
Abstract
Aims/hypothesis
Diabetogenic loci for type 2 diabetes have been mapped to mouse chromosome (Chr) 11 and 14 in the Nagoya–Shibata–Yasuda (NSY) mouse, an animal model of type 2 diabetes. We aimed to obtain direct evidence of these genes on each chromosome and to clarify their function and interaction in conferring susceptibility to type 2 diabetes.
Methods
We established three consomic strains homozygous for diabetogenic NSY-Chr11, NSY-Chr14 or both on the control C3H background (C3H-11NSY, C3H-14NSY and C3H-11NSY14NSY, respectively), and monitored diabetes-related phenotypes longitudinally. The glucokinase gene was sequenced as a positional candidate gene on Chr11.
Results
C3H-11NSY mice showed hyperglycaemia associated with impaired insulin secretion and age-dependent insulin resistance without obesity. C3H-14NSY mice exhibited hyperglycaemia mainly due to insulin resistance, with a slight increase in percentage body fat. C3H-11NSY14NSY double consomic mice showed marked hyperglycaemia and obesity, which was not observed in single consomic strains. Sequences of the glucokinase gene were allelically variant between NSY and C3H mice.
Conclusions/interpretation
These data provide direct evidence that Chr11 and Chr14 harbour major susceptibility genes for type 2 diabetes. These two chromosomes interact to cause more severe hyperglycaemia and obesity, which was not observed with the presence of either single chromosome, indicating different modes of gene–gene interaction depending on the phenotype. Marked changes in the phenotypes retained in the consomic strains will facilitate fine mapping and the identification of the responsible genes and their interaction with each other, other genes and environmental factors.
Similar content being viewed by others


Introduction
Type 2 diabetes is a multifactorial disease caused by a complex interaction of environmental and genetic factors, with the latter consisting of multiple susceptibility genes, making it difficult to clarify their functions and interactions in conferring susceptibility to diabetes in humans. Inbred animal models of diabetes are therefore invaluable in dissecting such a complex interaction.
The Nagoya–Shibata–Yasuda (NSY) mouse was established as an inbred animal model with spontaneous development of type 2 diabetes by selective breeding for glucose intolerance from the closed colony of Jcl:ICR mice [1]. The phenotypes of the mouse resemble human type 2 diabetes in that the onset is age dependent, the animal is moderately obese and both impaired insulin response to glucose and insulin resistance contribute to the disease development [2–8]. Two quantitative trait loci (QTLs) on chromosome (Chr) 11 (Nidd1n) and Chr14 (Nidd2n), which affect glucose tolerance, have been identified in crosses of NSY mice with control C3H mice [7]. Nidd1n and Nidd2n have been suggested to affect glucose tolerance through impaired insulin secretion and insulin resistance, respectively [7]. The peaks of the linkage curve for Nidd1n and Nidd2n have been positioned in the region between D11Mit236 (20.0 cM) and D11Mit195 (47.0 cM) and in the region near D14Mit59 (15.0 cM)/D14Mit5 (22.5 cM) [7]. The regions showing significant linkage for Nidd1n and Nidd2n were broad, however, suggesting the possibility that multiple genes on the same chromosome contribute to the linkage of the regions, as was evidenced by the contribution of multiple susceptibility genes on the same chromosome to susceptibility to diabetes in the NOD mouse model of type 1 diabetes [9–11]. In fact, in addition to Nidd1n in the central part of Chr11, the existence of another locus near D11Mit76 (2.0 cM), the most centromeric region on Chr11, distinct from the Nidd1n region, was suggested in our previous study [7].
The present study was performed to obtain direct evidence for susceptibility genes for type 2 diabetes on Chr11 and Chr14, and to clarify their function as well as interaction in conferring susceptibility to type 2 diabetes. To this end, we adopted a consomic approach [12], in which a whole chromosome of interest was introgressed onto the genetic background of the control strain. We first constructed two homozygous consomic strains, namely C3H-11NSY and C3H-14NSY mice, which carry an NSY-derived susceptible Chr11 or Chr14, respectively, on the control C3H background. Then, we established a double consomic strain, C3H-11NSY14NSY, containing both NSY-Chr11 and NSY-Chr14 in homozygous states on the C3H background. Various kinds of diabetes-related phenotypes of consomic strains have been monitored carefully and longitudinally. Finally, we performed sequence analysis of the glucokinase gene (Gck) on Chr11 (1.0 cM), as a functional candidate gene with peak linkage located in the centromeric region.
Methods
Animals
The NSY colony was maintained in the animal facilities of Osaka University Graduate School of Medicine. C3H/HeNcrj mice were purchased from Charles River Laboratories (Kanagawa, Japan). All mice had free access to tap water and a standard diet (CRF-1: Oriental Yeast, Tokyo, Japan) in an air-conditioned room (22–25°C) with a 12 h light–dark cycle (6:00–18:00 hours). Experimental designs were approved by the Osaka University Graduate School of Medicine Ethics Committee. Male mice were used for all experiments.
