
Abstract
Aims/hypothesis
Variation in fasting plasma glucose (FPG) within the normal range is a known risk factor for the development of type 2 diabetes. Several reports have shown that genetic variation in the genes for glucokinase (GCK), glucokinase regulatory protein (GCKR), islet-specific glucose 6 phosphatase catalytic subunit-related protein (G6PC2) and melatonin receptor type 1B (MTNR1B) is associated with FPG. In this study we examined whether these loci also contribute to type 2 diabetes susceptibility.
Methods
A random selection from the Dutch New Hoorn Study was used for replication of the association with FGP (2,361 non-diabetic participants). For the genetic association study we extended the study sample with 2,628 participants with type 2 diabetes. Risk allele counting was used to calculate a four-gene risk allele score for each individual.
Results
Variants of the GCK, G6PC2 and MTNR1B genes but not GCKR were associated with FPG (all, p ≤ 0.001; GCKR, p = 0.23). Combining these four genes in a risk allele score resulted in an increase of 0.05 mmol/l (0.04–0.07) per additional risk allele (p = 2 × 10−13). Furthermore, participants with less than three or more than five risk alleles showed significantly different type 2 diabetes susceptibility compared with the most common group with four risk alleles (OR 0.77 [0.65–0.93], p = 0.005 and OR 2.05 [1.50–2.80], p = 4 × 10−6 respectively). The age at diagnosis was also significantly associated with the number of risk alleles (p = 0.009).
Conclusions
A combined risk allele score for single-nucleotide polymorphisms in four known FPG loci is significantly associated with FPG and HbA1c in a Dutch population-based sample of non-diabetic participants. Carriers of low or high numbers of risk alleles show significantly different risks for type 2 diabetes compared with the reference group.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Variation in fasting plasma glucose (FPG) levels within the normal range are associated with an increased risk of developing type 2 diabetes and coronary heart disease [1, 2]. Furthermore, it is known that FPG is partially genetically determined [3]. Several loci influencing FPG levels have been identified. These loci encode glucokinase (GCK), glucokinase regulatory protein (GCKR) and islet-specific glucose 6 phosphatase catalytic subunit-related protein (G6PC2) [4–10]. Recently, the gene encoding melatonin receptor type 1B (MTNR1B) was identified as a fourth locus influencing FPG [11–13]. In this study we investigated the combined effect of these loci on FPG levels in the Netherlands and analysed their single and combined effects on the risk of type 2 diabetes.
Methods
Study samples
Study sample for continuous trait analysis
For this part of the study we used participants from the ongoing New Hoorn Study, a population-based cohort study in the Netherlands, which examines potential determinants of glucose intolerance and related disorders [14, 15]. From this study, 2,361 non-diabetic white participants (46% male, aged 53 ± 7 years) were selected from the original random sample of the population register of the town of Hoorn, the Netherlands. Glucose tolerance status was assessed with OGTTs using the 1999 WHO criteria [16].
Case–control sample for genetic association with type 2 diabetes
As a control sample we used all participants with normal glucose tolerance from the above-mentioned sample (n = 2,041). Subjects with impaired glucose tolerance (IGT) and/or impaired fasting glucose (IFG; n = 320) were excluded from the control group because they have an increased risk of type 2 diabetes.
For the case sample we used all known (n = 90) and newly identified (n = 90) cases from the New Hoorn Study. To improve power we added cases from Diabetes Care System West Friesland (DCS, n = 1,906) [17]. The DCS aims to improve diabetes care by coordinating diabetes care, involving all caregivers and providing education for patients in order to improve patient empowerment. Patients are referred to the DCS by their physicians and are from the same geographical region as those taking part in the New Hoorn Study. We also included 542 type 2 diabetes patients from the diabetes clinics at Leiden University Medical Centre (Leiden, the Netherlands) and VU University Medical Centre (Amsterdam, the Netherlands), who were referred to the clinic by their physicians. In total we selected 2,628 participants with type 2 diabetes (55% males, aged 64 ± 11 years) for the case–control study. All participants in our study were of white ethnicity. The study was approved by the appropriate medical ethics committees and was in accordance with the principles of the Declaration of Helsinki.
