
Abstract
Aims/hypothesis
Most of the known actions of angiotensin II have been considered primarily to be the result of angiotensin II subtype 1 receptor activation. However, recent data suggest that the angiotensin II subtype 2 receptor (AT2R) may modulate key processes linked to atherosclerosis. The aim of this study was to investigate the role of AT2R in diabetes-associated atherosclerosis using pharmacological blockade and genetic deficiency.
Methods
Aortic plaque deposition was assessed in streptozotocin-induced diabetic apolipoprotein E (Apoe) knockout (KO) and At 2 r (also known as Agtr2)/Apoe double-KO (DKO) mice. Control and diabetic Apoe-KO mice received an AT2R antagonist PD123319 (5 mg kg−1 day−1) via osmotic minipump for 20 weeks (n = 7–8 per group).
Results
Diabetes was associated with a sixfold increase in plaque area (diabetic Apoe-KO: 12.7 ± 1.4% vs control Apoe-KO: 2.3 ± 0.4%, p < 0.001) as well as a significant increase in aortic expression of the gene At 2 r (also known as Agtr2). The increase in plaque area with diabetes was attenuated in AT2R antagonist-treated diabetic Apoe-KO mice (7.1 ± 0.5%, p < 0.05) and in diabetic At 2 r/Apoe DKO mice (9.2 ± 1.3%, p < 0.05). These benefits occurred independently of glycaemic control or BP, and were associated with downregulation of a range of pro-inflammatory cytokines, adhesion molecules, chemokines and various extracellular matrix proteins.
Conclusions/interpretation
This study provides evidence for AT2R playing a role in the development of diabetes-associated atherosclerosis. These findings suggest a potential utility of AT2R blockers in the prevention and treatment of diabetic macrovascular complications.
Similar content being viewed by others
Introduction
Atherosclerosis is a major complication of diabetes, representing the predominant cause of morbidity and mortality in both type 1 and type 2 diabetic patients [1]. The renin–angiotensin system (RAS), and in particular its effector molecule, angiotensin II (AII), participates in the development of atherosclerosis, through the regulation of at least two key processes, inflammation and fibrosis [2]. As a result, pharmacological inhibition of the RAS has been proposed as a strategy for reducing atherosclerosis, beyond BP reduction. Pharmacological agents include ACE inhibitors (ACEi), which block the conversion of the pro-hormone angiotensin I to the active hormone AII, and selective AII subtype 1 receptor blockers (ARBs), which competitively inhibit the action of AII at the AT1 receptor subtype (AT1R) [3–6]. Both classes of compounds are widely used for reducing BP in patients with type 1 and type 2 diabetes. Furthermore, it has been suggested that there is a potentially important BP-independent protective effect on the risk of CHD for ACEi that has not been demonstrated for ARBs [7], although this possibility remains controversial [8].
AII binds with similar affinity to two major receptor subtypes, AT1R and the type 2 (AT2R) receptor [9]. While most of the well-known effects of AII are mediated via AT1R, there is growing interest in effects mediated via AT2R. The AT2R is ubiquitously produced in fetal tissues, but its production declines after birth [10]. In adults, AT2R production is detectable in the pancreas, heart, adrenals, brain, kidney and vasculature [9, 11, 12]. AT2R is overproduced in pathological situations involving tissue remodelling or inflammation, including kidney damage [13–15] and atherosclerosis [16, 17]. However, the role of AT2R in diabetes-associated atherosclerosis is not completely understood. It has been postulated that AT1R and AT2R have opposing actions on proliferation and apoptosis. The proliferative properties of AII have generally been considered to be associated with AT1R, whereas AT2R is viewed as promoting apoptosis and inhibiting cell growth [18]. However, the effects of AT2R activation are not always uniform and reproducible. Indeed, there is currently no consensus on the role of AT2R in atherosclerosis, with many studies, albeit in the non-diabetic setting, suggesting either a neutral or anti-atherosclerotic effect of this AII receptor subtype [17, 19–23]. In addition, recent data suggest that AT2R may promote cell growth and inflammation [24–26].
The present study examined the role of AT2R in the context of long-term diabetes in a well-characterised and widely used model of diabetic atherosclerosis, the streptozotocin-induced diabetic apolipoprotein E knockout (Apoe-KO) mouse [27–30]. The validity of this model has recently been confirmed as appropriate for the study of diabetes-associated atherosclerosis by the National Institutes of Health (NIH)/Juvenile Diabetes Research Foundation (JDRF)-supported Animal Models of Diabetic Complications Consortium [29]. Our aim was to determine whether disruption of the AT2R would protect these animals from developing vascular lesions in the presence of diabetes. Thus, two different approaches were employed: first, a pharmacological strategy using an AT2R antagonist, PD123319, and second, a genetic strategy involving the use of At 2 r-deficient rodents.
