
Abstract
Aims/hypothesis
The phosphatidylinositol 3-kinase (PI3K)/Akt pathway plays a critical role in promoting the survival of pancreatic beta cells. Akt becomes activated in isolated human islets following overnight culture despite significant levels of cell death. The aim of the current study was to identify the cause of the observed increase in Akt phosphorylation in isolated islets. We hypothesised that a factor secreted by the islets in culture was acting in an autocrine manner to activate Akt.
Methods
In order to identify the stimulus of the PI3K/Akt pathway in culture, we examined the effects of different culture conditions on Akt phosphorylation and islet survival during the immediate post-isolation period.
Results
We demonstrated that islet-conditioned medium induced Akt phosphorylation in freshly isolated human islets, whereas frequent medium replacement decreased Akt phosphorylation. Following overnight culture, islet-conditioned medium contained significantly elevated levels of insulin, indicating that insulin may be responsible for the observed increase in Akt phosphorylation. Indeed, treatment with an anti-insulin antibody or with inhibitors of insulin receptor/IGF receptor 1 kinase activity suppressed Akt phosphorylation, leading to decreased islet survival. In addition, dispersion of islets into single cells also suppressed Akt phosphorylation and induced islet cell death, indicating that islet integrity is also required for maximal Akt phosphorylation.
Conclusions/interpretation
Our findings demonstrate that insulin acts in an autocrine manner to activate Akt and mediate the survival of isolated human islets. These findings provide new information on how culturing islets prior to transplantation may be beneficial to their survival by allowing for autocrine activation of the pro-survival Akt pathway.
Similar content being viewed by others

Introduction
Although successful single-donor islet transplantations have been reported recently, the proportion of islet isolations producing sufficient viable islets for transplantation remains low [1]. Even with recent improvements to the isolation procedure, such as the use of the two-layer method and a less toxic iodixanol gradient, many experienced groups isolate enough islets for transplantation from only 50% of isolations. Furthermore, a significant number of cells are lost due to apoptosis and necrosis during the immediate post-isolation period [2, 3]. Thus, a better understanding of the mechanisms controlling isolated islet cell death may lead to the development of strategies to improve islet survival following isolation.
Akt (protein kinase B) is a Ser/Thr kinase that regulates several cellular processes, including cell cycle progression, transcription, glucose uptake and apoptosis. In particular, Akt plays a critical role in mediating cell survival in response to growth factor stimuli [4]. Akt is recruited to the plasma membrane following activation of phosphatidylinositol 3-kinase (PI3K) and is activated through phosphorylation by phosphoinositide-dependent kinases 1 and 2 [5, 6]. Activated Akt can directly phosphorylate several pro-apoptotic proteins, such as BAD, human caspase-9, apoptosis signal-regulating kinase-1 and forkhead transcription factor like-1, leading to suppression of apoptotic signals (reviewed in [7]). Transgenic mice expressing a constitutively active form of Akt1 under the control of the insulin promoter display increased beta-cell mass, improved glucose tolerance and resistance to streptozotocin (STZ)-induced diabetes [8, 9]. Adenovirus-mediated overexpression of constitutively active Akt1 in isolated human islets increased in vitro beta-cell proliferation, decreased apoptosis induced by serum/glucose withdrawal or high glucose, and decreased the number of transplanted islets required to reverse STZ-induced diabetes in mice [10]. In addition, PI3K/Akt signalling has been shown to mediate survival of isolated human islets [11, 12], canine islets [13] and insulinoma cell lines [14–17]. Taken together, these findings indicate that the PI3K/Akt pathway plays a critical role in promoting the survival of beta cells.