Genotyping and quality control
Based on previous publications, we selected the single-nucleotide polymorphisms (SNPs) rs1799884 in GCK [4], rs1260326 (P446L) in GCKR [7], rs560887 in G6PC2 [9] and rs10830963 in MTNR1B [11–13] for genotyping with Taqman SNP genotyping assay (Applied Biosystems, Foster City, CA, USA). All genotype frequencies were similar between the case subgroups. For quality control the allelic discrimination plots were visually observed for good clustering. Plates with bad clustering or a success rate below 95% were repeated. Next, we assessed Hardy–Weinberg equilibrium (p > 0.05) and genotyped approximately 5% duplicate samples, which all showed identical genotypes.
Statistical analysis
Differences in FPG and other clinical variables (HbA1c, 2 h glucose, triacylglycerol, LDL, HDL, total cholesterol, BMI and waist–hip ratio) were analysed in non-diabetic participants using linear regression, adjusted for BMI, age and sex as possible confounders. All analyses were performed using an additive model, because previous studies had shown that this model was the best fit. In order to combine the effects of all SNPs, risk alleles were counted and used as a sum score [18]. A risk allele was defined as an allele that results in an increased FPG as described in the literature. Differences in genotype distribution, allele frequency and risk allele scores between participants with normal glucose tolerance and those with type 2 diabetes were compared using standard contingency tables with Fisher’s exact test, and allelic ORs were calculated with logistic regression adjusted for age, sex and BMI. Subjects with either IGT or IFG were excluded from this analysis. Using Bonferroni correction for multiple hypothesis testing, p < 0.001 was considered statistically significant for association of FPG loci with clinical variables (36 tests). For the case–control study, p < 0.01 was considered significant (four tests). All statistics were calculated using SPSS 16.0 (SPSS, Chicago, IL, USA).
Power calculations
Power calculations were performed using Quanto [19]. We had an estimated power of 80% to detect a minimal per allele effect in clinical variables between 0.056 and 0.069 mmol/l, depending on allele frequency (α = 0.001). For the association study with type 2 diabetes we had an estimated power of 80% to detect a minimal OR between 1.13 and 1.16 for single gene effects (α = 0.01). For all power calculations we assumed an additive model.
Results
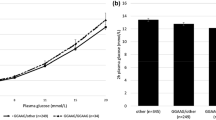
All SNPs passed quality control guidelines. Associations between SNPs and clinical variables were analysed in the non-diabetic participants only. Results of association with FPG levels were comparable to those reported in the literature (all p ≤ 0.001), except for GCKR, for which we could not detect a significant effect on FPG levels (p = 0.23; results shown in Table 1). However, GCKR showed nominal evidence for decreased 2 h glucose, but did not reach a formally significant p value (p = 0.008; see Electronic supplementary material [ESM] Table 1). Furthermore, GCK and G6PC2 showed increased HbA1c levels (p = 5 × 10−8 and 3 × 10−5; ESM Table 1). In line with our FPG results, GCKR was not associated with HbA1c levels (p = 0.50). However, we did confirm the previously reported association of the T allele of rs1260326 (GCKR) with increased triacylglycerol levels (p = 9 × 10−7; ESM Table 1) [5]. Other clinical variables were not associated with any of the analysed variants (ESM Table 1). We analysed the combined effect of all SNPs by calculating the risk allele score for each individual. We observed a combined effect of the risk alleles on FPG levels. The increase in FPG level per additional risk allele was 0.05 (0.04–0.07) mmol/l, p = 2 × 10−13 (Fig. 1a). A similar result was observed for HbA1c: 0.03% (0.02–0.04%) increase per additional risk allele, p = 5 × 10−10 (Fig. 1b). We also analysed whether the rate of the age-related increase in FPG was affected by the number of risk alleles. However, we did not observe any divergence in these rates between the different risk allele scores in our cross-sectional data set (ESM Fig. 1). Separate analysis of only the participants with normal glucose tolerance (n = 2,041) did not alter any of the results (data not shown).