Methods
Experimental model
Six-week-old male Apoe-KO mice (backcrossed 20 times onto a C57BL6/J background; Animal Resource Centre, Canning Vale, WA, Australia) and At 2 r (also known as Agtr2) double-KO (DKO) mice, confirmed by PCR genotyping and generated by backcrossing At 2 r-KO mice [31] on the C57BL6/J background (had ≥98% C57BL6/J background, genome scan by Jackson Laboratory; www.jax.org) into Apoe-KO mice for ten generations were housed at the Precinct Animal Centre (Baker IDI Heart and Diabetes Institute, Melbourne, VIC, Australia). The protocols followed for animal handling and experimentation were in accordance with ethical guidelines of the Alfred Medical Research and Education Precinct animal ethics committee and the National Health and Medical Research Council of Australia guidelines.
Mice were randomised to have diabetes induced via five daily i.p. injections of streptozotocin at 55 mg kg−1 day−1 (Boehringer, Mannheim, Germany) [2, 28] or sham injected with vehicle (citrate buffer, pH 4.5). On the sixth day, blood glucose was tested using Accu-Chek Advantage II test strips (Roche Diagnostics, Mannheim, Germany), and only mice with blood glucose >15 mmol/l were included as diabetic in the study (>90% of injected mice).
Control and diabetic Apoe-KO mice were then randomised to treatment with PD123319 (5 mg kg−1 day−1; Sigma-Aldrich, St Louis, MO, USA) [13, 32] or to no treatment for 20 weeks. The PD123319 drug and the vehicle (sterile Milli-Q water) were administrated s.c. via osmotic minipumps (Model 2004; Alzet, Cupertino, CA, USA), and these minipumps were replaced every 28 days. Animals were allowed access to standard mouse chow and water ad libitum. After 20 weeks, excised aortas (n = 15–20 per group) were placed in 10% neutral buffered formalin (10%, vol./vol.) and quantified for lesion area (n = 7–8 per group) before being embedded in paraffin for immunohistochemical analysis. In the remaining mice (n = 8–12 per group) aortas were snap-frozen in liquid nitrogen and stored at −80°C for subsequent RNA extraction [2, 28].
Metabolic variables and BP
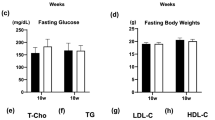
After 20 weeks, blood was collected from the left ventricle for the measurement of glycated haemoglobin (HbA1c) [33], fasting glucose, total cholesterol and triacylglycerol [34].
Systolic BP was measured at week 19 of the study period by a computerised, non-invasive tail cuff system in conscious prewarmed mice [35].
Plaque area quantification
Aortas removed from mice were cleaned of excess fat under a dissecting microscope and subsequently stained with Sudan IV-Herxheimer’s solution (0.5% wt/vol.) (Gurr; BDH, Poole, UK) as described previously [2, 28]. The aortas were divided into arch, thoracic and abdominal sections and then cut longitudinally. After pinning en face onto wax, images were acquired with a dissecting microscope (Olympus SZX10, Olympus Optical, Tokyo, Japan) with a videocamera (Q-capture Pro Version 5.1, Burnaby, BC, Canada). Total plaque area was quantified as a percentage area of the whole aorta stained by Sudan IV (Adobe Photoshop version 6.0). Aortas were subsequently embedded in paraffin and sections cut for cross-sectional analysis.
Real-time RT-PCR
Total RNA was extracted from the whole aorta by homogenising and treated with DNAse as described previously [2, 28]. cDNA was synthesised by reverse transcription (Pierce, Rockford, IL, USA). Quantitative real-time RT-PCR was performed using the Taqman System on an ABI Prism 7500 Sequence Detector (Applied Biosystems, Foster City, CA, USA) and analysed using Sequence Detection Software (SDS version 1.9; Applied Biosystems, Foster City, CA, USA). Gene expression was normalised to 18S rRNA (Applied Biosystems). Detailed information on probes is provided in the Electronic supplementary material (ESM) Table 1.
