminal kinase
- l-NAME:
-
N ω-nitro-l-arginine methyl ester
- NFκB:
-
Nuclear factor κB
- nNOS:
-
Neuronal nitric oxide synthase
- NO:
-
Nitric oxide
- NOS:
-
Nitric oxide synthase
- PERK:
-
Protein kinase-like ER-associated kinase
- UPR:
-
Unfolded protein response
- XBP1:
-
X-box binding protein-1
References
Poitout V, Robertson RP (2007) Glucolipotoxicity: fuel excess and beta-cell dysfunction. Endocr Rev 29:351–366
Kaiser N, Leibowitz G (2009) Failure of beta-cell adaptation in type 2 diabetes: lessons from animal models. Front Biosci 14:1099–1115
Eizirik DL, Cardozo AK, Cnop M (2008) The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29:42–61
Robertson R, Zhou H, Zhang T, Harmon JS (2007) Chronic oxidative stress as a mechanism for glucose toxicity of the beta cell in type 2 diabetes. Cell Biochem Biophys 48:139–146
Patil C, Walter P (2001) Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol 13:349–355
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Marciniak SJ, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86:1133–1149
Fonseca SG, Lipson KL, Urano F (2007) Endoplasmic reticulum stress signaling in pancreatic beta-cells. Antioxid Redox Signal 9:2335–2344
Ortsäter H, Sjöholm A (2007) A busy cell-endoplasmic reticulum stress in the pancreatic beta-cell. Mol Cell Endocrinol 277:1–5
Laybutt DR, Preston AM, Akerfeldt MC et al (2007) Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50:752–563
Lai E, Bikopoulos G, Wheeler MB, Rozakis-Adcock M, Volchuk A (2008) Differential activation of ER stress and apoptosis in response to chronically elevated free fatty acids in pancreatic beta-cells. Am J Physiol Endocrinol Metab 294:E540–E550
Karaskov E, Scott C, Zhang L, Teodoro T, Ravazzola M, Volchuk A (2006) Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 147:3398–3407
Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G (2009) Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS ONE 4:e4954
Kim I, Xu W, Reed JC (2008) Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 7:1013–1030
Gotoh T, Mori M (2006) Nitric oxide and endoplasmic reticulum stress. Arterioscler Thromb Vasc Biol 26:1439–1446
Oyadomari S, Takeda K, Takiguchi M et al (2001) Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA 98:10845–10850
Eizirik DL, Colli ML, Ortis F (2009) The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 5:219–226
Meidute Abaraviciene S, Lundquist I, Galvanovskis J, Flodgren E, Olde B, Salehi A (2008) Palmitate-induced beta-cell dysfunction is associated with excessive NO production and is reversed by thiazolidinedione-mediated inhibition of GPR40 transduction mechanisms. PLoS ONE 3:e2182
Henningsson R, Salehi A, Lundquist I (2002) Role of nitric oxide synthase isoforms in glucose-stimulated insulin release. Am J Physiol Cell Physiol 283:C296–C304
Qader SS, Jimenez-Feltstrom J, Ekelund M, Lundquist I, Salehi A (2007) Expression of islet inducible nitric oxide synthase and inhibition of glucose-stimulated insulin release after long-term lipid infusion in the rat is counteracted by PACAP27. Am J Physiol Endocrinol Metab 292:E1447–E1455
Salehi A, Ekelund M, Lundquist I (2003) Total parenteral nutrition-stimulated activity of inducible nitric oxide synthase in rat pancreatic islets is suppressed by glucagon-like peptide-1. Horm Metab Res 35:48–54
Salehi A, Meidute Abaraviciene S, Jimenez-Feltstrom J, Ostenson CG, Efendic S, Lundquist I (2008) Excessive islet NO generation in type 2 diabetic GK rats coincides with abnormal hormone secretion and is counteracted by GLP-1. PLoS ONE 3:e2165
Carlsson C, Borg LA, Welsh N (1999) Sodium palmitate induces partial mitochondrial uncoupling and reactive oxygen species in rat pancreatic islets in vitro. Endocrinology 140:3422–3428
Kharroubi I, Ladriere L, Cardozo AK, Dogusan Z, Cnop M, Eizirik DL (2004) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145:5087–5096
Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL (2005) Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2):S97–S107
Kaiser N, Nesher R, Donath MY et al (2005) Psammomys obesus, a model for environment–gene interactions in type 2 diabetes. Diabetes 54(Suppl 2):S137–S144
Attali V, Parnes M, Ariav Y, Cerasi E, Kaiser N, Leibowitz G (2006) Regulation of insulin secretion and proinsulin biosynthesis by succinate. Endocrinology 147:5110–5118
Kaiser N, Corcos AP, Tur-Sinai A, Ariav Y, Cerasi E (1988) Monolayer culture of adult rat pancreatic islets on extracellular matrix: long term maintenance of differentiated B cell function. Endocrinology 123:834–840
Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY (2003) Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 52:726–733
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126:131–138
Cunha DA, Hekerman P, Ladriere L et al (2008) Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J Cell Sci 121:2308–2318
Calfon M, Zeng H, Urano F et al (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96
Lee AH, Iwakoshi NN, Glimcher LH (2003) XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol 23:7448–7459
Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V (2004) Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes 53:2610–2616
Lupi R, Dotta F, Marselli L et al (2002) Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 51:1437–1442
Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 3:193–197
Rabkin SW, Klassen SS (2008) Palmitate-induced NO production has a dual action to reduce cell death through NO and accentuate cell death through peroxynitrite formation. Prostaglandins Leukot Essent Fatty Acids 78:147–155
