
Abstract
Aims/hypothesis
Pro-inflammatory cytokines cause beta cell secretory dysfunction and apoptosis—a process implicated in the pathogenesis of type 1 diabetes. Cytokines induce the expression of inducible nitric oxide (NO) synthase (iNOS) leading to NO production. NO contributes to cytokine-induced apoptosis, but the underlying mechanisms are unclear. The aim of this study was to investigate whether NO modulates signalling via mitogen-activated protein kinases (MAPKs) and Akt.
Materials and methods
MAPK activities in INS-1 cells and isolated islets were determined by immunoblotting and in vitro kinase assay. Apoptosis was determined by ELISA measurement of histone–DNA complexes present in cytoplasm.
Results
Apoptosis in INS-1 cells induced by IL-1β plus IFNγ was dependent on NO production as demonstrated by the use of the NOS blocker N G-methyl-l-arginine. Accordingly, an NO donor (S-nitroso-N-acetyl-d,l-penicillamine, SNAP) dose-dependently caused apoptosis in INS-1 cells. SNAP activated c-Jun N-terminal kinase (JNK) and p38 MAPK, but suppressed the activity of extracellular signal-regulated kinase MAPK. In rat islets, NOS inhibition decreased JNK and p38 activities induced by a 6-h exposure to IL-1β. Likewise, IL-1β-induced JNK and p38 activities were lower in iNOS(−/−) mouse islets than in wild-type islets. In human islets, SNAP potentiated IL-1β-induced JNK activation. The constitutive level of active, Ser473-phosphorylated Akt in INS-1 cells was suppressed by SNAP. IGF-I activated Akt and protected against SNAP-induced apoptosis. The anti-apoptotic effect of IGF-I was not associated with reduced JNK activation.
Conclusions/interpretation
We suggest that NO contributes to cytokine-induced apoptosis via potentiation of JNK activity and suppression of Akt.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In type 1 diabetes, the insulin-producing pancreatic beta cells are destroyed by an immune-mediated process. During this process, pro-inflammatory cytokines including IL-1β and IFNγ released by activated immune cells are believed to be key mediators of beta cell apoptosis [1]. In rodent beta cells, IL-1β alone or in combination with IFNγ induces the expression of inducible nitric oxide (NO) synthase (iNOS) resulting in production of NO. Using either pharmacological blockers of NO production or taking advantage of isolated islets from iNOS-deficient mice, several in vitro studies have shown that NO is a contributing factor in cytokine-induced impairment of beta cell secretory function and cell death [2–7]. Also, iNOS-deficient mice are significantly protected against chemically induced diabetes [7], further indicating that NO plays a role in the beta cell demise in type 1 diabetes. However, the exact molecular signalling mechanisms behind NO-mediated rodent beta cell cytotoxicity are unclear. Experiments with human islets have shown that despite an increase in NO production, cytokine-induced human islet functional impairment and cell death cannot be prevented by blockers of NO production [8–10]. Thus, the exact role and biological significance of NO for human beta cell destruction remain uncertain.
The c-Jun N-terminal kinase (JNK) is a mitogen-activated protein kinase (MAPK) that is mainly activated in response to cytokines and various environmental stresses including UV irradiation, oxidative stress and osmotic and heat shock [11, 12]. Activation of JNK is achieved by dual phosphorylation of Thr183 and Tyr185 via the MAPK kinases (MAPKKs) MKK7 and MKK4. These MAPKKs are phosphorylated by MAPKK kinases such as MEKK1 and MLK3. Following its activation, JNK phosphorylates a broad spectrum of target proteins including AP-1 transcription factors (e.g. c-Jun and ATF2) and members of the Bcl-2 family of apoptosis-regulating proteins [11, 12]. JNK is an important mediator of cytokine-induced beta cell death. Thus, blocking JNK signalling by overexpression of islet-brain (IB)-1, a JNK scaffold protein, or the JNK-binding domain of IB1 protects against IL-1β-induced apoptosis, whereas IB1 antisense treatment, leading to higher JNK activity, augments IL-1β-induced apoptosis in insulin-secreting cells [13–16]. Further, a small molecule JNK inhibitor delays cytokine-induced suppression in human islet cell viability [17]. Two other MAPKs, the extracellular signal-regulated kinase (ERK) and p38, are also involved in mediating deleterious cytokine effects in beta cells. Hence, both ERK and p38 are required for IL-1β-induced iNOS expression and NO production [18] and, individually, ERK and p38 have been shown to be involved in cytokine-induced beta cell apoptosis [19, 20].
The Ser/Thr kinase Akt, also known as protein kinase B (PKB), is widely involved in cell growth and survival [21, 22]. Akt is commonly activated in a phosphatidylinositol 3-kinase (PI3K)-dependent manner by phospholipid binding and phosphorylation at Thr308 and Ser473. Active Akt has been demonstrated to inhibit several pro-apoptotic components including BAD (a pro-apoptotic member of the Bcl-2 family), Forkhead transcription factors and the JNK pathway [21, 22]. Recently, cytokines were shown to decrease the level of Thr308-phosphorylated Akt in mouse beta cells [23]. Beta-cell-specific expression of mutant constitutively active Akt in mice is associated with increased beta cell mass, and confers protection against chemically induced diabetes [24]. Further, IGF-I, a strong activator of Akt, increases beta cell survival after cytokine exposure in vitro [2, 25–28]. IGF-I also has been shown to decrease and delay hyperglycaemia and improve beta cell mass in animal models of type 1 diabetes [29, 30]. Together, these findings suggest an important anti-apoptotic function of the PI3K–Akt signalling pathway in beta cells.
In the present study it was investigated whether NO modulates MAPK and Akt signalling in insulin-secreting cells and isolated intact islets.