Immunostaining
Serial 4 µm paraffin aortic cross-sections were stained with haematoxylin and eosin to evaluate the atherosclerotic lesion complexity, or with Picrosirius-Red with polarisation microscopy to detect collagen content [36, 37]. Serial 4 µm paraffin aortic cross-sections were immunostained for alpha-smooth muscle actin (α-SMA) (1:500; Serotec, Oxford, UK), vascular cell adhesion molecule-1 (VCAM-1) (1:50; Pharmingen, San Diego, CA, USA), monocyte chemotactic protein-1 (MCP-1) (1:500; R&D Systems, Minneapolis, MN, USA) and the macrophage marker F4/80 (1:50; Serotec) as previously described [34]. Results were quantified as per cent of positively stained tissue. Image analysis was performed using ImageJ (http://rsb.info.nih.gov/ij, accessed 3 March 2008) and ImagePro 6.0 software (Media Cybernetics, Inc., Bethesda, MD, USA).
Statistical analysis
Data were analysed by ANOVA using Statview (version 5.0) and post hoc analysis of group means was performed by Fisher’s least significant difference method. Data are expressed as means±SEM unless otherwise specified. p < 0.05 was considered to be statistically significant.
Results
Metabolic variables and BP
The induction of diabetes in both Apoe-KO and At 2 r/Apoe-DKO mice resulted in increased plasma glucose and HbA1c concentrations, which were comparable between the diabetic groups. Plasma total cholesterol concentrations were also significantly increased in both groups following the induction of diabetes. BP was not altered after 20 weeks of diabetes in either Apoe-KO or At 2 r/Apoe-DKO groups compared with the non-diabetic groups. PD123319 treatment in the diabetic Apoe-KO mice did not alter any of these variables (Table 1).
Atherosclerotic plaque area
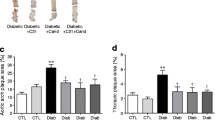
The induction of diabetes in Apoe-KO mice led to an approximately sixfold increase in atherosclerotic plaque area of the whole aorta compared with non-diabetic Apoe-KO mice (Fig. 1). In these diabetic animals lesions were predominantly complex fibrous plaques (Fig. 2). AT2R blocker (AT2RB) treatment significantly attenuated the extent and complexity of plaques in diabetic Apoe-KO mice. Plaque area was also significantly reduced in diabetic At 2 r/Apoe-DKO mice (Fig. 1). By contrast, no effect was detected in control Apoe-KO mice treated with AT2RB, nor did control At 2 r/Apoe-DKO mice show any difference in plaque area compared with control Apoe-KO mice alone.

a–f Representative en face photomicrographs of aortas with each box divided into aortic arch (left), thoracic (centre) and abdominal aorta (right) segments. g Total aortic plaque area expressed as the percentage of aortic area staining for Sudan IV Red (black, arch; white, thoracic; horizontal lines, abdominal). C, control Apoe-KO; C+PD, AT2RB-treated control Apoe-KO; C DKO, control At 2 r/Apoe-DKO; D, diabetic Apoe-KO; D+Veh, vehicle-treated diabetic Apoe-KO; D+PD, AT2RB-treated diabetic Apoe-KO; D DKO, diabetic At 2 r/Apoe-DKO. Means ± SEM. *p < 0.05 vs control Apoe-KO; † p < 0.05 vs diabetic Apoe-KO (n = 7–8 per group)

Representative thoracic aortic sections stained with Picrosirius Red with polarisation microscopy to evaluate the presence of collagen I and III in control Apoe-KO (C), AT2RB-treated control Apoe-KO (C+PD), control At 2 r/Apoe-DKO (C DKO), diabetic Apoe-KO (D), AT2RB-treated diabetic Apoe-KO (D+PD) and diabetic At 2 r/Apoe-DKO (D DKO). Magnification ×200
At2r expression
In diabetic Apoe-KO mice, aortic mRNA levels of the At 2 r gene were increased compared with control Apoe-KO mice and this increase in gene expression was decreased by AT2RB treatment (Fig. 3).