Construction of consomic strains (C3H-11NSY and C3H-14NSY) and double consomic strain (C3H-11NSY14NSY)
C3H-11NSY mice (Electronic supplementary material [ESM] Fig. 1) were produced by mating (NSY × C3H) F1 with C3H and selecting males that were heterozygous for the whole Chr11. These male mice were mated with C3H female mice, and their male progeny, heterozygous for the whole Chr11, were used for the next generation. In this process, we adopted a marker-assisted speed congenic method [13]. Namely, in every generation after the N3 generation, background genes were typed with polymorphic markers throughout the genome, and the best male mouse, which had the most substituted C3H genotype, was selected for breeding. This process was repeated until all the markers for background typing became homozygous for C3H genotypes (N6 or N7). Mice heterozygous for Chr11 were then intercrossed to obtain mice homozygous for Chr11. A total of four mice homozygous for NSY-derived Chr11 were obtained at the N6F1 (one out of 40 mice) and N7F1 (three out of 46 mice) generations. This line was maintained by brother–sister mating.
C3H-14NSY mice (ESM Fig. 1) were constructed in the same way as for C3H-11NSY mice. Five mice homozygous for NSY-derived Chr14 were obtained in the N8F1 generation (five out of 99 mice), and the line was maintained by brother–sister mating.
C3H-11NSY14NSY mice (ESM Fig. 1) were produced by mating (C3H-11NSY × C3H-14NSY) F1 with C3H-14NSY and selecting mice that were homozygous for the NSY-derived allele at all loci on Chr14 and heterozygous for the NSY-derived allele at all loci on Chr11 (4/62 mice; 6.5%). These mice were crossed with C3H-11NSY to obtain mice that were homozygous for the NSY-derived allele at all loci on Chr11 and heterozygous for the NSY-derived allele at all loci on Chr14 (25/109 mice; 22.9%). Offspring that were homozygous for the NSY-derived allele at all loci on Chr14 as well as at all loci on Chr11 were selected (10/89 mice; 11.2%) and maintained by brother–sister mating.
Genotype analysis and localisation of markers
Genomic DNA was extracted from the tail. Information on microsatellite markers was obtained from the Mouse Genome Database (www.informatics.jax.org). The markers were amplified using PCR with primers with or without labels with 6FAM, NED or HEX. A total of 79 informative marker loci spanning the whole genome were analysed (ESM Table 1). In particular, we used 16 markers on Chr11 (average spanning less than 5 cM) and ten markers on Chr14 (average spanning less than 7 cM) to confirm no recombination, and to confirm none of the C3H-derived genome on Chr11 (in C3H-14NSY) and Chr14 (in C3H-11NSY). The non-labelled PCR products were electrophoresed on 9% polyacrylamide gels and visualised by ethidium bromide staining. The labelled PCR products were electrophoresed using an ABI 3100 sequencer (Applied Biosystems, Foster City, CA, USA) with GENESCAN 350 ROX (Applied Biosystems) as a size standard.
Phenotypic analysis
Assessment of glucose tolerance
Glucose tolerance and body weight in NSY, C3H-11NSY, C3H-14NSY, C3H-11NSY14NSY and C3H mice were monitored longitudinally at 3, 6, 9 and 12 months of age. Glucose tolerance was assessed by intraperitoneal glucose tolerance test (ipGTT) (2 g glucose/kg body weight) in overnight-fasted mice, and blood glucose level was measured at 0, 30, 60, 90 and 120 min. The area under the glucose concentration curve (gAUC) was calculated according to the trapezoidal rule. Blood glucose level was measured directly by the glucose oxidase method using Glutest E (Kyoto Daiichi Kagaku, Kyoto, Japan).
Assessment of insulin secretion
Insulin secretion in response to glucose was assessed by the insulinogenic index. IpGTT (2 g glucose/kg body weight) was performed as described above, and plasma insulin level was measured at 0, 15 and 30 min. Plasma insulin level was measured by ELISA (Morinaga, Yokohama, Japan). Incremental AUC (ΣΔgAUC) and incremental AUC (ΣΔiAUC) were calculated according to the trapezoidal rule from the glucose and insulin measurements at baseline (0 min), 15 and 30 min. Insulinogenic index was calculated as ΣΔiAUC ÷ ΣΔgAUC.
Assessment of insulin resistance
HOMA-IR, as an indicator of insulin resistance, was calculated from the basal insulin and glucose concentrations (fasting glucose × fasting insulin).
Insulin tolerance test (ITT) was performed by injecting human insulin (0.25 U/kg) intraperitoneally in overnight-fasted mice at 3, 6, 9 and 12 months of age to evaluate insulin resistance longitudinally. Blood glucose level was measured at 0, 15, 30, 45 and 60 min.
Anatomical analysis
Anatomical phenotypes were studied at 1 year of age. After anaesthesia by i.p. injection of pentobarbital sodium (Dainippon, Osaka, Japan), body weight and anal–nasal length were measured. BMI was calculated as body weight in grams divided by the square of anal–nasal length in centimetres Mice were killed under sevoflurane, and the epididymal, mesenteric, retroperitoneal fat pads and interscapular brown adipose tissue (BAT) were dissected and weighed.