Although the original Edmonton protocol involved transplanting islets immediately following isolation, the recovery of islets in culture prior to transplantation has once again become common practice [1, 18]. One reason for culturing islets prior to transplantation is that a significant amount of islet cell death due to the isolation occurs during the 48 h following isolation, and thus it is advantageous to allow these cells to die off in culture so as not to provoke additional local inflammation at the graft site. Indeed, porcine islets that have been cultured for at least 24 h are significantly better at reversing hyperglycaemia when transplanted into diabetic nude mice compared with freshly isolated islets [19]. In addition, there is a much higher relative expression of inflammatory genes during the immediate post-isolation period when compared with islets cultured for 1 week [20]. Culturing islets also provides an opportunity for therapies aimed at improving islet survival, inducing islet expansion, and reducing islet immunogenicity. We and others have demonstrated that immediately following isolation, human islets display low levels of Akt phosphorylation [11, 12]. However, following overnight culture in either serum-containing or serum-free medium, Akt becomes highly phosphorylated in a PI3K-dependent manner [11–13]. This increased Akt phosphorylation affords islets a certain degree of protection against insults such as inflammatory cytokines [11]. The aim of the current study was to identify the cause of the observed increase in Akt phosphorylation in isolated human islets.
Materials and methods
Human islet isolation
Pancreata (n = 19) were retrieved from heart-beating cadaveric organ donors (mean age: 50.4 ± 2.9 years, mean BMI: 26.6 ± 1.0 kg/m2, mean blood glucose: 9.1 ± 0.8 mmol/l, mean amylase levels: 254.4 ± 67.8 U/l, mean pancreas weight: 86.8 ± 4.4 g) at the time of multi-organ harvest for transplantation. Consent for donation of tissues for research was obtained by the local organ procurement organisation (Quebec-Transplant). Warm ischaemia time was approximately 5 min and cold ischaemia time, using refrigerated University of Wisconsin solution perfusion, was 3–8 h. Islets were isolated using the method of Ricordi et al. [21] as described previously [11]. The average islet yield was 389,983 ± 48,809 islet equivalents (IEQs)/isolation. Glucose-stimulated insulin secretion was routinely assessed to ensure islet functionality.
Islet cell culture
Isolated islets (>90% purity) were cultured in CMRL 1066 medium (Gibco, Burlington, ON, Canada) containing 10% fetal bovine serum (FBS) (Montreal Biotech Inc., Montreal, QC, Canada) at 37°C in a humidified atmosphere of 5% CO2. Medium was changed every other day. In some experiments, medium was supplemented with insulin (Eli Lilly, Indianapolis, IN, USA) at the indicated concentration. Azide-free monoclonal anti-insulin (Acris Antibodies, Hiddenhausen, Germany) and anti-IGF-I (Upstate, Lake Placid, NY, USA) were added to culture medium to a final concentration of 1:50. Triciribine (TCN), Akti-1/2 and CB5233705 (all from Calbiochem, San Diego, CA, USA) were used at both 1 and 10 μmol/l. The insulin receptor (IR)/IGF receptor 1 (IGFR1) inhibitors AG1024 and AGL2263 (both from Calbiochem) were used at both 1 and 10 μmol/l. 3-Isobutyl-1-methylxanthine (IBMX; Sigma-Aldrich Canada Ltd, Oakville, ON, Canada) was used at 50 μmol/l. For all compounds prepared in DMSO, the final concentration of DMSO in the culture medium was kept below 0.1%. Vehicle controls were prepared for all treatments. In some cases, islets were dispersed by first washing with Dispersion Solution (Gibco) followed by a 10-min incubation at 37°C with trypsin/EDTA (Gibco). Trypsinised islets were washed with cold CMRL 1066 medium containing 10% FBS, pipetted gently, and resuspended in CMRL 1066 containing 10% FBS. The insulin content of islet medium was assessed using a commercially available insulin ELISA kit (ALPCO Diagnostics, Windham, NH, USA).
MTT assay
Mitochondrial metabolic activity was used as an indicator of islet viability [22], and was performed with an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay as described previously [11]. Four independent samples were analysed per experiment and each experiment was performed at least three times.