At 2 r gene expression quantified by real-time RT-PCR at the conclusion of the 20 week study in the whole aorta from the six experimental groups. C, control Apoe-KO; C+PD, AT2RB-treated control Apoe-KO; C DKO, control At 2 r/Apoe-DKO; D, diabetic Apoe-KO; D+PD, AT2RB-treated diabetic Apoe-KO; D DKO, diabetic At 2 r/Apoe-DKO. Means±SEM. *p < 0.05 vs control Apoe-KO; † p < 0.05 vs diabetic Apoe-KO. a.u., arbitrary units; ND, not detected
Inflammation
Inflammation plays a central role in the development and progression of atherosclerotic lesions. Thus, we assessed the regulation of nuclear factor-kappa B (NF-κB), a key mediator of inflammation, by measuring aortic expression of the gene encoding NF-κB subunit p65. Diabetes induced a greater than threefold increase in the expression in Apoe-KO aorta. This upregulation was significantly attenuated by both AT2RB treatment and At 2 r gene deletion (Table 2). MCP-1, an NF-κB-dependent chemokine, is considered a key mediator in AII-induced progression of atherosclerosis [38]. Indeed, in this study, diabetes was associated with a significant increase in levels of this chemokine. AT2RB treatment and At 2 r gene deletion significantly decreased the aortic production of MCP-1 (Table 2, Figs 4 and 5) as well as suppressing the induction of pro-inflammatory cytokines such as TNF-α (Table 2) and cell adhesion molecules such as VCAM-1 (Table 2, Figs 4 and 5). Furthermore, macrophage accumulation, as detected by staining with the F4/80 marker, was increased in diabetic Apoe-KO mice compared with control Apoe-KO mice and also reduced by both AT2RB treatment and At 2 r gene deletion (Fig. 5). Macrophage staining was detected in both the media and the fibrous cap of the atherosclerotic lesions, particularly in diabetic Apoe-KO mice (Fig. 5).

Aortic protein abundance quantified by immunohistochemistry (% stained area) for MCP-1 (a), VCAM-1 (b), collagen (c) and α-SMA (d) at the conclusion of the 20 week study in control Apoe-KO (C), AT2RB-treated control Apoe-KO (C+PD), control At 2 r/Apoe-DKO (C DKO), diabetic Apoe-KO (D), AT2RB-treated diabetic Apoe-KO (D+PD) and diabetic At 2 r/Apoe-DKO (D DKO). Means±SEM. *p < 0.05 vs control Apoe-KO; †p < 0.05 vs diabetic Apoe-KO

Representative aortic sections stained for MCP-1, VCAM-1 and F4/80 in control Apoe-KO (C), diabetic Apoe-KO (D), AT2RB-treated diabetic Apoe-KO (D+PD) and diabetic At 2 r/Apoe-DKO (D DKO). Magnification ×200 for MCP-1 and VCAM-1; ×400 for F4/80
Smooth muscle cell recruitment and extracellular matrix accumulation
Atherosclerosis in diabetic Apoe-KO mice involves the proliferation and migration of medial vascular smooth muscle cells into the vessel intima, as demonstrated by increased gene and protein expression of α-SMA compared with control Apoe-KO mice. The atherosclerotic plaque in AT2RB-treated diabetic Apoe-KO mice and diabetic At 2 r/Apoe-DKO mice was significantly less complex, with reduced α-SMA production compared with diabetic Apoe-KO mice (Table 2, Fig. 4).
Extracellular matrix accumulation is a dominant feature of the diabetic plaque. Aortic expression of genes encoding fibronectin (Fn1), collagen I (Col1a1) and collagen III (Col3a1) were significantly increased in diabetic Apoe-KO mice compared with non-diabetic Apoe-KO mice. mRNA levels for these variables were decreased in AT2RB-treated diabetic Apoe-KO mice and in diabetic At 2 /Apoe-DKO mice compared with diabetic Apoe-KO mice (Table 2). Aortic fibrillar collagen content was also assessed by summing the percentage of orange-red (type I collagen) and yellow-green (type III collagen) fibres with polarisation microscopy. Collagen content was significantly increased in diabetic Apoe-KO mice compared with non-diabetic Apoe-KO mice. AT2RB treatment and At 2 r gene deletion significantly reduced collagen content compared with untreated diabetic Apoe-KO mice (Fig. 2).
Discussion
The current study has demonstrated in vivo that both AT2R blockade using a pharmacological approach and At 2 r deficiency induced by gene deletion attenuated to a similar degree diabetes-associated atherosclerosis. This occurred despite similar HbA1c and BP levels between diabetic mice with and without interruption of AT2R. Furthermore, this attenuation of diabetes-associated atherosclerosis, as a result of modulation of AT2R, was associated with reduced levels of pro-inflammatory and profibrotic molecules.