Sequence analysis of Gck as candidate gene on chromosome 11
Genomic DNA was extracted from the livers of NSY and C3H mice. Fourteen pairs of primers (ESM Table 2) were designed so that the whole ten exons and exon–intron boundaries of Gck were covered. Gck produces two isoforms, beta cell-specific and liver-specific isoforms with alternative splicing, which are different in the promoter and first exon [14], so the primer pairs were designed separately for both. Genomic DNA was amplified by PCR with these primers, and the products were purified using a Wizard PCR Preps DNA Purification System (Promega, Madison, WI, USA). The sequencing reaction was performed using Big Dye Terminator (Applied Biosystems) according to the manufacturer’s protocol, and the products were directly sequenced using an ABI 3100 sequencer (Applied Biosystems). To detect transcription binding sites, we used the TFSEARCH program (www.cbrc.jp/research/db/TFSEARCHJ.html) [15].
Statistical analysis
All results are expressed as mean ± SEM. Statistical analysis was performed by unpaired t test or one-way ANOVA. p < 0.05 was regarded as significant.
Results
Longitudinal phenotypes of C3H-11NSY mice
C3H-11NSY mice had significantly higher blood glucose levels after fasting (p < 0.0001) and at all time points after a glucose challenge (p < 0.0001) than those in C3H mice at 12 months (Fig. 1a). No significant difference in body weight was observed between the two strains at any age, except at 3 months (Fig. 1b). In the longitudinal analysis of glucose tolerance, C3H-11NSY mice showed significantly higher blood glucose levels than C3H mice at all ages studied (p < 0.0001) (Fig. 1c). These results indicate that introduction of a single NSY-Chr11 can convert normoglycaemic C3H mice to hyperglycaemic mice without a change in body weight.

Phenotypic analyses of C3H-11NSY. a IpGTT at 12 months of age in C3H-11NSY (n = 28; black squares) and C3H mice (n = 38; white squares). b Growth curve in C3H-11NSY (n = 17–56; black squares) and C3H mice (n = 21–39; white squares). Body weight was measured after fasting (3, 6, 9 and 12 months) and under ad libitum feeding (7 and 13 months). c–f Longitudinal analyses of: (c) glucose tolerance (assessed by gAUC on ipGTT; n = 28–56 C3H-11NSY and n = 33–39 C3H mice); (d) insulinogenic index (n = 15–33 C3H-11NSY and n = 18–32 C3H mice); (e) insulin resistance (assessed by decrease in glucose area in insulin tolerance test; n = 14–18 C3H-11NSY and n = 16–21 C3H mice); and (f) insulin resistance (assessed by HOMA-IR; n = 15–33 C3H-11NSY and n = 18–32 C3H mice). C3H-11NSY mice, grey bars; C3H mice, white bars. * p < 0.05, ** p < 0.01, *** p < 0.0001 compared with C3H
To better understand the mechanism of hyperglycaemia observed in C3H-11NSY mice, we evaluated insulin secretion in response to glucose and insulin resistance longitudinally. The insulinogenic indices were significantly lower in C3H-11NSY than in C3H mice at all ages (Fig. 1d). The glucose-lowering effect of insulin during ITT progressively worsened with age in C3H-11NSY mice (Fig. 1e). The HOMA-IR value was significantly higher in C3H-11NSY than in C3H mice at and after 6 months (Fig. 1f). These results suggest that both impaired insulin secretion in response to glucose and insulin resistance contribute to hyperglycaemia in C3H-11NSY mice. Furthermore, the results suggest that impaired insulin secretion in response to glucose begins as early as at 3 months and remains constant thereafter, whereas insulin resistance begins at 6 months and gets worse with age in the C3H-11NSY strain.
To further clarify the relationship of insulin resistance with body weight and abdominal fat accumulation in C3H-11NSY, anatomical analysis was performed. As shown in Table 1, body weight and BMI in C3H-11NSY mice were not significantly different from those in C3H mice. Fat-pad weight and the percentage of fat-pad weight/body weight were not significantly different either, suggesting that the age-dependent insulin resistance in C3H-11NSY mice was independent of obesity and changes in fat accumulation.
Longitudinal phenotypes of C3H-14NSY mice
C3H-14NSY mice exhibited significantly higher blood glucose levels after fasting (p < 0.0001) and at all time points after a glucose challenge (p < 0.0001) than those in C3H mice at 12 months (Fig. 2a). Body weight was slightly heavier in C3H-14NSY than in C3H mice at 3 and 6 months, but no significant difference was observed at and after 7 months (Fig. 2b). Longitudinal analysis of glucose tolerance during ipGTT showed significantly impaired glucose tolerance in C3H-14NSY compared with that in C3H mice at all ages (Fig. 2c). No significant difference in the insulinogenic index was observed at any age between the two strains (Fig. 2d), suggesting that impaired insulin secretion in response to glucose did not contribute to hyperglycaemia in C3H-14NSY mice. In contrast, the glucose-lowering effect of insulin during ITT was markedly and significantly impaired in C3H-14NSY compared with that in C3H mice at and after 6 months (Fig. 2e). HOMA-IR was also significantly higher in C3H-14NSY than in C3H mice at and after 6 months (Fig. 2f). These observations suggest that insulin resistance, but not impaired insulin secretion, contributed to glucose intolerance in the C3H-14NSY strain.