Immunofluorescence
Samples were stored at 4°C in phosphate-buffered formalin until cell pellets were embedded in 2% agarose. Following routine processing and embedding, 4-μm sections were cut and dewaxed in xylene and petroleum ether. Slides were then incubated in blocking buffer (Zymed, San Francisco, CA, USA) for 15 min before being exposed overnight at 4°C to an anti-insulin antibody (Dako, Carpinteria, CA, USA) diluted 1:100 in blocking buffer. Following washing in PBS, slides were incubated with a broad-spectrum biotinylated secondary antibody (Zymed) for 30 min at room temperature, washed, and re-incubated with a streptavidin–rhodamine conjugate (Molecular Probes Inc., Eugene, OR, USA) diluted 1:200 in blocking buffer for 30 min at room temperature. Slides were washed again and re-probed with an anti-phospho-Akt antibody (Cell Signaling, Danvers, MA, USA) diluted 1:100 in blocking buffer overnight at 4°C. An FITC-conjugated anti-rabbit secondary antibody (Molecular Probes) was used at 1:200 in blocking buffer for 30 min at room temperature. Slides were coverslipped with mounting medium containing 4,6-diamidino-2-phenylindole (DAPI) (Vector, Burlingame, CA, USA), and analysed using Northern Eclipse software (Empix Imaging, Mississauga, ON, USA). Confocal microscopy was performed using a Zeiss LSM 510 Confocal Laser Scanning Microscope (Carl Zeiss Canada Ltd, Toronto, ON, Canada).
Propidium iodide staining
Whole islets were dispersed (see above) and resuspended in PBS containing 2 μmol/l propidium iodide (PI) (Molecular Probes) and 0.67 μmol/l fluorescein diacetate (FDA) (Molecular Probes). The stained islets were placed on glass slides with cover-slips and incubated for 15 min at room temperature in a humid foil-covered container. Slides were visualised under a fluorescent Olympus BX60 microscope connected via a digital video camera to a PC. Five hundred cells were counted per slide, with three slides per group for at least three independent experiments. The percentage of PI-positive cells was determined by dividing the number of PI-positive cells by the total number of cells counted.
Western blotting
Western blotting was performed as described previously [11]. Anti-Akt and anti-phospho-Akt (Ser-473) (both from Cell Signaling) were used at a dilution of 1:1,000. Densitometric analysis was performed using Scion Image (Scion Corp., Frederick, MD, USA).
Statistical analysis
All results are expressed as means ± SEM. Statistical significance was determined using a one-way ANOVA with a post-hoc Bonferroni test as well as the Student’s t test using SYSTAT 9 (SPSS Inc., Chicago, IL, USA). Differences were considered significant at p < 0.05.
Results
Akt phosphorylation is detectable 3 h after isolation and reaches maximal levels after 12 h
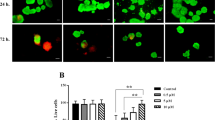
We have previously demonstrated that Akt becomes highly phosphorylated in isolated human and canine islets after 12–16 h in culture [11, 13]. In order to determine the kinetics of this signal, we examined the levels of Akt phosphorylation in human islets over the course of the first 24 h following isolation in CMRL 1066 medium (5.5 mmol/l glucose) containing 10% FBS. Using western blot analysis, an increase in Akt phosphorylation was detectable as early as 3 h following isolation (Fig. 1a). The level of Akt phosphorylation reached a maximum by 12 h, consistent with our previous findings [11, 13]. To examine whether the Akt phosphorylation detected in whole-cell lysates was occurring in insulin-producing islets, we co-stained islets cultured overnight for phospho-Akt and insulin. Indeed, significant colocalisation was observed between insulin and phospho-Akt staining (Fig. 1b), indicating that Akt is phosphorylated in insulin-producing cells of the islet.