Subjects, materials and methods
Materials
Recombinant human or mouse IL-1β was obtained from the National Cancer Institute (Bethesda, MD, USA), Novo Nordisk A/S (Bagsværd, Denmark) or BD Pharmingen (San Diego, CA, USA). Recombinant rat IFNγ and recombinant human IGF-I were from R&D Systems (Oxon, UK). S-Nitroso-N-acetyl-d,l-penicillamine (SNAP) was from Calbiochem (San Diego, CA, USA). All reagents for SDS-PAGE were from Invitrogen (Carlsbad, CA, USA). Antibodies against procaspase-3, caspase-3, Thr183/Tyr185-phosphorylated JNK1/2, total JNK1/2, Thr202/Tyr204-phosphorylated ERK1/2, total ERK1/2, Thr180/Tyr182-phosphorylated p38, total p38, Ser473-phosphorylated Akt and total Akt were all obtained from Cell Signaling (Beverly, MA, USA). Anti-iNOS antibody was obtained from BD Pharmingen. Anti-β-tubulin and anti-actin antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). GST-c-Jun (amino acids 1–79) was obtained from Calbiochem. Recombinant murine heat shock protein-25 (Hsp25) was from Stressgen (Victoria, Canada). GST-Elk-1 was synthesised by standard GST fusion protein expression and purification procedures using the RediPack GST Purification Module kit (Amersham Biosciences, Amersham, Bucks, UK). [γ32P]ATP (3000 Ci/mmol) was purchased from Amersham Biosciences. NOS blockers N G-nitro-l-arginine-methyl ester (NAME) or N G-methyl-l-arginine (NMA) were obtained from Sigma (St Louis, MO, USA). 8-Bromo-3′,5′-cGMP (8-Br-cGMP), LY294002, LY83583, PD98059, SB203580 and KT5823 were obtained from Calbiochem. All other reagents were from Sigma or Merck (Darmstadt, Germany).
Cell culture and islet isolation
INS-1 cells were maintained in RPMI 1640 medium (11 mmol/l glucose) supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin (all from Life Technologies, Inc.). In addition, the medium contained 50 μmol/l β-mercaptoethanol. Cells were trypsinised and passaged weekly. For preparation of cell extracts, 0.5×106 cells/well were seeded in 12-well culture dishes (Nunc, Roskilde, Denmark). For ELISA apoptosis measurements, 0.5×105 cells/well were seeded in 48-well culture dishes (Nunc). Cells were precultured for 2 days prior to the treatment with desired agents. Rat pancreatic islets of Langerhans (Charles River, Sulzfeldt, Germany) were isolated from neonatal rats and cultured as previously described [18]. On the day of experimentation, either 150 islets/well were placed in four-well dishes (Nunc) or 400–500 islets/well were placed in 12-well dishes (Nunc) in 300 μl or 1 ml, respectively, of RPMI 1640 medium supplemented with 0.5% human serum. Islets from male wild-type or iNOS-deficient mice (8–12 weeks of age) [31] were isolated by a collagenase digestion procedure, and islets were picked by hand with a braking pipette [32]. Groups of islets were precultured 6–8 days in RPMI 1640 medium (Sigma) containing 11.1 mmol/l glucose, supplemented with 10% FCS, penicillin (100 U/ml) and streptomycin (100 μg/ml). Medium was changed every second day. On the day of experimentation, 150 mouse islets were transferred to new culture dishes containing 2 ml medium supplemented with 10% FCS. Animal care was carried out according to national and international law and ethical standards. Human islets from two different donors (female aged 61 years, BMI=19 kg/m2; and male aged 66 years, BMI >30 kg/m2) with normal beta cell function were isolated and cultured as described [33].
Cell and islet experiments and lysis
For experimentation in INS-1 cells, the medium was removed and fresh medium with or without SNAP, cytokines, inhibitors and/or IGF-I was added. Following exposure for the desired period of time, the medium was removed and cells washed in cold PBS and lysed for 30 min on ice in 100 μl lysis buffer containing 20 mmol/l Tris–acetate, pH 7.0, 0.27 mol/l sucrose, 1 mmol/l EDTA, 1 mmol/l EGTA, 50 mmol/l NaF, 1% v/v Triton X-100, 5 mmol/l sodium pyrophosphate, 10 mmol/l sodium glycerophosphate, 1 mmol/l benzamidine, 1 mmol/l dithiothreitol, 1 mmol/l Na3VO4 and 4 μg/ml leupeptin. Rat or mouse islets were exposed to the desired conditions for 3 or 6 h. For 6-h rat islet experiments, arginine-free medium was used in combination with 1 mmol/l NAME. Following the treatments, islets were carefully transferred to 1.5 ml tubes and mildly centrifuged. Medium was removed and islets lysed in 30–50 μl lysis buffer on ice for 30 min. Detergent-insoluble material was pelleted by centrifugation at 15,000×g for 5 min at 4°C. The supernatants (whole-cell extracts) were stored at −80°C until assay. Protein concentration in lysates was determined by the Bradford method using the dye reagent concentrate (Bio-Rad, Richmond, CA, USA).
Western blotting
Whole-cell lysates were mixed with 4×LDS sample buffer (Invitrogen), boiled for 10 min and subjected to 10% SDS-PAGE. Proteins were then electrotransferred to nitrocellulose filter membranes. Non-specific protein binding was blocked by incubating the filter membrane in blocking buffer (1×TBS, pH 7.6, 0.1% Tween-20, 5% non-fat dry milk) for 1 h. Membranes were incubated with primary antibodies diluted either in TBS with 0.1% containing 5% BSA or in blocking buffer. Filter membranes were then incubated with secondary horseradish peroxidase-conjugated antibodies. Immune complexes were detected by chemiluminescence using LumiGLO (Cell Signaling).
In vitro kinase assay
Phosphotransferase activities toward GST-c-Jun, GST-Elk-1 and Hsp25 were measured by a whole-cell lysate in vitro kinase assay as described elsewhere [18] except that 2 μg GST-c-Jun (amino acids 1–79) were used instead of activating transcription factor 2. Phosphorylated substrates were visualised by autoradiography and quantified by PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA, USA).
Apoptosis measurements
Apoptotic cell death was determined by the detection of DNA–histone complexes present in the cytoplasmic fraction of cells using Cell Death Detection ELISAPLUS (Roche, Basel, Switzerland) as described by the manufacturer. Briefly, following exposure of cells to the desired reagents, the culture medium was removed and cells lysed in 400 μl lysis buffer for 30 min at room temperature. Lysates were centrifuged for 10 min at 200×g and 20 μl supernatant and 80 μl immunoreagent (anti-DNA–POD antibody and anti-histone–biotin antibody) were added to streptavidin-coated microtitre plates and incubated for 2 h under shaking conditions (300 rev/min) at room temperature. The solution was then removed and each well washed three times with 250 μl incubation buffer, after which 100 μl ABTS solution was added. Absorbance was measured at 405 and 490 (reference) nm. The induction of apoptosis was calculated as described by the manufacturer.
Statistical analysis
In bar graphs, data are means±SE. Student’s t-test or ANOVA was used for statistical analyses where appropriate (Microsoft Excel). A p value of <0.05 was considered significant.