Phenotypic analyses of C3H-14NSY. a IpGTT at 12 months of age in C3H-14NSY (n = 32; black squares) and C3H mice (n = 38; white squares). b Growth curve in C3H-14NSY (n = 15–36; black squares) and C3H mice (n = 21–39; white squares). Body weight was measured after fasting (3, 6, 9 and 12 months) and under ad libitum feeding (7 and 13 months). c–f Longitudinal analyses of: (c) glucose tolerance (assessed by gAUC on ipGTT; n = 26–36 C3H-14NSY and n = 33–39 C3H mice); (d) insulinogenic index (n = 15–30 C3H-14NSY and n = 18–32 C3H mice); (e) insulin resistance (assessed by decrease in glucose area in insulin tolerance test; n = 15–20 C3H-14NSY and n = 16–21 C3H mice); and (f) insulin resistance (assessed by HOMA-IR; n = 15–30 C3H-14NSY and n = 18–32 C3H mice). C3H-14NSY mice, grey bars; C3H mice, white bars. * p < 0.05, ** p < 0.01, *** p < 0.0001 compared with C3H
To clarify the cause of insulin resistance in C3H-14NSY, anatomical analysis was performed (Table 1). No significant difference was observed in body weight, BMI and fat-pad weight between C3H-14NSY and C3H mice. The percentage of fat-pad weight/body weight, however, was slightly but significantly elevated in C3H-14NSY mice compared with C3H mice (Table 1), suggesting that an increase in the percentage of body fat may play a role in insulin resistance in C3H-14NSY mice.
Longitudinal phenotypes of double consomic strain (C3H-11NSY14NSY)
C3H-11NSY14NSY mice showed significantly higher blood glucose levels after fasting and after a glucose challenge than C3H mice (p < 0.0001) (Fig. 3a) as well as single consomics, C3H-11NSY (p < 0.05) and C3H-14NSY mice (p < 0.01). Hyperglycaemia in C3H-11NSY14NSY, however, was not as severe as in NSY mice (p < 0.0001 at 12 months) (Fig. 3a). Longitudinal analysis of glucose tolerance revealed that gAUC in C3H-11NSY14NSY was significantly higher than in C3H mice at all ages (p < 0.0001), but not as high as in NSY mice (Fig. 3c). The insulinogenic index in C3H-11NSY14NSY mice was significantly lower than in C3H mice, and was similar to that in NSY mice at all ages (Fig. 3d). The glucose-lowering effect of insulin in C3H-11NSY14NSY mice was significantly impaired as compared with C3H mice at all ages (Fig. 3e). Insulin resistance in C3H-11NSY14NSY mice, however, was not as severe as in NSY mice (Fig. 3e, f). These results indicate that the major components of genetic susceptibility to hyperglycaemia in NSY were located on NSY-Chr11 and NSY-Chr14, but that other component(s) are also necessary for full reconstitution of the NSY phenotypes.

Phenotypic analyses of C3H-11NSY14NSY. a IpGTT at 12 months of age in NSY (n = 26; black triangles), C3H-11NSY14NSY (n = 23; black squares) and C3H mice (n = 38; white squares). b Growth curve in NSY (n = 9–26; black triangles), C3H-11NSY14NSY (n = 15–24; black squares) and C3H mice (n = 21–39; white squares). Body weight was measured after fasting (3, 6, 9 and 12 months) and under ad libitum feeding (7 and 13 months). c–f Longitudinal analyses of: c glucose tolerance (assessed by gAUC on ipGTT; n = 9–26 NSY, n = 23–26 C3H-11NSY14NSY and n = 33–39 C3H mice); d insulinogenic index (n = 8–20 NSY, n = 16–22 C3H-11NSY14NSY and n = 18–32 C3H mice); e insulin resistance (assessed by decrease in glucose area in insulin tolerance test; n = 9–19 NSY, n = 20–23 C3H-11NSY14NSY and n = 16–21 C3H mice); and f insulin resistance (assessed by HOMA-IR; n = 8–20 NSY, n = 16–22 C3H-11NSY14NSY and n = 18–32 C3H mice). NSY mice, black bars; C3H-11NSY14NSY mice, grey bars; C3H mice, white bars. * p < 0.05, ** p < 0.01, *** p < 0.0001 compared with C3H
C3H-11NSY14NSY mice showed significantly higher body weight than C3H mice at all ages (p < 0.01) (Fig. 3b), in contrast to no significant change in body weight in single consomics, C3H-11NSY and C3H-14NSY. These results provide direct evidence for a genetic interaction between NSY-Chr11 and NSY-Chr14, leading to obesity.