Islet-conditioned medium contains a heat-labile factor that induces Akt phosphorylation in freshly isolated human islets. a Whole-cell lysates were taken at the indicated times following islet isolation and analysed by western blotting. Densitometric analysis was performed and the mean ratio ± SEM of phospho-Akt (P-Akt) to total Akt is shown for four independent experiments. b Islets cultured for 24 h were stained for phospho-Akt (green) and insulin (red) as described in Materials and methods. Yellow staining indicates regions co-staining for both phospho-Akt and insulin. Nuclei were visualised by DAPI staining (blue). The bar represents 50 μm. c Following isolation, human islets were cultured for 1 h, after which the medium was replaced with either fresh medium (Ctrl), islet-conditioned medium (CM) or heat-inactivated conditioned medium (HCM). After 30 min of treatment, whole-cell lysates were prepared and analysed by western blotting. Densitometric analysis was performed and the mean ratio ± SEM of phospho-Akt to total Akt is shown for three independent experiments. d Isolated islets were placed in culture and the culture medium was replaced every 6 h (MR) or control cells were replated with the same medium (Ctrl). Whole-cell lysates were prepared after 24 h and analysed by western blotting. Densitometric analysis was performed and the mean ratio ± SEM of phospho-Akt to total Akt is shown for three independent experiments. *p < 0.05 compared with untreated controls
Islet-conditioned medium induces Akt phosphorylation in freshly isolated islets
If isolated islets were secreting a factor that was acting in an autocrine manner to induce Akt phosphorylation, then addition of islet-conditioned medium to freshly isolated islets would be expected to induce Akt phosphorylation. To address this possibility, conditioned medium was collected from islets that had been cultured for 24 h, a point at which Akt is highly phosphorylated. Islets from the ensuing isolation were then treated with conditioned medium within 1 h after isolation and Akt phosphorylation was examined after 30 min. Islets treated with conditioned medium showed increased Akt phosphorylation compared with islets which received fresh, untreated medium (Fig. 1c). Conditioned medium that was heated to 65°C for 30 min lost the ability to induce Akt phosphorylation, suggesting that a heat-labile factor was responsible for augmenting Akt phosphorylation.
Regular medium replacement decreases Akt phosphorylation
Since Akt becomes highly phosphorylated after 12–16 h in culture, it is possible that the autocrine factor must accumulate in the medium to have its maximal effect. We therefore examined whether frequent medium replacement would prevent the accumulation of the secreted factor in the medium, resulting in lower Akt phosphorylation. Replacing the islet culture medium every 6 h over a 24-h period resulted in decreased Akt phosphorylation compared with islets which were replated with the same medium (Fig. 1d). This finding supports the notion that a local secreted factor is indeed responsible for the increase in Akt phosphorylation in isolated islets.
Pharmacological inhibition of the Akt pathway leads to cell death in isolated human islets
In order to demonstrate a pro-survival role for Akt signalling in freshly isolated islets, we employed three small molecule inhibitors of the Akt pathway. TCN (Akt/protein kinase B signalling inhibitor-2) is a tricyclic nucleoside that inhibits the activation of all three Akt isoforms, possibly by interacting with their pleckstrin homology (PH) domains [23]. Akti-1/2 is a novel selective allosteric inhibitor of both Akt1 and Akt2, but not Akt3, which also acts via interactions with the PH domains of Akt1/2 [24]. A benzimidazole compound, ChemBridge 5233705 (CB5233705), also blocks Akt pathway activation by targeting an unknown kinase upstream of Akt [25]. All three inhibitors decreased islet viability and caused an increase in cell death after 24 h (Fig. 2), indicating that Akt activation following isolation mediates islet cell survival. One inhibitor, CB5233705, caused greater cell death than the others, perhaps due to the fact that it blocks an upstream kinase of Akt and is not specific for Akt per se.