It remains controversial as to what is the best model to study diabetes-associated atherosclerosis. As recently reviewed [29], there are strengths and limitations of the various models. Multiple injections of low-dose streptozotocin has been used to induce diabetes-associated atherosclerosis in Apoe-KO mouse, and this is one of the more popular approaches to be employed by investigators [27, 28, 30]. One of the potentially confounding factors of this model is the increase in lipids seen in the diabetic mice. However, importantly in this study, despite diabetic Apoe-KO mice having elevated cholesterol levels, the diabetic At 2 r/Apoe-DKO mice and AT2RB-treated diabetic Apoe-KO mice had similar elevations in serum cholesterol, yet demonstrated reduced plaque accumulation.
AT2R is produced at very low levels in the cardiovascular system of the adult. Its rate of production changes according to age [39], vessel type and the presence of pathophysiological states associated with tissue remodelling or inflammation, including diabetes. In several models of renal and vascular damage characterised by an inflammatory response, AT2R overproduction has been described [13–15]. In this study, we have reported that At 2 r gene expression is indeed increased in the aorta from diabetic Apoe-KO mice.
The role of AT2R in the vasculature is not well defined, but it has been conventionally considered to act in an opposite manner with respect to the trophic responses mediated by the AT1R subtype. The vasodilator, antigrowth and apoptotic actions of AT2R are postulated to act in a counter-regulatory manner to those conferred by AT1R. Thus, the effects of stimulation of AT2R on the cardiovascular system have been viewed as beneficial and thus no harm has been assumed as a result of increased activation of these receptors. However, there is recent evidence to suggest that activation of AT2R could, in certain contexts, exert growth stimulatory and pro-inflammatory effects that result in complementary rather than opposite effects to AT1R activation [22]. In the context of diabetes, our findings suggest that AT2R may have deleterious effects in the development and progression of atherosclerosis. We have used two disparate approaches to interrupt AT2R-dependent pathways, first a pharmacological blocker and second the use of At 2 r/Apoe-DKO diabetic mice. While no clear anti-atherosclerotic effects of AT2RB treatment or At 2 r deficiency were observed in control animals, as reported previously by other groups [17, 20], interrupting AT2R showed clear anti-atherosclerotic effects in diabetic mice. This is consistent with the view that AT2R activation may enhance vascular injury in certain contexts, particularly in pathological conditions such as diabetes.
Our group has previously shown that the atherosclerotic plaques observed in these animals resemble the complex morphology seen in patients with diabetes, including enhanced accumulation of macrophages, foam cells and cholesterol clefts within the fatty atheroma in association with increased levels of inflammatory markers, pro-inflammatory cytokines and chemokines [2, 40, 41]. In line with these findings, Levy et al. observed that chronic AT2R inhibition by PD123319 in AII-induced hypertensive rats was associated with a reduction in aortic collagen accumulation, hypertrophy and fibrosis [42]. Although the findings from the present study suggest that AT2R plays a pro-atherogenic role in the diabetic setting, this has not been a universal finding. For example, in AII-infused or non-diabetic high-fat fed mice, two different models of atherosclerosis, blockade of AT2R action, either using an antagonist such as PD123319 [20, 43] or deletion of this receptor subtype [17, 19], showed no effect on atherosclerosis, or possibly even an increase.
It remains unexplained why in the context of diabetes we observed a potential pro-atherogenic role for AT2R in this study. Other studies in the atherosclerotic setting were performed in different contexts such as AII infusion [43] and high-fat diet [17] and therefore it is possible that AT2R plays a different role in the various experimental settings that have been examined. However, in keeping with these previously reported findings, the atherosclerosis seen in non-diabetic Apoe-KO mice was not significantly influenced by AT2R blockade or deletion in this study. Indeed, the mechanisms examined in this study that may be responsible for the development of atherosclerosis in the context of high-fat diet and diabetes may be different, although this has not been extensively investigated. This issue clearly needs to be fully clarified with the advent of AT2R agonists such as C21, which has been shown to improve cardiac function in post-myocardial infarction [44]. However, the role of such an agonist has not been assessed in models of atherosclerosis, particularly with concomitant diabetes.