Diabetes-related phenotypes in single consomics, C3H-11NSY and C3H-14NSY, and double consomic, C3H-11NSY14NSY, in comparison with parental strains, NSY and C3H mice
As shown in Table 1, non-fasting blood glucose and insulin in C3H-11NSY14NSY and C3H-11NSY mice were significantly higher than those in C3H mice, whereas no significant difference was observed between C3H-14NSY and C3H mice. C3H-11NSY14NSY mice showed significantly higher body weight and BMI compared with C3H mice, in contrast to no change in single consomic strains, C3H-11NSY and C3H-14NSY mice. Fat-pad weight and BAT in C3H-11NSY14NSY mice were significantly greater than in C3H mice, whereas C3H-11NSY and C3H-14NSY mice were not significantly different from C3H mice in these respects. Histologically, the heavier BAT showed deposition of fat, resembling white adipose tissue (data not shown), as was previously reported for mice with diet-induced obesity [16]. The percentage of fat-pad weight/body weight in C3H-11NSY14NSY and C3H-14NSY mice was significantly higher than in C3H mice, whereas no significant difference was observed between C3H-11NSY and C3H mice.
As shown in Table 2, C3H-11NSY14NSY showed hyperglycaemia, which appeared to be simply an additive, not synergistic, effect of NSY-Chr11 and NSY-Chr14. The insulinogenic index in C3H-11NSY14NSY mice was similar to that in C3H-11NSY and parental NSY mice at all ages (Figs. 1d, 3d and Table 2), whereas that in C3H-14NSY mice was similar to that in C3H mice at all ages (Fig. 2d and Table 2), suggesting that a major component(s) for impaired insulin secretion in response to glucose in NSY mice is located on Chr11. Insulin resistance in C3H-11NSY14NSY mice was greater than in C3H-11NSY and C3H-14NSY mice, but less than in NSY mice (Table 2). This suggests that major components for insulin resistance in NSY are located on both Chr11 and Chr14, but that these two chromosomes are not sufficient to fully reconstitute the phenotypes of the parental NSY mice.
DNA sequence of Gck as candidate gene on chromosome 11
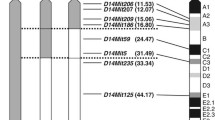
The nucleotide sequences of Gck, spanning the 5′ upstream region, 5′ untranslated region (UTR), coding region and 3′ UTR, were determined in NSY (Accession number AB255658) and C3H (Accession number AB255659) mice. As shown in Fig. 4, a total of eight variants—seven single nucleotide polymorphisms (SNPs) and one insertion/deletion—were identified between NSY and C3H mice. The SNPs in introns were not located in exon–intron boundaries. Using the TFSEARCH program, the substitution in the 5′ upstream region was shown to be not located in the known cis element. When the variants identified between NSY and C3H mice were compared with reference sequences, six out of eight polymorphisms found in NSY mice were identical to those in C57BL/6 mice, and five out of eight polymorphisms were identical among NSY, 129SV and C57BL/6 mice, but they were different from those in C3H mice. The insertion polymorphism in C3H mice in the 3′ UTR was not found in the other three strains.

Difference in Gck sequence between NSY and C3H. Exon 1P and exon 1L represent exon 1 of mRNA expressed in the pancreas and liver, respectively. Numbers represent exons. Black circles indicate different points in the sequence between NSY and C3H. Information on 129SV (Accession number L41631) and C57BL/6J strain were derived from an online database (www.ensembl.org/Mus_musculus). Dashes represent no base
Discussion
This study clearly demonstrated that substitution of a single Chr11 or Chr14 from the diabetes-resistant C3H strain to the diabetes-susceptible NSY strain caused marked changes in diabetes-related phenotypes. The mechanisms of inducing hyperglycaemia, however, appeared to be different between C3H-11NSY and C3H-14NSY mice. NSY-Chr11 affects both insulin secretion and insulin sensitivity, whereas NSY-Chr14 affects insulin sensitivity but not insulin secretion. Interestingly, impaired insulin secretion in C3H-11NSY was as severe as that in NSY mice, indicating that impaired insulin secretion in the NSY mouse could be accounted for mostly, if not totally, by Chr11. Despite the marked insulin resistance observed in C3H-11NSY mice, no significant difference was observed in adiposity and obesity between C3H-11NSY and C3H mice, suggesting that insulin resistance in C3H-11NSY mice is independent of adiposity and obesity. In contrast, C3H-14NSY mice showed a slight but significant increase in percentage fat-pad weight/body weight, suggesting that insulin resistance in C3H-14NSY mice is at least in part associated with an increase in adiposity.
Type 2 diabetes develops when the pancreatic beta cell cannot compensate for insulin resistance [17]. The capacity of insulin secretion deteriorates with age, whereas insulin resistance increases with age, at least in common forms of type 2 diabetes under polygenic control. In this study, C3H-11NSY mice showed that insulin secretion was impaired as early as at 3 months and continued to be impaired thereafter at any age. In contrast, insulin resistance worsened in an age-dependent manner. C3H-14NSY mice did not show impaired insulin secretion at any age, but showed an age-related change in insulin resistance. These results demonstrate that each susceptibility gene(s) on Chr11 and Chr14 interacts with the ageing process in the development of insulin resistance, but not of insulin secretion.