Akt inhibition leads to human islet cell death. Freshly isolated islets were treated immediately following isolation with either 1 or 10 μmol/l of the indicated inhibitor (triciribine [TCN]; Akti-1/2, a novel selective allosteric inhibitor of both Akt1 and Akt2, but not Akt3; or CB5233705 [CB52], which blocks Akt pathway activation by targeting an unknown kinase upstream of Akt). a Islet viability was assessed by the MTT assay after 24 h. Bars represent the mean MTT reduction expressed as a percentage of the control ± SEM for quadruplicate samples from four independent experiments. b Membrane permeability was assessed following 24 h in culture using PI/FDA staining. The number of PI-positive cells was expressed as a percentage of the total cells counted. Bars represent the mean values ± SEM for three independent experiments. **p < 0.01, ***p < 0.001 compared with untreated controls (Ctrl)
Treatment with an anti-insulin antibody decreases Akt phosphorylation and human islet survival
Since insulin is secreted by beta cells, is a strong activator of Akt [26] and is heat-sensitive [27], we examined whether insulin was the factor responsible for the observed rise in Akt phosphorylation. Using an insulin ELISA, we found that after 24 h, islet-conditioned medium contained significantly elevated levels of insulin (conditioned medium 59.2 ± 9.6 mU/ml; fresh medium 1.08 ± 0.062 μU/ml) (Fig. 3a). Freshly isolated human islets were then cultured with or without an anti-insulin antibody to sequester insulin secreted into the islet culture medium. Addition of anti-insulin antibody significantly decreased Akt phosphorylation after 24 h in culture (Fig. 3b). However, treatment with a similar concentration of an anti-IGF antibody, which binds both IGF-I and IGF-II, had no effect on Akt phosphorylation (Fig. 3b). Treatment with anti-insulin antibody also resulted in a 27% reduction in islet viability (Fig. 3c). On the other hand, an anti-IGF antibody had no significant effect on islet viability. Furthermore, addition of the anti-insulin antibody to the islet culture medium also caused an almost twofold increase in the number of PI-positive cells (Fig. 3d). Thus, isolated islets release high levels of insulin into the surrounding medium, which activates Akt and improves islet survival.

Anti-insulin antibody decreases Akt phosphorylation and induces cell death of isolated human islets. a Insulin content of fresh medium (Ctrl) and medium from islets cultured for 24 h was assessed by ELISA. Bars represent the mean insulin content ± SEM for duplicate samples from three independent experiments. b Immediately following isolation, islets were cultured with or without the addition of anti-insulin antibody (α-ins) (1:50) or anti-IGF (α-IGF) (1:50) to the culture medium. Whole-cell lysates were prepared after 24 h and assessed by western blot analysis. Densitometric analysis was performed and the mean ratio of phospho-Akt to total Akt is shown ± SEM for three independent experiments. c Islet viability was assessed by the MTT assay after 24 h. Bars represent the mean MTT reduction expressed as a percentage of the control ± SEM for quadruplicate samples from three independent experiments. d Membrane permeability was assessed following 24 h in culture using PI/FDA staining. The number of PI-positive cells was expressed as a percentage of the total cells counted. Bars represent the mean values ± SEM for three independent experiments. *p < 0.05, **p < 0.01 compared with untreated controls
Inhibition of IR/IGFR1 suppresses Akt phosphorylation and induces cell death in isolated human islets
In order to confirm that insulin signalling is responsible for the observed increase in Akt phosphorylation in cultured islets, we examined whether inhibition of insulin reception affected Akt phosphorylation and islet survival. Since at high concentrations insulin can also activate IGFR1 [28], we employed two inhibitors of both IR and IGFR1 tyrosine kinase activity: AGL2263 and AG1024. AGL2263 inhibits both IR and IGFR1 activity with an IC50 of 0.4 μmol/l and is 100-fold more specific for these receptors than for Akt itself [29]. AG1024 can also inhibit both IR and IGFR1, but appears to inhibit IGFR1 activity to a greater extent [30]. Addition of either inhibitor immediately following isolation led to decreased islet viability and increased cell death after 24 h in culture (Fig. 4a,b). Furthermore, IR/IGFR1 inhibition abolished Akt phosphorylation after 24 h in culture (Fig. 4c).