Increasingly, it is appreciated that the biological actions of AT2R involve the participation of AT2R-interacting proteins and promyelocytic zinc finger proteins, but the status of these proteins in the various contexts have not yet been fully examined [45]. Other possibilities for the different results among the various studies include the differences in mouse strains from which the At 2 r/Apoe-DKO mice were generated [31, 46]. For example, in a recent study of At 2 r/Apoe-DKO mice, the At 2 r-KO mice used were on a predominantly FVB background, which is a different background from that of the Apoe-KO mouse [17]. By contrast, in the present study the At 2 r-KO mice were on a C57BL6/J background, the same background as the Apoe-KO mice that were used to generate the At 2 r/Apoe-DKO mice. Finally, in previous reports PD123319 was employed at the lower dose of 3 mg kg−1 day−1 rather than the dose of 5 mg kg−1 day−1 used in the present study and this lower dose could explain the neutral effects seen with the AT2R antagonist in these other studies [20, 43]. The dose chosen in the present study was based on pilot studies and previous experiments using in vitro autoradiography to define a dose effectively inhibiting AT2R without influencing AT1R [13, 32].
AT2R activation is involved not only in the acceleration of atherosclerotic lesion formation but also in promoting pro-inflammatory pathways considered to play an important role in diabetes-accelerated atherosclerosis. Indeed, the diabetic plaque is characterised by increased production of inflammatory cytokines, chemokines and adhesion molecules that promote leucocyte infiltration and foam cell accumulation. Gene and protein expression of VCAM-1 were upregulated in aortas from diabetic Apoe-KO mice and these variables were attenuated in both diabetic AT2RB-treated and diabetic At 2 r/Apoe-DKO mice. In addition, although activation of NF-κB by AII has been reported to be AT1R dependent, there is also a significant body of literature implicating the AT2R subtype in AII-induced NF-κB activation, as reported by Ruiz-Ortega et al. in vascular smooth muscle cells [25]. In the present study, there was upregulation of expression of genes encoding the pivotal NF-κB subunit p65 in association with upregulation of the well-characterised NF-κB-dependent chemokine, MCP-1. Production of both of these proteins was attenuated in diabetic Apoe-KO mice with either gene deletion of At 2 r or with treatment with PD123319. Furthermore, other proteins such as TNF-α that are also pro-inflammatory and linked to NF-κB activation were also modified by AT2R blockade. This prominent inflammatory phenotype previously reported by several groups in diabetic Apoe-KO mice [34] remains to be fully investigated, but it appears that these changes, including macrophage infiltration, may be more prominent in the diabetic setting than in various other pro-atherosclerotic contexts such as high-fat feeding. It is hoped that ultimately more comprehensive elucidation of diabetes-specific mechanisms of atherosclerosis with a particular emphasis on inflammation may lead to a more targeted approach to reduce cardiovascular burden in diabetes.
In this study there was a reduction in extracellular matrix accumulation with blockade or deletion of At 2 r. It remains to be determined if this is a truly beneficial effect with the possibility, albeit unproven in experimental models, that a reduction in collagen content may lead to enhanced plaque rupture. Various other approaches have been reported to reduce matrix accumulation, including blockade of the RAS [2, 28] and molecular and pharmacological strategies to interrupt the advanced glycation pathway [30, 34, 41]. Indeed, some of these strategies that reduce vascular matrix accumulation, when adapted to the clinical context, have been shown to reduce cardiovascular events [47].
The potential clinical significance of AT2R as a target for reducing atherosclerosis in diabetic patients currently remains controversial. One must be very cautious in extrapolating these positive findings linking AT2R to diabetes-associated atherosclerosis to the clinical context. Nevertheless, a recent meta-analysis has suggested the possibility of superiority of an ACEi over an AT1R blocker in reducing cardiovascular events [7]. However, the recent ONTARGET study [8] suggested no advantage of one approach to interrupt the RAS over the other, albeit that that study was performed predominantly in non-diabetic individuals who already had established cardiovascular disease at the time of commencement of the study. Thus, given that currently no AT2RBs are available for clinical use, it remains to be fully clarified whether blockade of AT2R could be a beneficial therapeutic strategy in diabetic patients, particularly since they are at a high risk of premature cardiovascular disease as a result of accelerated atherosclerosis.