Combined effect of GCK, GCKR, G6PC2 and MTNR1B on FPG and HbA1c in non-diabetic participants from the New Hoorn Study. a Fasting plasma glucose. Numbers within the bars are numbers of participants per allele group. The per allele effect was 0.05 (0.04–0.07) mmol/l, p = 2 × 10−13). Error bars represent 95% CI. b HbA1c. Numbers within the bars represent the number of participants per allele group. The per allele effect was 0.03% (0.02–0.04%), p = 5 × 10−10 Error bars represent 95% CI
Next, we analysed the association of the single gene variants and the risk allele score with type 2 diabetes susceptibility. Only rs10830963 (MTNR1B) and rs1799884 (GCK) showed weak evidence for association with type 2 diabetes (p = 0.02 and p = 0.06 respectively; Table 1 and ESM Table 2). Risk allele scores were calculated for the participants with normal glucose tolerance and those with type 2 diabetes and all risk allele groups were compared with the reference group having four risk alleles, since this was the most common group (31%). The lower risk allele groups showed a protective effect on type 2 diabetes, while the risk allele groups with more than four risk alleles showed an increased risk of type 2 diabetes (Table 2). Those with fewer than three risk alleles had a significantly reduced risk of type 2 diabetes (OR 0.77 [0.65–0.93], p = 0.005) whereas those with more than five had a significantly increased risk of type 2 diabetes compared with the reference group (OR 2.05 [1.50–2.80], p = 4 × 10−6). Adjustment for age, sex and BMI did not alter the results.
We also noted a significant correlation with the age at diagnosis of type 2 diabetes in our study sample. We observed a per allele effect of −0.46 (−0.80 to −0.11) years in age at diagnosis per additional risk allele (p = 0.009; Table 2). At the extremes of the distribution, i.e. zero or one vs six to eight risk alleles, there was a difference of almost 4.5 years in age at diagnosis between the two groups (p = 0.002; Table 2).
Discussion
Several studies have shown that SNPs in GCK, GCKR, G6PC2 and MTNR1B are associated with FPG levels [4–7, 9, 11–13, 20]. In this study we replicated these findings in a Dutch population, with the exception of the association of GCKR with FPG. However, our results for GCKR are in the same direction as those of most other studies and it should be noted that some other recent publications reported considerable variability in effect size between different samples [8] or failed to replicate this observation [11]. GCK and G6PC2 were associated with HbA1c in our study, which confirms previous observations [13, 21].
We observed a significant combined effect of all variants on FPG levels. This confirms a recent observation in a French study [13]. The association of FPG levels with the risk allele count was also reflected in increased HbA1c levels, arguing against previous findings in which it was suggested that FPG and HbA1c have independent underlying risk loci [22, 23]. Our cross-sectional data suggest that these loci cause a physiological disturbance of glucose homeostasis by raising the set point of insulin secretion, leading to an elevation of FPG depending on the number of risk alleles present, which is not further affected by ageing. However, longitudinal studies and a wider age span would be needed to confirm this observation.
To our knowledge, this is the first report showing that the analysed loci have a combined effect on type 2 diabetes susceptibility, although the contribution of each individual variant to the risk of type 2 diabetes is very low or undetectable (Table 1). Our data show that carriers of fewer than three risk alleles are at decreased risk of type 2 diabetes whereas those with more than five risk alleles have increased susceptibility to type 2 diabetes compared with the most common risk allele group of four risk alleles. We also noted a significantly different age at diagnosis between the different groups, indicating that the number of risk alleles also influences the age at which the disease becomes manifest. This might also have implications for the development of complications. If replicated, our results imply that these loci not only influence FPG levels, probably through an altered set point for glucose at which an insulin response is elicited, but also jointly increase the risk of type 2 diabetes and the age at diagnosis.
In conclusion, we replicated the combined effect of GCK, GCKR, G6PC2 and MTNR1B risk alleles with FPG. Furthermore, we showed that the risk allele score is also associated with HbA1c and that carriers of a low or high number of risk alleles have significantly different susceptibilities to the development of type 2 diabetes and age at diagnosis of the disease.