Inhibition of IR/IGFR1 suppresses Akt phosphorylation and induces cell death in isolated human islets. Freshly isolated islets were treated immediately following isolation with either 1 or 10 μmol/l of the indicated inhibitor (AGL2263 or AG1024). a Islet viability was assessed by the MTT assay after 24 h. Bars represent the mean MTT reduction expressed as a percentage of the control ± SEM for quadruplicate samples from four independent experiments. b Membrane permeability was assessed following 24 h in culture using PI/FDA staining. The number of PI-positive cells was expressed as a percentage of the total cells counted. Bars represent the mean values ± SEM for three independent experiments. c Whole-cell lysates were prepared after 24 h and analysed by western blotting. Densitometric analysis was performed and the mean ratio of phospho-Akt (P-Akt) to total Akt is shown±SEM for three independent experiments. *p < 0.05, **p < 0.01 compared with untreated controls (Ctrl)
Insulin, but not glucose, leads to increased Akt phosphorylation in isolated human islets
Immediately following isolation, Akt phosphorylation levels are low, while c-jun NH2-terminal kinase (JNK) is highly phosphorylated. Over the ensuing 24 h, JNK phosphorylation levels decrease concomitantly with a rise in Akt phosphorylation [11, 31]. Since JNK is known to suppress insulin signalling [32], it is possible that the insulin signalling pathway is suppressed in islets immediately following isolation and Akt only becomes activated following suppression of the JNK pathway. We therefore examined whether exogenous insulin could induce Akt phosphorylation in islets immediately following isolation, at a time when JNK is highly activated. Within 1 h after isolation, human islets were treated with increasing concentrations of insulin for 15 min and Akt phosphorylation was assessed. Insulin was able to cause an increase in Akt phosphorylation at a concentration of 100 nmol/l (Fig. 5a), which is within the concentration range found in islet-conditioned medium (59.2 mU/ml, ∼360 nmol/l). Therefore, signalling to Akt via the IR is functional in isolated human islets, supporting our previous finding that, in fact, the PI3K/Akt pathway suppresses JNK activation [11]. This point is further demonstrated by the finding that the treatment of isolated islets with insulin decreases JNK phosphorylation [33].

Exogenous insulin leads to increased Akt phosphorylation in freshly isolated human islets but does not improve islet survival after 48 h in culture. a Isolated islets were cultured for 1 h following isolation and then treated with increasing concentrations of insulin. After 15 min of treatment, whole-cell lysates were prepared and analysed by western blotting. Densitometric analysis was performed and the mean ratio of phospho-Akt (P-Akt) to total Akt is shown ±SEM for three independent experiments. b Freshly isolated islets were cultured for 1 h, after which they were treated for 15 min with glucose (22.2 mmol/l) or glucose in combination with IBMX (50 μmol/l). Whole-cell lysates were prepared and examined by western blot analysis. Densitometric analysis was performed and the mean ratio of phospho-Akt to total Akt is shown ±SEM for three independent experiments. c Immediately following isolation, islets were cultured with the indicated concentration of insulin. Islet viability was assessed by the MTT assay after 48 h. Bars represent the mean MTT reduction expressed as a percentage of the control ± SEM for quadruplicate samples from five independent experiments. d Immediately following isolation, islets were cultured with the indicated concentration of insulin. Membrane permeability was assessed following 24 h in culture using PI/FDA staining. The number of PI-positive cells was expressed as a percentage of the total cells counted. Bars represent the mean values ± SEM for four independent experiments. *p < 0.05 compared with untreated controls
Previous studies have suggested that glucose-induced activation of intracellular signalling pathways is mediated by the autocrine action of secreted insulin. Treatment of βTC3 cells for 2 min with 15 mmol/l glucose resulted in increased IR tyrosine phosphorylation and increased association of IRS-1 with the 85-kDa subunit of PI3K [34], and similar findings were observed in isolated rat islets [35]. Furthermore, 16.7 mmol/l glucose was able to activate Akt after only 5 min in the HIT-T15 beta-cell line [36]. Based on our finding that basal insulin secretion activates Akt following isolation, we examined whether inducers of insulin secretion can accelerate Akt phosphorylation following isolation. Treatment of freshly isolated human islets with 22.2 mmol/l glucose for 15 min had no effect on Akt phosphorylation when compared with islets cultured in serum-containing CMRL 1066 medium, which contains 5.5 mmol/l glucose (Fig. 5b). Similarly, a combination of 22.2 mmol/l glucose and IBMX, an inhibitor of cAMP phosphodiesterase that causes elevated intracellular cAMP levels that lead to increased exocytosis of insulin [37], had no significant effect on Akt phosphorylation. Therefore, in our hands, glucose was unable to induce Akt phosphorylation in human islets immediately following isolation. Similarly to these findings, treatment of isolated rat islets with high glucose had no effect on Akt phosphorylation, although 100 nmol/l insulin did [38, 39].