Results
Cytokine-induced apoptosis in INS-1 cells is mediated by NO
We first examined the contribution of NO to cytokine-induced apoptosis in rat insulin-secreting INS-1 cells. Following 24 h of exposure to a mixture of IL-1β and IFNγ in the presence or absence of 1 mmol/l NOS blocker NMA, apoptotic cell death was evaluated by the ELISA determination of histone–DNA complexes present in the cytoplasm. Cytokines increased apoptosis by 15.2±2.9-fold compared with untreated control cells (Fig. 1a). NMA reduced cytokine-induced apoptosis to 2.7±0.7-fold corresponding to a ∼90% inhibition. Measurement of cytokine-induced NO formation as determined by accumulated nitrite in the culture medium revealed that cytokines increased medium nitrite from <1 μmol/l to 14.1±1.4 μmol/l (n=4) during 24 h, which was blocked to <1 μmol/l by NMA. As expected, cytokine-stimulated cells showed expression of iNOS, which was unaffected by NMA (Fig. 1b). These results indicate that NO production in response to cytokines is essential for cytokine-induced apoptosis in INS-1 cells. In line with this, application of the NO-donor SNAP to the culture medium for 24 h dose-dependently induced apoptosis (Fig. 1c). Western blot analysis of caspase-3 further demonstrated an apoptotic process in INS-1 cells treated with SNAP (Fig. 1d).

NMA reduces cytokine-induced apoptosis. a INS-1 cells were left untreated or exposed to IL-1β (530 pg/ml) plus IFNγ (36 ng/ml) in the presence or absence of 1 mmol/l NMA. Apoptotic cell death was determined after 24 h by the Cell Death Detection ELISAPLUS. Results are means±SE of n=3. *p<0.05 vs. untreated. b Cell extracts from INS-1 cells treated as in (a) were analysed by western blotting for expression of iNOS. A western blot of β-tubulin was used as loading control. Blots shown are representative of n=4. c Apoptosis in INS-1 cells left untreated, treated with vehicle (DMSO) or exposed to the indicated concentrations of SNAP for 24 h. Results are means±SE of n=4. d Cell extracts from INS-1 cells left untreated or exposed to 200 μmol/l SNAP for 24 h were analysed by western blotting for procaspase-3, caspase-3 and actin. Blots shown are representative of two experiments
NO-donor SNAP activates JNK and p38, but suppresses ERK
To investigate if NO modulates signalling via JNK, which plays an essential role in cytokine-induced apoptosis in insulin-secreting cells, INS-1 cells were treated with increasing concentrations of SNAP for 3 h and JNK activation in cell extracts was assessed by western blotting using phospho-specific antibodies directed against dually phosphorylated (Thr183 and Tyr185), active JNK. Figure 2a shows that SNAP dose-dependently induced phosphorylation of JNK1/2. Because it has previously been reported that NO suppresses the enzymatic activity of JNK by nitrosylation of a critical cysteine residue [34], it was important to confirm the effect of NO on JNK at the enzymatic level. We therefore assayed the cell extracts for in vitro kinase activity towards the specific JNK substrate GST-c-Jun (amino acids 1–79) containing the two main JNK phosphoacceptor sites Ser63 and Ser73. SNAP dose-dependently stimulated an increase in the phosphorylation of GST-c-Jun (Fig. 2b), confirming an increase in the enzymatic activity of JNK in response to SNAP. A time-course analysis revealed that NO caused phosphorylation of JNK1/2 within 30 min which persisted for several hours (Fig. 2c). It was also observed that JNK1/2 phosphorylation induced by a 1-h exposure to IL-1β in INS-1 cells was potentiated by SNAP, reaching a level much higher than just an additive effect of each treatment alone (data not shown). We then examined the effects of NO on p38 and ERK MAPKs. The activity of these two kinases was assessed by western blotting with phospho-specific antibodies against the activated forms of the kinases. A 3-h exposure to SNAP induced phosphorylation of p38 (Fig. 3a). In contrast, SNAP caused a striking suppression of the constitutive phosphorylation of ERK1/2 (Fig. 3a). Again, to confirm these findings at the enzymatic level of the kinases, cell extracts from INS-1 cells treated with SNAP for 0–6 h were assayed for in vitro kinase activity towards the substrates Hsp25 and GST-Elk-1. Hsp25 is a substrate of MAPKAPK2 which is specifically activated by p38, and GST-Elk-1 is a substrate of ERK in insulin-secreting cells [18]. The experiment revealed that SNAP stimulated a sustained increase in the phosphotransferase activity towards Hsp25 with kinetics similar to that for phosphorylated JNK1/2, whereas the constitutive kinase activity towards GST-Elk-1 was reduced by SNAP (Fig. 3b). These findings indicate that NO activates JNK and p38, but suppresses constitutive ERK activity in INS-1 cells.

SNAP activates JNK. a INS-1 cells were left untreated or exposed to the indicated concentrations of SNAP for 3 h. Cell extracts were analysed by western blotting for phosphorylated JNK (P-JNK1/2) and total JNK (JNK1/2). Blots shown are representative of n=3. b Cell extracts from INS-1 cells treated as in (a) were analysed for in vitro kinase activity towards GST-c-Jun in the presence of [32P]ATP. Phosphorylated GST-c-Jun was visualised by autoradiography and quantified by PhosphoImager analysis. Results are means±SE of n=3. A representative autoradiogram is shown. c INS-1 cells were left untreated or exposed to 500 μmol/l SNAP for the indicated time periods. Cell extracts were analysed by western blotting for phosphorylated and total JNK. Blots shown are representative of two experiments

SNAP activates p38, but suppresses ERK. a INS-1 cells were left untreated or exposed to 500 μmol/l SNAP for 3 h. Cell extracts were analysed by western blotting for phosphorylated p38 and ERK (P-p38 and P-ERK1/2) and total p38 and ERK (p38 and ERK1/2). Blots shown are representative of n=3. b INS-1 cells were left untreated or exposed to 500 μmol/l SNAP for the indicated time periods. Cell extracts were analysed for in vitro kinase activity towards GST-Elk-1 and Hsp25 in the presence of [32P]ATP. Phosphorylated substrates were visualised by autoradiography. Autoradiograms shown are representative of two experiments