In our previous mapping, F2 mice homozygous for NSY alleles at both Nidd1n and Nidd2n showed hyperglycaemia comparable with that in the parental NSY mouse [7]. These data, together with the marked phenotypic changes in each of the single consomic strains observed in the present study, suggest that type 2 diabetes in NSY may be oligogenic, with Nidd1n and Nidd2n accounting for most of the phenotypic changes in NSY. To ascertain whether or not Nidd1n and Nidd2n are sufficient to reconstitute most of the NSY phenotypes, a double consomic strain, C3H-11NSY14NSY with both Nidd1n and Nidd2n, was produced. Although the degree of hyperglycaemia in C3H-11NSY14NSY was greater than that observed in each single consomic, it was not as severe as that in NSY. These results suggest that additional genes on other chromosomes are necessary for the full expression of diabetes-related phenotypes of NSY mice. Moreover, in contrast to the absence of obvious obesity in each single consomic strain, C3H-11NSY14NSY mice showed significant obesity, demonstrating that genetic interaction between the two chromosomes plays a role in causing obesity. Thus, the present study clearly demonstrated that major components of genetic susceptibility to type 2 diabetes in NSY are located or clustered on Chr11 and Chr14, which can account for the majority of the phenotypic difference between NSY and C3H mice, but significant interaction between these two chromosomes as well as between these and other chromosomes is necessary for full reconstitution of the phenotypes.
The syntenic region on mouse Chr11 and Chr14 is human Chr17, 5q, 7p (for Nidd1n) and Chr3p, 10q, 8p, 13q (for Nidd2n). These regions are, therefore, considered to be candidate regions containing diabetogenic genes in humans. In fact, loci associated with type 2 diabetes have been mapped by whole-genome screening in humans [18–24]. More recently, many genome-wide or large-scale association studies revealed several candidate genes for type 2 diabetes and fasting glucose, such as KCNJ11, KCNQ1, IGF2BP2, TCF7L2, MTNR1B, G6PC2, and GCKR [25–29], although the orthologues of these genes are not located on mouse Chr11 and Chr14. In mice, linkages with type 2 diabetes were also reported on Chr11 [30–33] and Chr14 [34–37] in several independent crosses (ESM Table 3).
We previously reported sequence analyses of the genes for hepatocyte nuclear factor-1β, GLUT4 and nucleoredoxin [6, 7, 38–40]. In this study, we determined the nucleotide sequences of Gck, which is mapped in the centromeric region of Chr11. Although other positional candidate genes, such as genes for insulin-like growth factor binding protein (Igfbp) 1 and 3, are also located in the centromeric region of Chr11, Gck, which encodes glucokinase, a main glucose-phosphorylating enzyme acting as a glucose sensor of pancreatic beta cells, is a good functional candidate gene for QTL, which is located in the centromeric region of Chr11, because the QTL was linked to glucose/insulin ratio as well as hyperglycaemia [7]. Heterozygous mutations in the gene for human glucokinase, GCK, have been identified in patients with MODY [41, 42]. In the general population, a polymorphism in the beta cell-specific GCK promoter is associated with hyperglycaemia [43]. In mice, impaired insulin secretion and normal histology of pancreatic islets, as observed in C3H-11NSY mice in the present study, were reported in mice with pancreatic beta cell-specific targeted disruption of Gck [44]. In a large scale mutagenesis project using N-ethyl-nitrosourea, it was reported that a number of mutations in Gck were identified in mice with the type 2 diabetes phenotype [45–47]. We found seven SNPs and one insertion/deletion polymorphism between NSY and C3H mice. The NSY allele is similar to that in C57BL/6 mice, but different from that in C3H mice. Interestingly, inbred control strains of mice have been reported to exhibit marked differences in glucose tolerance, with C57BL/6 mice having the worst and C3H mice the best glucose tolerance [48]. It is therefore reasonable to speculate that the combination of variants with weaker effects in the non-coding SNPs in Gck results in susceptibility to common forms of type 2 diabetes, whereas functional mutations in exons cause a more severe form of diabetes, as in the case of MODY in humans [42]. Functional studies, including studies on insulin secretion in isolated islets, are necessary to clarify whether or not a variant of Gck is the cause of the insulin secretory defect in NSY, C3H-11NSY and C3H-11NSY14NSY mice.
In summary, the present study clearly provides direct evidence that Chr11 and Chr14 harbour diabetogenic genes in the NSY mouse. Introgression of each single chromosome onto control mice led to marked changes in phenotype. These two chromosomes interact to cause a more severe phenotype (hyperglycaemia) or a phenotype that was not observed in a single chromosome (obesity), suggesting a different mode of gene–gene interaction depending on the phenotype. The present study indicated the usefulness of the consomic strategy in proving the localisation as well as studying the functions and interactions of susceptibility genes for multifactorial diseases in general and diabetes-related phenotypes in particular, by dissecting disease-related phenotypes into each component. Marked changes in the phenotypes retained in the consomic strain will facilitate the fine mapping and the identification of the genes responsible and their interactions. The consomic strains established in the present study are also useful to study the interaction of genes on each chromosome with environmental factors in conferring susceptibility to diabetes. These studies are now under way.