Addition of exogenous insulin to islet culture medium had no significant effect on islet viability
Based on the finding that autocrine insulin action improves islet survival, we examined whether addition of exogenous insulin immediately after isolation could improve cultured human islet survival. However, islets treated with up to 100 nmol/l insulin displayed no significant improvement in survival after 48 h in culture (Fig. 5c,d).
Dispersion of islets into single cells suppresses Akt phosphorylation and induced islet cell death
Although diminished, Akt phosphorylation was still observed in the presence of anti-insulin antibody, suggesting that the local concentration of insulin within the islet might be higher than in the surrounding medium and that anti-insulin antibody is not able to completely sequester intra-islet insulin. We therefore examined whether dispersion of isolated human islets into single cells affects Akt phosphorylation. Isolated human islets were trypsinised and dispersed into single cells immediately after isolation. An identical number of IEQs was cultured in both the dispersed and control cultures (1,000 IEQs/ml medium). Therefore, the dispersed islet culture (1×) contained the same number of cells/ml as the undispersed control islets. Dispersion of islets into single cells significantly decreased Akt phosphorylation and increased cell death (Fig. 6). If the decrease in Akt phosphorylation caused by dispersion was due to reduced local concentration of insulin, then increasing the cell density would increase the insulin concentration and Akt phosphorylation. However, increasing the cell density up to eightfold had no significant effect on Akt phosphorylation (Fig. 6a). These findings demonstrate that islet integrity is required for maximal Akt activation and cell survival.

Dispersion of islets into single cells suppresses Akt phosphorylation and induced islet cell death. Immediately following isolation, islets were dispersed into single cells (see Materials and methods). Dispersed cells were cultured at the indicated density, where the lowest density (1×) corresponds to the same number of cells/ml as the undispersed control islets (Ctrl). a Whole-cell lysates were prepared after 12 h in culture and examined by western blot analysis. Densitometric analysis was performed and the mean ratio of phospho-Akt (P-Akt) to total Akt is shown ±SEM for three independent experiments. b Membrane permeability was assessed following 24 h in culture using PI/FDA staining. The number of PI-positive cells was expressed as a percentage of the total cells counted. Bars represent the mean values ± SEM for three independent experiments. Disp, dispersed. *p < 0.05 compared with untreated controls (Ctrl)
Discussion
We previously demonstrated that the rise in Akt phosphorylation in cultured islets is dependent on PI3K activity and that inhibition of PI3K induced islet cell death [11, 13]. Here we present evidence that insulin released by the islets in culture activates Akt in an autocrine manner to mediate islet survival. We demonstrate that treatment with an anti-insulin antibody decreased Akt phosphorylation and induced cell death, similar to findings in dispersed rat islets where treatment with anti-insulin antibody increased the amount of TUNEL (TdT-mediated dUTP-biotin nick end labelling)-positive cells [40]. We also demonstrate that inhibition of insulin reception blocks Akt phosphorylation and leads to cell death. Finally, we confirm that inhibition of Akt induces cell death in islets. Taken together, these findings outline an autocrine survival pathway in isolated human islets where secreted insulin improves islet survival by activating Akt.
The current findings provide new information on how culturing islets prior to transplantation may improve graft survival. Indeed, treatments which activate Akt during islet culture can improve graft survival [12], indicating that elevated Akt activity could render islets less susceptible to injury during the immediate post-transplantation period. Therefore, it is plausible that culturing islets prior to transplantation allows for secreted insulin to activate Akt in an autocrine fashion, yielding islets which are more likely to survive the insults encountered immediately following transplantation. Moreover, it is tempting to speculate that the negative effects of calcineurin inhibitor-based immunosuppressants on islet survival are due to the fact that these compounds suppress insulin secretion [41], thus decreasing autocrine insulin-mediated islet survival.