Endogenous NO production causes JNK and p38 activation in isolated islets
To investigate the potential contribution of endogenous NO production for beta-cell MAPK regulation, isolated intact rat pancreatic islets were exposed to IL-1β for 6 h in the presence or absence of 1 mmol/l NOS inhibitor NAME. Islet extracts were assessed for in vitro kinase activity towards GST-Elk-1, GST-c-Jun and Hsp25. Extracts from islets treated with IL-1β contained increased phosphotransferase activity towards GST-c-Jun and Hsp25, which was reduced by NAME (Fig. 4a). Quantitative assessment revealed that NAME blocked IL-1β-induced GST-c-Jun and Hsp25 phosphorylations by 76 and 80%, respectively. There was no detectable increase in GST-Elk-1 phosphorylation following IL-1β stimulation (Fig. 4a), suggesting that IL-1β activation of ERK is more transient as compared with JNK and p38. Measurement of accumulated nitrite in culture media confirmed that NAME effectively blocked IL-1β-induced rat islet NO production (data not shown). These results suggest that endogenous NO contributes to JNK and p38 activation in IL-1β-exposed rat islets. We also determined if NAME affects IL-1β-induced rat islet MAPK activity at earlier time points. Although the effects of NAME were more partial, NAME also reduced GST-c-Jun and Hsp25 phosphorylation induced by a 20-min or 1.5-h exposure to IL-1β (data not shown), i.e. prior to iNOS expression. Short-term stimulation of rat islets with IL-1β also induced phosphorylation of GST-Elk-1. The increase in GST-Elk-1 phosphorylation induced by a 1.5-h, but not 20-min exposure to IL-1β was reduced by co-incubation with NAME (data not shown). That NAME decreased the IL-1β (20 min)-stimulated GST-c-Jun and Hsp25, but not GST-Elk-1 phosphorylation suggests that the inhibitory effects of NAME on GST-c-Jun and Hsp25 phosphorylation were not due to unspecific effects. These findings may therefore point towards a role for NO produced by the constitutively expressed NOS form. The importance of NO for IL-1β-stimulated sustained JNK and p38 activation was further established using islets obtained from iNOS−/− mice. As seen in Fig. 4b, the increase in kinase activity towards GST-c-Jun and Hsp25 induced by a 6-h exposure to IL-1β was reduced in iNOS−/− islets as compared with control (wild-type) mouse islets. Quantitative analysis showed that IL-1β-induced phosphorylations of GST-c-Jun and Hsp25 were reduced by ∼35 and ∼65%, respectively, in iNOS−/− islets as compared with wild-type islets. Notably, IL-1β caused a ∼65% reduction in the constitutive GST-Elk-1 phosphorylation in wild-type islets, but not in iNOS−/− islets (Fig. 4b). Western blot analysis of the MAPKs confirmed that IL-1β-induced phosphorylations of JNK1/2 and p38 were reduced in iNOS−/− islets vs control islets, and that basal ERK1/2 phosphorylation was suppressed in control, but not in iNOS−/− islets (Fig. 4b). Western blot analysis of iNOS verified IL-1β-induced expression of iNOS in wild-type, but not in iNOS-deficient mouse islets (Fig. 4b). Finally, we examined the effect of SNAP on JNK in human islets. Using isolated human islets from two different donors, we found that a 3-h exposure to SNAP induced phosphorylation of JNK1/2 mainly in the islets from donor #1 (Fig. 5). IL-1β appeared to stimulate JNK1/2 phosphorylation in both islet preparations but, interestingly, the combination of SNAP and IL-1β resulted in higher JNK1/2 phosphorylation than each treatment alone in islets from both donors (Fig. 5).

NAME blocks IL-1β-induced rat islet in vitro phosphorylation of GST-c-Jun and Hsp25. a Rat islets were left untreated or exposed to IL-1β (150 pg/ml) in the presence or absence of 1 mmol/l NAME for 6 h. Islet extracts were analysed for in vitro kinase activities toward GST-c-Jun, Hsp25 and GST-Elk-1 in the presence of [32P]ATP. Phosphorylated substrates were visualised by autoradiography and quantified by PhosphorImager analysis. Results are means±SE of n=8–10. Representative autoradiograms are shown. *p<0.002 vs. untreated; # p<0.002 vs. IL-1β. b Islets from iNOS−/− or wild-type mice (wt) were left untreated or exposed to IL-1β (320 pg/ml) for 6 h. Islet extracts were subjected to in vitro kinase assay as above and to western blot analysis of phosphorylated JNK, p38 and ERK (P-JNK1/2, P-p38 and P-ERK1/2) and total JNK (JNK1/2). Autoradiograms and blots shown are representative of two experiments

SNAP potentiates IL-1β-induced JNK activation in human islets. Human islets isolated from two different donors were left untreated or exposed to SNAP (500 μmol/l) in the presence or absence of IL-1β (160 pg/ml) for 3 h. Cell extracts were analysed by western blotting for phosphorylated JNK (P-JNK1/2) and total JNK (JNK1/2)
cGMP is not involved in mediating NO effects on JNK
A known target of NO is guanylyl cyclase, which upon activation by NO produces cGMP. NO has previously been shown to stimulate guanylyl cyclase activity in insulin-producing cells [35]. In other cells, a cGMP–protein kinase G (PKG) pathway has been shown to activate JNK [36, 37]. cGMP could therefore potentially mediate the effects of NO on MAPKs in INS-1 cells. To test this hypothesis, we examined the effect of the membrane-permeable cGMP analogue 8-Br-cGMP on JNK activity. Exposure of INS-1 cells for 3 h to increasing concentrations of 8-Br-cGMP stimulated modest phosphorylation of JNK1/2 (Fig. 6a). We then examined the effect of a guanylyl cyclase inhibitor (LY83583) on NO-induced JNK phosphorylation. However, LY83583 (1 μmol/l) only modestly decreased SNAP-induced phosphorylation of JNK1/2 (Fig. 6b). Finally, the effect of KT5823, an inhibitor of PKG, on JNK activation by NO was tested (Fig. 6c). KT5823 (1 μmol/l) failed to affect NO-induced JNK1/2 phosphorylation. From these findings it appears that the guanylyl cyclase–cGMP–PKG pathway is not involved in mediating NO effects on JNK MAPK in insulin-secreting cells. Despite this, a PKG-dependent mechanism could still be involved in signalling NO-induced apoptosis; however, we found no effect of KT5823 on SNAP-induced apoptosis (Fig. 6d).