Abbreviations
- BAT:
-
Brown adipose tissue
- Chr:
-
Chromosome
- gAUC:
-
Area under the glucose concentration curve
- ipGTT:
-
Intraperitoneal glucose tolerance test
- ITT:
-
Insulin tolerance test
- NSY:
-
Nagoya–Shibata–Yasuda
- QTL:
-
Quantitative trait locus
- SNP:
-
Single nucleotide polymorphism
References
Shibata M, Yasuda B (1980) New experimental congenital diabetic mice (NSY mice). Tohoku J Exp Med 130:139–142
Hamada Y, Ikegami H, Ueda H et al (2001) Insulin secretion to glucose as well as nonglucose stimuli is impaired in spontaneously diabetic Nagoya–Shibata–Yasuda mice. Metabolism 50:1282–1285
Ikegami H, Fujisawa T, Ogihara T (2004) Mouse models of type 1 and type 2 diabetes derived from the same closed colony: genetic susceptibility shared between two types of diabetes. ILAR J 45:268–277
Itoi-Babaya M, Ikegami H, Fujisawa T et al (2007) Fatty liver and obesity: phenotypically correlated but genetically distinct traits in a mouse model of type 2 diabetes. Diabetologia 50:1641–1648
Ueda H, Ikegami H, Kawaguchi Y et al (2000) Paternal–maternal effects on phenotypic characteristics in spontaneously diabetic Nagoya–Shibata–Yasuda mice. Metabolism 49:651–656
Ueda H, Ikegami H, Kawaguchi Y et al (2000) Age-dependent changes in phenotypes and candidate gene analysis in a polygenic animal model of Type II diabetes mellitus; NSY mouse. Diabetologia 43:932–938
Ueda H, Ikegami H, Kawaguchi Y et al (1999) Genetic analysis of late-onset type 2 diabetes in a mouse model of human complex trait. Diabetes 48:1168–1174
Ueda H, Ikegami H, Yamato E et al (1995) The NSY mouse: a new animal model of spontaneous NIDDM with moderate obesity. Diabetologia 38:503–508
Ghosh S, Palmer SM, Rodrigues NR et al (1993) Polygenic control of autoimmune diabetes in nonobese diabetic mice. Nat Genet 4:404–409
Todd JA, Aitman TJ, Cornall RJ et al (1991) Genetic analysis of autoimmune type 1 diabetes mellitus in mice. Nature 351:542–547
Wicker LS, Todd JA, Prins JB, Podolin PL, Renjilian RJ, Peterson LB (1994) Resistance alleles at two non-major histocompatibility complex-linked insulin-dependent diabetes loci on chromosome 3, Idd3 and Idd10, protect nonobese diabetic mice from diabetes. J Exp Med 180:1705–1713
Singer JB, Hill AE, Burrage LC et al (2004) Genetic dissection of complex traits with chromosome substitution strains of mice. Science 304:445–448
Markel P, Shu P, Ebeling C et al (1997) Theoretical and empirical issues for marker-assisted breeding of congenic mouse strains. Nat Genet 17:280–284
Magnuson MA, Shelton KD (1989) An alternate promoter in the glucokinase gene is active in the pancreatic beta cell. J Biol Chem 264:15936–15942
Heinemeyer T, Wingender E, Reuter I et al (1998) Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res 26:362–367
Watanabe M, Houten SM, Mataki C et al (2006) Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 439:484–489
Kahn BB (1998) Type 2 diabetes: when insulin secretion fails to compensate for insulin resistance. Cell 92:593–596
Cai G, Cole SA, Freeland-Graves JH, MacCluer JW, Blangero J, Comuzzie AG (2004) Genome-wide scans reveal quantitative trait Loci on 8p and 13q related to insulin action and glucose metabolism: the San Antonio Family Heart Study. Diabetes 53:1369–1374
Demenais F, Kanninen T, Lindgren CM et al (2003) A meta-analysis of four European genome screens (GIFT Consortium) shows evidence for a novel region on chromosome 17p11.2-q22 linked to type 2 diabetes. Hum Mol Genet 12:1865–1873
Duggirala R, Blangero J, Almasy L et al (1999) Linkage of type 2 diabetes mellitus and of age at onset to a genetic location on chromosome 10q in Mexican Americans. Am J Hum Genet 64:1127–1140
Mitchell BD, Cole SA, Hsueh WC et al (2000) Linkage of serum insulin concentrations to chromosome 3p in Mexican Americans. Diabetes 49:513–516
Parker A, Meyer J, Lewitzky S et al (2001) A gene conferring susceptibility to type 2 diabetes in conjunction with obesity is located on chromosome 18p11. Diabetes 50:675–680
Pezzolesi MG, Nam M, Nagase T et al (2004) Examination of candidate chromosomal regions for type 2 diabetes reveals a susceptibility locus on human chromosome 8p23.1. Diabetes 53:486–491
Reynisdottir I, Thorleifsson G, Benediktsson R et al (2003) Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34–q35.2. Am J Hum Genet 73:323–335
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C et al (2009) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 41:89–94