Abbreviations
- DCS:
-
Diabetes Care System West Friesland
- FPG:
-
Fasting plasma glucose
- SNP:
-
Single-nucleotide polymorphism
References
de Vegt F, Dekker JM, Ruhe HG et al (1999) Hyperglycaemia is associated with all-cause and cardiovascular mortality in the Hoorn population: the Hoorn Study. Diabetologia 42:926–931
de Vegt F, Dekker JM, Jager A et al (2001) Relation of impaired fasting and postload glucose with incident type 2 diabetes in a Dutch population: The Hoorn Study. JAMA 285:2109–2113
Snieder H, Boomsma DI, van Doornen LJ, Neale MC (1999) Bivariate genetic analysis of fasting insulin and glucose levels. Genet Epidemiol 16:426–446
Weedon MN, Frayling TM, Shields B et al (2005) Genetic regulation of birth weight and fasting glucose by a common polymorphism in the islet cell promoter of the glucokinase gene. Diabetes 54:576–581
The Diabetes Genetics Initiative of the Broad Institute of MIT and Harvard and Lund University and Novartis Institutes for BioMedical Research (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Sparso T, Andersen G, Nielsen T et al (2008) The GCKR rs780094 polymorphism is associated with elevated fasting serum triacylglycerol, reduced fasting and OGTT-related insulinaemia, and reduced risk of type 2 diabetes. Diabetologia 51:70–75
Vaxillaire M, Cavalcanti-Proenca C, Dechaume A et al (2008) The common P446L polymorphism in GCKR inversely modulates fasting glucose and triglyceride levels and reduces type 2 diabetes risk in the DESIR prospective general French population. Diabetes 57:2253–2257
Orho-Melander M, Melander O, Guiducci C et al (2008) Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes 57:3112–3121
Bouatia-Naji N, Rocheleau G, van Lommel L et al (2008) A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 320:1085–1088
Chen JF, Guo NN, Li T, Wang ED, Wang YL (2000) CP1 domain in Escherichia coli leucyl-tRNA synthetase is crucial for its editing function. Biochemistry 39:6726–6731
Prokopenko I, Langenberg C, Florez JC et al (2009) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41:77–81
Lyssenko V, Nagorny CL, Erdos MR et al (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet 41:82–88
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C et al (2009) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 41:89–94
van ’t Riet E, Rijkelijkhuizen JM, Nijpels G, Dekker JM (2008) Limited agreement between HbA1c and glucose in the general Dutch population: The New Hoorn Study. Diabetologia 51:S164 abstract
Reiling E, van Vliet-Ostaptchouk JV, van ’t Riet E et al (2009) Genetic association analysis of 13 nuclear-encoded mitochondrial candidate genes with type II diabetes mellitus: the DAMAGE Study. Eur J Hum Genet. doi:10.1038/ejhg.2009.4
World Health Organization: Definition, diagnosis and classification of Diabetes Mellitus, Report of a WHO Consultation, Part 1: Diagnosis and classification of Diabetes Mellitus, WHO/NCD/NCS/99.2, Geneva. 1-1-1999
Welschen L (2008) Disease management for patients with type 2 diabetes: towards patient empowerment. Int J Integr Care 8:e69
Weedon MN, McCarthy MI, Hitman G et al (2006) Combining information from common type 2 diabetes risk polymorphisms improves disease prediction. PLoS Med 3:e374
Gauderman WJ (2002) Sample size requirements for association studies of gene–gene interaction. Am J Epidemiol 155:478–484
Chen WM, Erdos MR, Jackson AU et al (2008) Variations in the G6PC2/ABCB11 genomic region are associated with fasting glucose levels. J Clin Invest 118:2620–2628
Pare G, Chasman DI, Parker AN et al (2008) Novel association of HK1 with glycated hemoglobin in a non-diabetic population: a genome-wide evaluation of 14,618 participants in the Women’s Genome Health Study. PLoS Genet 4:e1000312
Snieder H, Sawtell PA, Ross L, Walker J, Spector TD, Leslie RD (2001) HbA(1c) levels are genetically determined even in type 1 diabetes: evidence from healthy and diabetic twins. Diabetes 50:2858–2863
Simonis-Bik AM, Eekhoff EM, Diamant M et al (2008) The heritability of HbA1c and fasting blood glucose in different measurement settings. Twin Res Hum Genet 11:597–602
Acknowledgements
The authors would like to acknowledge all participants for their cooperation. This project was funded by The Netherlands Organization for Scientific Research, ZonMW RIDE program and the Dutch Diabetes Research Foundation.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 21 kb)
ESM Table 2
(PDF 11 kb)
ESM Fig. 1
(PDF 30 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Reiling, E., van ’t Riet, E., Groenewoud, M.J. et al. Combined effects of single-nucleotide polymorphisms in GCK, GCKR, G6PC2 and MTNR1B on fasting plasma glucose and type 2 diabetes risk. Diabetologia 52, 1866–1870 (2009). https://doi.org/10.1007/s00125-009-1413-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1413-9