8-Br-cGMP, LY83583 or KT5823 do not affect SNAP-induced JNK activation. a INS-1 cells were left untreated or exposed to the indicated concentrations of 8-Br-cGMP for 3 h. Cell extracts were analysed by western blotting for phosphorylated JNK (P-JNK1/2) and total JNK (JNK1/2). b, c INS-1 cells were left untreated or exposed to 500 μmol/l SNAP in the presence or absence of 1 μmol/l LY83583 or 1 μmol/l KT5823 for 3 h. Cell extracts were analysed by western blotting for phosphorylated JNK and total JNK. All blots shown are representative of n=2–6. d INS-1 cells were left untreated or exposed to the indicated concentrations of SNAP in the presence or absence of 1 μmol/l KT5823. Apoptotic cell death was determined after 24 h by the Cell Death Detection ELISAPLUS. Results are means±SE of n=4
NO decreases Ser473 phosphorylation of Akt
Signalling via the anti-apoptotic kinase Akt is important for cell growth and survival, and Akt has been shown to act as a repressor of JNK activation, suggesting that NO effects on JNK/MAPKs might potentially involve modulation of Akt activity. Thus, we investigated if NO affected the PI3K–Akt signalling pathway. Exposure of INS-1 cells to increasing concentrations of SNAP caused a dose- and time-dependent reduction in the cellular content of active, Ser473-phosphorylated Akt as shown by western blotting (Fig. 7a,b). To determine the functional role of impaired Akt activity in NO-induced apoptosis, we examined the effect of IGF-I, a known stimulator of the Akt pathway, on SNAP-induced cell death. Figure 8a shows that IGF-I (100 nmol/l) decreased NO-induced apoptosis—an effect correlating with an increase in phosphorylation of Akt on Ser473 (Fig. 8b). To specifically address the dependence of PI3K–Akt for the survival effect of IGF-I on NO-induced apoptosis, the PI3K blocker LY294002 was applied. The addition of LY294002 (1 or 10 μmol/l) to the culture medium abolished the protective effect of IGF-I on NO-induced apoptosis (Fig. 8c). LY294002 effectively blocked constitutive Ser473 phosphorylation of Akt (Fig. 8d), verifying the efficacy of the inhibitor. These results suggest that NO causes suppression of the PI3K–Akt survival signalling pathway in insulin-secreting cells. To investigate if cytokines also lead to a reduction in the cellular content of phosphorylated Akt via NO, cells were exposed to cytokines for 24 h. Cytokines decreased the phosphorylation of Akt in an NO-dependent manner (Fig. 9a), and IGF-I reduced cytokine-induced apoptosis (Fig. 9b)—an effect that was not associated with changes in cytokine-induced iNOS protein expression (Fig. 9c) or NO production (13.2±1.2 vs 13.6±1.6 μmol/l nitrite for cytokines alone or in combination with IGF-I, respectively). Finally, to investigate if Akt is an upstream negative modulator of JNK activity, we examined if Akt activation by IGF-I affects SNAP-induced JNK phosphorylation. However, phosphorylation of JNK1/2 by SNAP was not affected by cotreatment with IGF-I (Fig. 9d) in INS-1 cells. Similarly, IGF-I failed to affect NO regulation of ERK activity (data not shown), suggesting that NO effects on MAPKs in general are insensitive to IGF-I treatment. Consistent with these findings, activation of JNK1/2 in response to SNAP, IL-1β or IL-1β+SNAP in rat islets was unaffected by co-incubation with IGF-I (Fig. 9e). These data suggest that IGF-I-mediated, Akt-dependent protection against NO-induced apoptosis is not associated with repression of the JNK pathway.

SNAP decreases Ser473 phosphorylation of Akt. a INS-1 cells were left untreated or exposed to the indicated concentrations of SNAP for 3 h. Cell extracts were analysed by western blotting for Ser473 phosphorylated Akt (P-Akt) and total Akt (Akt). Blots shown are representative of n=4. b INS-1 cells were left untreated or exposed to 500 μmol/l SNAP for the indicated time periods. Cell extracts were analysed as in (a). Blots shown are representative of two experiments

IGF-I reduces SNAP-induced apoptosis. a INS-1 cells were left untreated or exposed to the indicated concentrations of SNAP in the presence or absence of 100 nmol/l IGF-I. Apoptotic cell death was determined after 24 h by the Cell Death Detection ELISAPLUS. Results are means±SE of n=4. b INS-1 cells were left untreated or exposed to 100 nmol/l IGF-I in the presence or absence of 500 μmol/l SNAP for 3 h. Cell extracts were analysed by western blotting for Ser473 phosphorylated Akt (P-Akt) and total Akt (Akt). c INS-1 cells were left untreated or exposed to 100 μmol/l SNAP in the presence or absence of 100 nmol/l IGF-I with or without the indicated concentrations of LY294002. Apoptotic cell death was determined after 24 h by the Cell Death Detection ELISAPLUS. Results are means±SE of n=5. d INS-1 cells were left untreated or exposed to 10 μmol/l LY294002 for 3 h. Cell extracts were analysed by western blotting for phosphorylated Akt and total Akt. *p<0.001 vs SNAP; # p<0.001 vs SNAP; noso non-significant vs SNAP

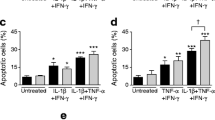
Cytokines decrease Ser473 phosphorylation of Akt via NO. a INS-1 cells were left untreated or exposed to IL-1β (530 pg/ml) plus IFNγ (36 ng/ml) in the presence or absence of 1 mmol/l NMA for 24 h. Cell extracts were analysed by western blotting for Ser473 phosphorylated Akt (P-Akt) and β-tubulin. Blots shown are representative of n=5. b INS-1 cells were left untreated or exposed to cytokines in the presence or absence of 100 nmol/l IGF-I. Apoptotic cell death was determined after 24 h by the Cell Death Detection ELISAPLUS. Results are means±SE of n=4. c INS-1 cells were left untreated or exposed to cytokines in the presence or absence of 100 nmol/l IGF-I for 24 h. Cell extracts were analysed by western blotting for iNOS and β-tubulin. Blots shown are representative of n=4. d INS-1 cells were left untreated or exposed to 500 μmol/l SNAP in the presence or absence of 100 nmol/l IGF-I for 3 h. Cell extracts were analysed by western blotting for phosphorylated JNK (P-JNK1/2) and total JNK (JNK1/2). Blots shown are representative of n=5. e Rat islets were left untreated or exposed to SNAP (500 μmol/l), IL-1β (150 pg/ml) or SNAP+IL-1β in the presence or absence of 100 nmol/l IGF-I for 3 h. Islet extracts were analysed by western blotting for P-JNK1/2, JNK1/2 and actin. Blots shown are representative of three experiments. *p<0.05 vs untreated; # p<0.05 vs IL-1β+IFNγ
Discussion
In beta cells, pro-inflammatory cytokines induce iNOS expression and NO production, leading to impairment of secretory function and cell death. The molecular signalling mechanisms by which NO contributes to beta cell death are, however, poorly understood. In the present study, we asked if NO might modulate signalling via MAPKs and Akt/PKB. The results obtained suggested that both exogenously applied NO, in the form of NO-donor SNAP, and endogenous production of NO by iNOS stimulate activation of JNK and p38 MAPK, whereas ERK MAPK activity was suppressed or unaffected.