Lyssenko V, Nagorny CL, Erdos MR et al (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet 41:82–88
Prokopenko I, Langenberg C, Florez JC et al (2009) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41:77–81
Prokopenko I, McCarthy MI, Lindgren CM (2008) Type 2 diabetes: new genes, new understanding. Trends Genet 24:613–621
Reiling E, van’t Riet E, Groenewoud MJ et al (2009) Combined effects of single-nucleotide polymorphisms in GCK, GCKR, G6PC2 and MTNR1B on fasting plasma glucose and type 2 diabetes risk. Diabetologia 52:1866–1870
Hirayama I, Yi Z, Izumi S et al (1999) Genetic analysis of obese diabetes in the TSOD mouse. Diabetes 48:1183–1191
Leiter EH, Reifsnyder PC, Flurkey K, Partke HJ, Junger E, Herberg L (1998) NIDDM genes in mice: deleterious synergism by both parental genomes contributes to diabetogenic thresholds. Diabetes 47:1287–1295
Takeshita S, Moritani M, Kunika K, Inoue H, Itakura M (2006) Diabetic modifier QTLs identified in F2 intercrosses between Akita and A/J mice. Mamm Genome 17:927–940
Toye AA, Lippiat JD, Proks P et al (2005) A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 48:675–686
Almind K, Kahn CR (2004) Genetic determinants of energy expenditure and insulin resistance in diet-induced obesity in mice. Diabetes 53:3274–3285
Almind K, Kulkarni RN, Lannon SM, Kahn CR (2003) Identification of interactive loci linked to insulin and leptin in mice with genetic insulin resistance. Diabetes 52:1535–1543
Reifsnyder PC, Churchill G, Leiter EH (2000) Maternal environment and genotype interact to establish diabesity in mice. Genome Res 10:1568–1578
Suto J, Matsuura S, Imamura K, Yamanaka H, Sekikawa K (1998) Genetic analysis of non-insulin-dependent diabetes mellitus in KK and KK-Ay mice. Eur J Endocrinol 139:654–661
Babaya N, Ikegami H, Fujisawa T et al (2005) Susceptibility to streptozotocin-induced diabetes is mapped to mouse chromosome 11. Biochem Biophys Res Commun 328:158–164
Ueda H, Ikegami H, Kawaguchi Y et al (2001) Mapping and promoter sequencing of HNF-1beta gene in diabetes-prone and -resistant mice. Diabetes Res Clin Pract 53:67–71
Yamada K, Ikegami H, Kawaguchi Y et al (2001) Sequence analysis of candidate genes for common susceptibility to type 1 and type 2 diabetes in mice. Endocr J 48:241–247
Froguel P, Vaxillaire M, Sun F et al (1992) Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 356:162–164
Gloyn AL (2003) Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia of infancy. Hum Mutat 22:353–362
Rose CS, Ek J, Urhammer SA et al (2005) A −30G > A polymorphism of the beta-cell-specific glucokinase promoter associates with hyperglycemia in the general population of whites. Diabetes 54:3026–3031
Terauchi Y, Sakura H, Yasuda K et al (1995) Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J Biol Chem 270:30253–30256
Aigner B, Rathkolb B, Herbach N, Hrabe de Angelis M, Wanke R, Wolf E (2008) Diabetes models by screen for hyperglycemia in phenotype-driven ENU mouse mutagenesis projects. Am J Physiol Endocrinol Metabol 294:E232–E240
Inoue M, Sakuraba Y, Motegi H et al (2004) A series of maturity onset diabetes of the young, type 2 (MODY2) mouse models generated by a large-scale ENU mutagenesis program. Hum Mol Genet 13:1147–1157
Toye AA, Moir L, Hugill A et al (2004) A new mouse model of type 2 diabetes, produced by N-ethyl-nitrosourea mutagenesis, is the result of a missense mutation in the glucokinase gene. Diabetes 53:1577–1583
Kaku K, Fiedorek FT Jr, Province M, Permutt MA (1988) Genetic analysis of glucose tolerance in inbred mouse strains. Evidence for polygenic control. Diabetes 37:707–713
Acknowledgements
We thank M. Moritani for her skilful technical assistance and M. Shibata for his contribution to establishing the NSY colony and the discussion. This study was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. 1
Schematic illustration of genomes of consomic strains (C3H-11NSY, C3H-14NSY) and double consomic strain (C3H-11NSY14NSY), which carry NSY-derived susceptible chromosome(s) on a C3H-derived resistance background. Chromosomes from the NSY strain are shown in grey and chromosomes from the C3H strain are shown in white (PDF 44 kb)
Table 1
(PDF 48 kb)
Table 2
(PDF 41 kb)
Table 3
(PDF 52 kb)
Rights and permissions
About this article
Cite this article
Babaya, N., Fujisawa, T., Nojima, K. et al. Direct evidence for susceptibility genes for type 2 diabetes on mouse chromosomes 11 and 14. Diabetologia 53, 1362–1371 (2010). https://doi.org/10.1007/s00125-010-1737-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-010-1737-5