Abbreviations
- ACEi:
-
ACE inhibitors
- AII:
-
Angiotensin II
- AT1R:
-
Angiotensin II subtype 1 receptor
- AT2R:
-
Angiotensin II subtype 2 receptor
- AT2RB:
-
Angiotensin II subtype 2 receptor blocker
- DKO:
-
Double knockout
- KO:
-
Knockout
- MCP-1:
-
Monocyte chemoattractant protein-1
- NF-κB:
-
Nuclear factor-kappa B
- RAS:
-
Renin–angiotensin system
- α-SMA:
-
Alpha-smooth muscle actin
- VCAM-1:
-
Vascular cell adhesion molecule-1
References
Juutilainen A, Lehto S, Ronnemaa T, Pyorala K, Laakso M (2008) Similarity of the impact of type 1 and type 2 diabetes on cardiovascular mortality in middle-aged subjects. Diabetes Care 31:714–719
Candido R, Allen TJ, Lassila M et al (2004) Irbesartan but not amlodipine suppresses diabetes-associated atherosclerosis. Circulation 109:1536–1542
Brenner BM, Cooper ME, de Zeeuw D et al (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 345:861–869
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD (1993) The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 329:1456–1462
Lewis EJ, Hunsicker LG, Clarke WR et al (2001) Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 345:851–860
Parving HH, Lehnert H, Brochner-Mortensen J, Gomis R, Andersen S, Arner P (2001) The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med 345:870–878
Turnbull F, Neal B, Pfeffer M et al (2007) Blood pressure-dependent and independent effects of agents that inhibit the renin-angiotensin system. J Hypertens 25:951–958
Yusuf S, Teo KK, Pogue J et al (2008) Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med 358:1547–1559
de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 52:415–472
Nahmias C, Strosberg AD (1995) The angiotensin AT2 receptor: searching for signal-transduction pathways and physiological function. Trends Pharmacol Sci 16:223–225
Cao Z, Bonnet F, Candido R et al (2002) Angiotensin type 2 receptor antagonism confers renal protection in a rat model of progressive renal injury. J Am Soc Nephrol 13:1773–1787
Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM (1997) Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension 30:1238–1246
Cao Z, Kelly DJ, Cox A et al (2000) Angiotensin type 2 receptor is expressed in the adult rat kidney and promotes cellular proliferation and apoptosis. Kidney Int 58:2437–2451
Hakam AC, Siddiqui AH, Hussain T (2006) Renal angiotensin II AT2 receptors promote natriuresis in streptozotocin-induced diabetic rats. Am J Physiol Renal Physiol 290:F503–F508
Ruiz-Ortega M, Esteban V, Suzuki Y et al (2003) Renal expression of angiotensin type 2 (AT2) receptors during kidney damage. Kidney Int Suppl:S21–S26
Johansson ME, Fagerberg B, Bergstrom G (2008) Angiotensin type 2 receptor is expressed in human atherosclerotic lesions. J Renin Angiotensin Aldosterone Syst 9:17–21
Sales VL, Sukhova GK, Lopez-Ilasaca MA, Libby P, Dzau VJ, Pratt RE (2005) Angiotensin type 2 receptor is expressed in murine atherosclerotic lesions and modulates lesion evolution. Circulation 112:3328–3336
Carey RM, Wang ZQ, Siragy HM (2000) Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension 35:155–163
Iwai M, Chen R, Li Z et al (2005) Deletion of angiotensin II type 2 receptor exaggerated atherosclerosis in apolipoprotein E-null mice. Circulation 112:1636–1643
Johansson ME, Wickman A, Fitzgerald SM, Gan LM, Bergstrom G (2005) Angiotensin II, type 2 receptor is not involved in the angiotensin II-mediated pro-atherogenic process in ApoE−/− mice. J Hypertens 23:1541–1549
Kim MP, Zhou M, Wahl LM (2005) Angiotensin II increases human monocyte matrix metalloproteinase-1 through the AT2 receptor and prostaglandin E2: implications for atherosclerotic plaque rupture. J Leukoc Biol 78:195–201
Levy BI (2004) Can angiotensin II type 2 receptors have deleterious effects in cardiovascular disease? Implications for therapeutic blockade of the renin-angiotensin system. Circulation 109:8–13
Steckelings UM, Kaschina E, Unger T (2005) The AT2 receptor—a matter of love and hate. Peptides 26:1401–1409
Esteban V, Lorenzo O, Ruperez M et al (2004) Angiotensin II, via AT1 and AT2 receptors and NF-kappaB pathway, regulates the inflammatory response in unilateral ureteral obstruction. J Am Soc Nephrol 15:1514–1529
Ruiz-Ortega M, Ruperez M, Esteban V et al (2006) Angiotensin II: a key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol Dial Transplant 21:16–20
Wolf G (2002) ‘The road not taken’: role of angiotensin II type 2 receptor in pathophysiology. Nephrol Dial Transplant 17:195–198