In INS-1 cells, IL-1β+IFNγ-induced apoptosis was almost completely abrogated by the NOS blocker NMA, indicating a strong dependence on NO for cytokine-induced INS-1 cell death, thus confirming previous studies that NO mediates cytokine-induced rodent beta-cell death [2–7]. The finding that even relatively small concentrations of the NO-donor SNAP induced apoptosis indicates that an increase in cellular NO is a sufficient stimulus to promote an apoptotic response. Although it cannot be excluded that NO also induced necrotic cell death of INS-1 cells, the finding that NO caused cleavage of the 32 kDa procaspase-3 form into the active 17 kDa form indicates that NO specifically causes activation of the classic apoptosis pathway in insulin-secreting cells.
SNAP stimulated activation of JNK and p38 MAPK in a dose- and time-dependent manner as demonstrated by western blotting and the in vitro kinase assay using GST-c-Jun and Hsp25 as substrates. These findings are consistent with observations in other cells, where NO donors also induce activation of JNK and p38 and to a lesser extent ERK [38–44], thus supporting our findings. Further, the NO-donor sodium (Z)-1(N,N-diethylamino) diazen-1-ium-1,2-diolate at 500 μmol/l induced phosphorylation of p38 and JNK1/2 during 10- or 30-min treatments in insulin-secreting rat RINm5F cells [35]. However, in contrast to our findings, NO also induced phosphorylation of ERK in that [35] and another study [45] in RINm5F cells. The underlying cause of the discrepancy between effects of NO donors on ERK in INS-1 vs RINm5F cells is not clear, but it might be due to the specific nature of the NO donor used, cell clonal differences and/or differences in experimental conditions.
We observed that blocking rat islet endogenous NO production induced by a 6-h exposure to IL-1β inhibited IL-1β-induced in vitro kinase activity towards GST-c-Jun and Hsp25, suggesting that IL-1β-induced prolonged JNK and p38 activity is mediated via NO. Interestingly, there was no increase in phosphorylation of the ERK substrate GST-Elk-1 after 6 h of exposure of rat islets to IL-1β, suggesting that IL-1β-induced ERK activation is more transient than that of JNK and p38. The lack of prolongation of ERK activation in rat islets indirectly supports the data on INS-1 cells indicating that NO is not a major activator of ERK. A potential role for NO in beta cell MAPK modulation was further substantiated by the use of islets from iNOS−/− mice. Exposure of iNOS−/− islets to IL-1β for 6 h caused less activation of p38 MAPK and JNK as compared with control mouse islets determined both by western blotting and in vitro phosphotransferase activity towards GST-c-Jun and Hsp25. Additionally, and consistent with the notion that NO did not activate ERK in INS-1 cells or rat islets, ERK phosphorylation as well as kinase activity towards GST-Elk-1 in wild-type mouse islets exposed to IL-1β was decreased to below basal levels, whereas this was not the case in iNOS−/− islets. Hence, using both a pharmacological and a genetic approach to block iNOS activity, our data provide evidence for a role of IL-1β-induced NO production in the sustained activation of JNK and p38 MAPK. Because JNK among the MAPK members appears most important for IL-1β-induced apoptosis [13–16, 19, 20], NO prolongation of, in particular, JNK activity is likely to play a role in NO-dependent cytotoxicity. Thus, the fact that iNOS−/− mouse islets are more resistant to the toxic effects of cytokines than wild-type islets in vitro [4, 6, 7] might be explained by the lower level of and more transient JNK activation in response to IL-1β. Although JNK is mainly known to be pro-apoptotic, JNK has recently been suggested to play a role in beta cell recovery after short-term exposure to NO followed by washing and addition of fresh culture medium [35]. Based on this, JNK might therefore signal both protective and deleterious effects in response to NO in beta cells, but in the continuous presence of NO the deleterious effects prevail. In human islets from two different donors, it was observed that SNAP potentiated IL-1β-induced JNK phosphorylation. These findings suggest NO, as observed in rodent beta cells, may aggravate cytokine signalling via pro-apoptotic JNK in human beta cells. As NO is dispensable for cytokine-induced human beta cell death [8–10], the physiological relevance of NO as an amplifier of cytokine-induced JNK activity can be questioned. Nevertheless, NO may be one of several factors that, in synergy, tip the ‘life–death’ balance towards apoptosis in human beta cells.
Because NO stimulates the activation of guanylyl cyclase in many cells including beta cells [35, 46], leading to generation of cGMP, the effects of NO on MAPKs could involve cGMP. However, exposing INS-1 cells to increasing concentrations of 8-Br-cGMP for 3 h only weakly induced JNK1/2 phosphorylation, and application of the guanylyl cyclase inhibitor LY85583 only slightly reduced SNAP-induced JNK phosphorylation, suggesting that generation of cGMP is neither sufficient nor necessary for NO-induced JNK MAPK activation in INS-1 cells. Further, the PKG inhibitor KT5823 did not affect NO-stimulated JNK phosphorylation. In other cells, a cGMP–PKG-dependent pathway that leads to activation of JNK has been established [36, 37]. Apparently, this mechanism of JNK activation therefore seems to be cell-type specific.
We found that SNAP suppressed the constitutive cellular level of Ser473-phosphorylated Akt in a dose- and time-dependent fashion in INS-1 cells. Using IGF-I to reverse the inhibitory effect of NO on Akt Ser473 phosphorylation and by the use of the PI3K blocker LY294002, we established that the suppression of Akt activity induced by NO is a possible contributing factor for NO-induced apoptosis. Cytokines also caused a reduction in the phosphorylation of Akt—an effect preventable by NMA. Further, IGF-I decreased cytokine-induced apoptosis, but without affecting cytokine-induced expression of iNOS. Given that cytokine-induced NO production is essential for the induction of apoptosis in INS-1 cells, these findings may therefore suggest that the protective effect of IGF-I on apoptosis is afforded at a point downstream from iNOS expression and NO formation. We added IGF-I and cytokines simultaneously. In a previous report, describing a protective effect of IGF-I on cytokine-induced rat islet beta-cell apoptosis, islets had to be pre-incubated for 24 h with IGF-I to obtain the protective effect [2]. Also, in that study IL-1β-induced rat islet iNOS expression was inhibited by IGF-I pretreatment. Similarly, adenovirus-mediated gene transfer of IGF-I to human islets blocked both IL-1β-induced apoptosis and NO formation [27]. However, islets were transduced with the IGF-I gene days prior to IL-1β exposure. In another study, IGF-I blocked NO formation induced by IFNγ+lipopolysaccharide, but not that induced by IL-1β+IFNγ+/−TNF-α [26]. Thus, whether IGF-I protects against cytokine-stimulated beta cell apoptosis up- or downstream (or both) from the induction of iNOS is not completely clear. However, as IGF-I potently protected INS-1 cells against SNAP-induced apoptosis via Akt, IGF-I appears to have the potential to block apoptosis downstream from the production of NO.