Pacher P, Beckman JS, Liaudet L (2007) Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87:315–424
Storling J, Binzer J, Andersson AK et al (2005) Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia 48:2039–2050
Kitiphongspattana K, Khan TA, Ishii-Schrade K, Roe MW, Philipson LH, Gaskins HR (2007) Protective role for nitric oxide during the endoplasmic reticulum stress response in pancreatic beta-cells. Am J Physiol Endocrinol Metab 292:E1543–E1554
Nakamura T, Lipton SA (2009) Cell death: protein misfolding and neurodegenerative diseases. Apoptosis 14:455–468
Akerfeldt MC, Howes J, Chan JY et al (2008) Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling. Diabetes 57:3034–3044
Xu L, Eu JP, Meissner G, Stamler JS (1998) Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science 279:234–237
Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C (1999) Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem J 340:657–669
Uehara T, Nakamura T, Yao D et al (2006) S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 441:513–517
Noguchi A, Takada M, Nakayama K, Ishikawa T (2008) cGMP-independent anti-apoptotic effect of nitric oxide on thapsigargin-induced apoptosis in the pancreatic beta-cell line INS-1. Life Sci 83:865–870
Acknowledgements
This study was supported by grants from the Israel Science Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. 1
Effect of palmitate on nNOS production in INS-1E beta cells. INS-1E cells were incubated for 24 h with different concentrations of palmitate at 11.1 mmol/l glucose. nNOS production was assessed by western blotting. Representative gels showing the production of nNOS and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and quantification of nNOS production normalised to GAPDH are presented. Results are expressed as fold nNOS production in INS-1E cells incubated in the absence of palmitate (n = 4) (PDF 76 kb)
Fig. 2
NOS activity in rat islets exposed to glucolipotoxicity and in rat cerebellum (a), and nitrite production of beta cells treated with palmitate, l-NAME and cytokines (b). a Rat islets were incubated for 16 h at 5.5 mmol/l glucose or 22.2 mmol/l glucose+0.5 mmol/l palmitate (Palm.). NOS isoform activity was analysed in islets and in cerebellum as described in the Methods. b INS-1E cells (5 × 104 cells) were incubated for 16 h at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate and 1 mmol/l or 10 mmol/l l-NAME and with a cocktail of cytokines (described in the legend of Fig. 1) at 11.1 mmol/l glucose. Medium nitrite concentration was determined using the Greiss reagent. Results in b are expressed as mean±SEM of three individual experiments. ^p < 0.001 for the difference between the indicated groups or between these and untreated cells at the same glucose concentration (PDF 412 kb)
Fig. 3
Effects of nNos knockdown on production of nNOS and stress markers and apoptosis in INS-1E beta cells. INS-1E cells were transfected with RNAi oligos for nNOS (Si-2) as described in Methods. The sequence targeted corresponded to bases 2824–2846 of nNos mRNA. The sense and antisense oligonucleotides were: 5′-UGGCCAAUGUGAGGUUCUCAGUGUU and 5′-AACACUGAGAACCUCACAUUGGCCA, respectively. Note that the RNAi oligos in this experiment differed from those used for nNOS silencing in the experiments shown in Fig. 4. The cells were incubated for 16 h at 3.3 and 22.2 mmol/l glucose with and without 0.5 mmol/l palmitate. a nNOS production, phosphorylated and total JNK, CHOP production, cleaved and non-cleaved caspase 3 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were analysed by western blotting. Relative band intensity is shown underneath each blot. b Apoptosis was analysed using the apoptosis ELISA assay. *p < 0.05, **p < 0.01, ^p < 0.001 for the difference between the indicated groups and untreated cells at the same glucose concentration AU, arbitrary units (PDF 114 kb)
Fig. 4
Effects of nNos knockdown in INS-1E beta cells on nNOS nitrite production. INS-1E cells were transfected with RNAi oligos for nNOS (black bars) or with control, scramble RNA oligos (white bars) as described in the Methods. Cells were then incubated for 16 h at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate (P). Medium nitrite concentration was determined using the Greiss reagent. *p < 0.05, ^p < 0.001 for the difference between the indicated groups (PDF 25 kb)
Fig. 5
Effect of nNOS inhibition by l-NAME on IRE1α production and phosphorylation and PERK phosphorylation in INS-1E beta cells. INS-1E cells were incubated at 3.3 and 27.8 mmol/l glucose with and without 0.5 mmol/l palmitate and 1 or 10 mmol/l l-NAME for 16 h. IRE1α production and phosphorylation and PERK phosphorylation were analysed by western blotting (see Figs 5 and 6). Quantification of IRE1α production and phosphorylation and PERK phosphorylation is shown. Each experiment was performed in triplicate. Results are normalised to GAPDH and expressed as mean ± SEM of three individual experiments. *p < 0.05, **p < 0.01, ^p < 0.001 for the difference between the indicated groups or between these and untreated cells at the same glucose concentration (PDF 555 kb)
Rights and permissions
About this article
Cite this article
Bachar, E., Ariav, Y., Cerasi, E. et al. Neuronal nitric oxide synthase protects the pancreatic beta cell from glucolipotoxicity-induced endoplasmic reticulum stress and apoptosis. Diabetologia 53, 2177–2187 (2010). https://doi.org/10.1007/s00125-010-1833-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1833-6