Bucciarelli LG, Wendt T, Qu W et al (2002) RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 106:2827–2835
Candido R, Jandeleit-Dahm KA, Cao Z et al (2002) Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 106:246–253
Hsueh W, Abel ED, Breslow JL et al (2007) Recipes for creating animal models of diabetic cardiovascular disease. Circ Res 100:1415–1427
Park L, Raman KG, Lee KJ et al (1998) Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 4:1025–1031
Ichiki T, Labosky PA, Shiota C et al (1995) Effects on blood pressure and exploratory behaviour of mice lacking angiotensin II type-2 receptor. Nature 377:748–750
Cao Z, Dean R, Wu L, Casley D, Cooper ME (1999) Role of angiotensin receptor subtypes in mesenteric vascular proliferation and hypertrophy. Hypertension 34:408–414
Cefalu WT, Wang ZQ, Bell-Farrow A, Kiger FD, Izlar C (1994) Glycohemoglobin measured by automated affinity HPLC correlates with both short-term and long-term antecedent glycemia. Clin Chem 40:1317–1321
Soro-Paavonen A, Watson AM, Li J et al (2008) Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes 57:2461–2469
Krege JH, Hodgin JB, Hagaman JR, Smithies O (1995) A noninvasive computerized tail-cuff system for measuring blood pressure in mice. Hypertension 25:1111–1115
Crisby M, Nordin-Fredriksson G, Shah PK, Yano J, Zhu J, Nilsson J (2001) Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation 103:926–933
Junqueira LC, Bignolas G, Brentani RR (1979) Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J 11:447–455
Aiello RJ, Bourassa PA, Lindsey S et al (1999) Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 19:1518–1525
Pinaud F, Bocquet A, Dumont O et al (2007) Paradoxical role of angiotensin II type 2 receptors in resistance arteries of old rats. Hypertension 50:96–102
Calkin AC, Forbes JM, Smith CM et al (2005) Rosiglitazone attenuates atherosclerosis in a model of insulin insufficiency independent of its metabolic effects. Arterioscler Thromb Vasc Biol 25:1903–1909
Forbes JM, Yee LT, Thallas V et al (2004) Advanced glycation end product interventions reduce diabetes-accelerated atherosclerosis. Diabetes 53:1813–1823
Levy BI, Benessiano J, Henrion D et al (1996) Chronic blockade of AT2-subtype receptors prevents the effect of angiotensin II on the rat vascular structure. J Clin Invest 98:418–425
Daugherty A, Manning MW, Cassis LA (2001) Antagonism of AT2 receptors augments angiotensin II-induced abdominal aortic aneurysms and atherosclerosis. Br J Pharmacol 134:865–870
Kaschina E, Grzesiak A, Li J et al (2008) Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin-angiotensin system in myocardial infarction? Circulation 118:2523–2532
Mogi M, Iwai M, Horiuchi M (2007) Emerging concepts of regulation of angiotensin II receptors: new players and targets for traditional receptors. Arterioscler Thromb Vasc Biol 27:2532–2539
Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK (1995) Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature 377:744–747
Heart Outcomes Prevention Evaluation Study Investigators (2000) Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet 355:253–259
Acknowledgements
We thank K. Sheehy, M. Arnstein and K. Gilbert for excellent technical assistance. This work was supported by the Juvenile Diabetes Research Foundation (JDRF), the National Institutes of Health (RO1HL083452) and the National Heart Foundation of Australia. A. Koïtka was supported by an Association de Langue Française pour l’Etude du Diabète et des Maladies Métaboliques fellowship and the Société Française d’Hypertension Artérielle. A. M. D. Watson is supported by a National Health and Medical Research Council (NHMRC) of Australia Postdoctoral Biomedical Fellowship. K. A. M. Jandeleit-Dahm and T. J. Allen are recipients of NHMRC Senior Research Fellowships. M. E. Cooper is a recipient of an NHMRC Australia Fellowship and a JDRF Scholars Award.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Author information
Authors and Affiliations
Corresponding author
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
ESM Table 1
Real time RT-PCR probe and primer designs (PDF 125 kb)
Rights and permissions
About this article
Cite this article
Koïtka, A., Cao, Z., Koh, P. et al. Angiotensin II subtype 2 receptor blockade and deficiency attenuate the development of atherosclerosis in an apolipoprotein E-deficient mouse model of diabetes. Diabetologia 53, 584–592 (2010). https://doi.org/10.1007/s00125-009-1619-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-009-1619-x