A putative mechanism by which Akt suppresses cell death is via inhibition of JNK signalling [47, 48], presumably via phosphorylation and inhibition of the MAP3K apoptosis-signal-regulating kinase 1 [49]. The finding that IGF-I did not affect SNAP-induced phosphorylation of JNK1/2 in INS-1 cells suggests that IGF-I-afforded, Akt-dependent protection against NO-induced apoptosis is via a mechanism not involving JNK repression. Therefore, we propose that the decrease in Ser473 phosphorylation of Akt caused by NO is not exclusively responsible for JNK activation following exposure to SNAP. The mechanisms responsible for the anti-apoptotic effect of Akt in NO-mediated beta cell apoptosis as well as the mechanisms involved in mediating NO effects on MAPKs remain to be clarified. In our study we have been using both insulin-secreting cells and isolated rodent and human islets. It cannot be excluded that there are species differences in the specific signalling pathways and how they are modulated by cytokines and NO, and caution should be exerted before generalising this concept. More experiments elucidating NO-dependent and -independent effects of cytokines in different beta cell models may clarify this further.
In conclusion, the present study provides evidence that NO contributes to cytokine-induced apoptosis via potentiation of JNK activity and suppression of Akt. These data may add to our understanding of the interrelationship between NO-dependent and -independent pathways leading to beta cell destruction.
Duality of interest statement
T. Mandrup-Poulsen is employed by Novo Nordisk A/S, Bagsværd and has employee stock shares in Novo Nordisk. Novo Nordisk manufactures and markets pharmaceuticals related to the treatment of diabetes but T. Mandrup-Poulsen declares that he has no conflict of interest relevant to this present manuscript.
Abbreviations
- 8-Br-cGMP:
-
8-bromo-3′,5′-cGMP
- ERK:
-
extracellular signal-regulated kinase
- Hsp25:
-
heat shock protein-25
- IB1:
-
islet-brain 1
- iNOS:
-
inducible nitric oxide synthase
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- MAPKK:
-
MAPK kinase
- NAME:
-
N G-nitro-l-arginine-methyl ester
- NMA:
-
N G-methyl-l-arginine
- NO:
-
nitric oxide
- PI3K:
-
phosphatidylinositol 3-kinase
- PKB:
-
protein kinase B
- PKG:
-
protein kinase G
- SNAP:
-
S-nitroso-N-acetyl-d,l-penicillamine
References
Eizirik DL, Mandrup-Poulsen T (2001) A choice of death—the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia 44:2115–2133
Mabley JG, Belin V, John N, Green IC (1997) Insulin-like growth factor I reverses interleukin-1beta inhibition of insulin secretion, induction of nitric oxide synthase and cytokine-mediated apoptosis in rat islets of Langerhans. FEBS Lett 417:235–238
Ling Z, Van de Casteele M, Dong J et al (2003) Variations in IB1/JIP1 expression regulate susceptibility of beta-cells to cytokine-induced apoptosis irrespective of C-Jun NH2-terminal kinase signaling. Diabetes 52:2497–2502
Liu D, Pavlovic D, Chen MC, Flodstrom M, Sandler S, Eizirik DL (2000) Cytokines induce apoptosis in beta-cells isolated from mice lacking the inducible isoform of nitric oxide synthase (iNOS−/−). Diabetes 49:1116–1122
Zumsteg U, Frigerio S, Hollander GA (2000) Nitric oxide production and Fas surface expression mediate two independent pathways of cytokine-induced murine beta-cell damage. Diabetes 49:39–47
Andersson AK, Flodstrom M, Sandler S (2001) Cytokine-induced inhibition of insulin release from mouse pancreatic beta-cells deficient in inducible nitric oxide synthase. Biochem Biophys Res Commun 281:396–403
Flodstrom M, Tyrberg B, Eizirik DL, Sandler S (1999) Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes 48:706–713
Rabinovitch A, Suarez-Pinzon WL, Strynadka K et al (1994) Human pancreatic islet beta-cell destruction by cytokines is independent of nitric oxide production. J Clin Endocrinol Metab 79:1058–1062
Eizirik DL, Sandler S, Welsh N et al (1994) Cytokines suppress human islet function irrespective of their effects on nitric oxide generation. J Clin Invest 93:1968–1974
Eizirik DL, Pavlovic D (1997) Is there a role for nitric oxide in beta-cell dysfunction and damage in IDDM? Diabetes/Metab Rev 13:293–307
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103:239–252
Chen YR, Tan TH (2000) The c-Jun N-terminal kinase pathway and apoptotic signaling. Int J Oncol 16:651–662
Bonny C, Oberson A, Steinmann M, Schorderet DF, Nicod P, Waeber G (2000) IB1 reduces cytokine-induced apoptosis of insulin-secreting cells. J Biol Chem 275:16466–16472
Ammendrup A, Maillard A, Nielsen K et al (2000) The c-Jun amino-terminal kinase pathway is preferentially activated by interleukin-1 and controls apoptosis in differentiating pancreatic beta-cells. Diabetes 49:1468–1476
Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF (2001) Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes 50:77–82
Haefliger JA, Tawadros T, Meylan L et al (2003) The scaffold protein IB1/JIP-1 is a critical mediator of cytokine-induced apoptosis in pancreatic beta cells. J Cell Sci 116:1463–1469
Aikin R, Maysinger D, Rosenberg L (2004) Cross-talk between phosphatidylinositol 3-kinase/AKT and c-Jun NH2-terminal kinase mediates survival of isolated human islets. Endocrinology 145:4522–4531
Larsen CM, Wadt KA, Juhl LF et al (1998) Interleukin-1beta-induced rat pancreatic islet nitric oxide synthesis requires both the p38 and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinases. J Biol Chem 273:15294–15300
Pavlovic D, Andersen NA, Mandrup-Poulsen T, Eizirik DL (2000) Activation of extracellular signal-regulated kinase (ERK)1/2 contributes to cytokine-induced apoptosis in purified rat pancreatic beta-cells. Eur Cytokine Netw 11:267–274
Saldeen J, Lee JC, Welsh N (2001) Role of p38 mitogen-activated protein kinase (p38 MAPK) in cytokine- induced rat islet cell apoptosis. Biochem Pharmacol 61:1561–1569
Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13:2905–2927
Brazil DP, Park J, Hemmings BA (2002) PKB binding proteins. Getting in on the Akt. Cell 111:293–303
Jambal P, Masterson S, Nesterova A et al (2003) Cytokine-mediated down-regulation of the transcription factor cAMP-response element-binding protein in pancreatic beta-cells. J Biol Chem 278:23055–23065
Tuttle RL, Gill NS, Pugh W et al (2001) Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 7:1133–1137
Harrison M, Dunger AM, Berg S et al (1998) Growth factor protection against cytokine-induced apoptosis in neonatal rat islets of Langerhans: role of Fas. FEBS Lett 435:207–210
Castrillo A, Bodelon OG, Bosca L (2000) Inhibitory effect of IGF-I on type 2 nitric oxide synthase expression in Ins-1 cells and protection against activation-dependent apoptosis: involvement of phosphatidylinositol 3-kinase. Diabetes 49:209–217
Giannoukakis N, Mi Z, Rudert WA, Gambotto A, Trucco M, Robbins P (2000) Prevention of beta cell dysfunction and apoptosis activation in human islets by adenoviral gene transfer of the insulin-like growth factor I. Gene Ther 7:2015–2022
Hill DJ, Petrik J, Arany E, McDonald TJ, Delovitch TL (1999) Insulin-like growth factors prevent cytokine-mediated cell death in isolated islets of Langerhans from pre-diabetic non-obese diabetic mice. J Endocrinol 161:153–165
Kaino Y, Hirai H, Ito T, Kida K (1996) Insulin-like growth factor I (IGF-I) delays the onset of diabetes in non-obese diabetic (NOD) mice. Diabetes Res Clin Pract 34:7–11
George M, Ayuso E, Casellas A, Costa C, Devedjian JC, Bosch F (2002) Beta cell expression of IGF-I leads to recovery from type 1 diabetes. J Clin Invest 109:1153–1163
Andersson AK, Thorvaldson L, Carlsson C, Sandler S (2004) Cytokine-induced PGE(2) formation is reduced from iNOS deficient murine islets. Mol Cell Endocrinol 220:21–29
Sandler S, Andersson A, Hellerstrom C (1987) Inhibitory effects of interleukin 1 on insulin secretion, insulin biosynthesis, and oxidative metabolism of isolated rat pancreatic islets. Endocrinology 121:1424–1431
Lehmann R, Weber M, Berthold P et al (2004) Successful simultaneous islet-kidney transplantation using a steroid-free immunosuppression: two-year follow-up. Am J Transplant 4:1117–1123
Park HS, Huh SH, Kim MS, Lee SH, Choi EJ (2000) Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc Natl Acad Sci U S A 97:14382–14387
Scarim AL, Nishimoto SY, Weber SM, Corbett JA (2003) Role for c-Jun N-terminal kinase in beta-cell recovery from nitric oxide-mediated damage. Endocrinology 144:3415–3422
Soh JW, Mao Y, Liu L, Thompson WJ, Pamukcu R, Weinstein IB (2001) Protein kinase G activates the JNK1 pathway via phosphorylation of MEKK1. J Biol Chem 276:16406–16410
Soh JW, Mao Y, Kim MG et al (2000) Cyclic GMP mediates apoptosis induced by sulindac derivatives via activation of c-Jun NH2-terminal kinase 1. Clin Cancer Res 6:4136–4141
Lander HM, Jacovina AT, Davis RJ, Tauras JM (1996) Differential activation of mitogen-activated protein kinases by nitric oxide-related species. J Biol Chem 271:19705–19709
Jun CD, Oh CD, Kwak HJ et al (1999) Overexpression of protein kinase C isoforms protects RAW 264.7 macrophages from nitric oxide-induced apoptosis: involvement of c-Jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and CPP-32 protease pathways. J Immunol 162:3395–3401
Huwiler A, Pfeilschifter J (1999) Nitric oxide stimulates the stress-activated protein kinase p38 in rat renal mesangial cells. J Exp Biol 202:655–660
Pfeilschifter J, Huwiler A (1996) Nitric oxide stimulates stress-activated protein kinases in glomerular endothelial and mesangial cells. FEBS Lett 396:67–70
Callsen D, Brune B (1999) Role of mitogen-activated protein kinases in S-nitrosoglutathione-induced macrophage apoptosis. Biochemistry 38:2279–2286
Browning DD, Windes ND, Ye RD (1999) Activation of p38 mitogen-activated protein kinase by lipopolysaccharide in human neutrophils requires nitric oxide-dependent cGMP accumulation. J Biol Chem 274:537–542
Kim H, Shim J, Han PL, Choi EJ (1997) Nitric oxide modulates the c-Jun N-terminal kinase/stress-activated protein kinase activity through activating c-Jun N-terminal kinase kinase. Biochemistry 36:13677–13681
Bernabe JC, Tejedo JR, Rincon P, Cahuana GM, Ramirez R, Sobrino F, Bedoya FJ et al (2001) Sodium nitroprusside-induced mitochondrial apoptotic events in insulin-secreting RINm5F cells are associated with MAP kinases activation. Exp Cell Res 269:222–229
Corbett JA, Sweetland MA, Wang JL, Lancaster JR Jr, McDaniel ML (1993) Nitric oxide mediates cytokine-induced inhibition of insulin secretion by human islets of Langerhans. Proc Natl Acad Sci U S A 90:1731–1735
Cerezo A, Martinez A, Lanzarot D, Fischer S, Franke TF, Rebollo A (1998) Role of Akt and c-Jun N-terminal kinase 2 in apoptosis induced by interleukin-4 deprivation. Mol Biol Cell 9:3107–3118
Okubo Y, Blakesley VA, Stannard B, Gutkind S, Le Roith D (1998) Insulin-like growth factor-I inhibits the stress-activated protein kinase/c-Jun N-terminal kinase. J Biol Chem 273:25961–25966
Kim AH, Khursigara G, Sun X, Franke TF, Chao MV (2001) Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol 21:893–901
Acknowledgements
The technical assistance of Fie Hillesø and Hanne Foght is gratefully acknowledged. This work was conducted in collaboration with and supported by the Juvenile Diabetes Research Foundation Center for Prevention of Beta cell Destruction in Europe under grant no. 4-2002-457. It was also supported by grants from the Danish Diabetes Association, the Sehested-Hansen Foundation, the Eva and Hans Carl Adolf Holm foundation, the Danish Medical Research foundation/JC Moltons foundation, Novo Nordisk A/S, the Swedish Research Council, and the Swedish Diabetes Association.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Størling, J., Binzer, J., Andersson, A.K. et al. Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia 48, 2039–2050 (2005). https://doi.org/10.1007/s00125-005-1912-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-1912-2