Addition of exogenous insulin immediately following isolation was not able to improve short-term islet survival. Despite the fact that exogenous insulin activated Akt immediately following isolation, Akt becomes highly activated in untreated islet after 3 h and is maximally activated after 12–16 h. Thus, the additional Akt activity afforded by exogenous insulin is minimal and would explain the lack of any observed effect on islet survival. However, this finding does not rule out the possibility that exogenous insulin would have a beneficial effect in islets cultured for a longer period, where insulin expression and secretion could be diminished. In addition, it is possible that insulin signalling is partially suppressed immediately following isolation, since 100 nmol/l insulin only induced a mild increase in Akt phosphorylation. A combination of JNK inhibition and insulin treatment immediately following islet isolation may further increase Akt activation and survival.
We did not observe any increase in Akt phosphorylation following treatment with high levels of glucose. Similarly to our findings, treatment of isolated rat islets with high glucose had no effect on Akt phosphorylation, but 100 nmol/l insulin did [38, 39]. In addition, no effect of glucose on Akt was observed in INS-1 cells treated for under 1 h [15, 39, 42]. However, high glucose has been shown to activate Akt after 1 h in INS-1 cells [42, 43], and after 2 h in MIN6 cells and mouse islets [17], which does not coincide with the kinetics of glucose-stimulated insulin release. Taken together, these data suggest that the ability of glucose to activate Akt in beta cells after at least 1 h may be independent of glucose-stimulated insulin secretion [38]. It is also possible that during glucose-stimulated insulin release, the insulin signalling pathway is suppressed in beta cells for a short period of time to avoid over stimulation of insulin-mediated signalling.
Our findings demonstrate that dispersion of human islets into single cells suppresses Akt phosphorylation and induces islet cell death. Previous studies have also demonstrated high levels of cell death in dispersed islet cultures [40, 44]. One explanation for the increase in cell death upon dispersion of islets into single cells is the loss of contact with the extracellular matrix (ECM), which is mediated by the integrin receptor family. Integrin ligation is known to mediate survival through activation of Akt [45], and the loss of ECM contact upon islet dispersion could explain the observed decrease in Akt (Fig. 6). Concordantly, culturing dispersed islets in the presence of ECM proteins decreases apoptosis [46]. It is also possible that dispersed islets secrete less insulin, since aggregates of beta cells have been shown to secrete more insulin than free beta cells [47]. Alternatively, it is possible that the local concentration of insulin within the islet could be higher than in the surrounding medium. However, if the decrease in Akt phosphorylation caused by dispersion was due to reduced local concentration of insulin, then increasing the local insulin concentration by increasing the cell density would restore Akt phosphorylation, which it did not (Fig. 6). It is also possible that synergism between growth factor and integrin signalling could be affected in dispersed islets. Overall, the specific reasons for the poor survival of dispersed islets remain unclear, but it is probably due to the disruption of the islet microenvironment.
It is unclear why cell death is still occurring despite high levels of Akt activation following overnight culture of isolated islets [2, 11, 12]. This could be due to the energy requirements of Akt-mediated survival. Indeed, Akt-mediated survival in the absence of growth factors is dependent on the presence of glucose [48]. Furthermore, Akt cannot prevent apoptosis under hypoxic conditions [49]. Islets are highly vascularised structures whose function depends greatly on proper blood supply. Following isolation, islets are avascular, and revascularisation is not complete until 10–14 days following transplantation [50]. During that time, oxygen delivery to the islets is limited to diffusion. Islets are exposed to varying degrees of ischaemia throughout the isolation procedure, and culture techniques that are suitable for dispersed cell cultures may not be adequate for proper oxygen delivery throughout the entire islet. Therefore, efforts to improve oxygen delivery during culture should further improve islet survival.
Abbreviations
- DAPI:
-
4,6-diamidino-2-phenylindole
- FBS:
-
fetal bovine serum
- FDA:
-
fluorescein diacetate
- IBMX:
-
3-isobutyl-1-methylxanthine
- IEQ:
-
islet equivalent
- IGFR1:
-
insulin-like growth factor receptor 1
- IR:
-
insulin receptor
- JNK:
-
c-jun NH2-terminal kinase
- MTT:
-
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PI:
-
propidium iodide
- PI3K:
-
phosphatidylinositol 3-kinase
- TCN:
-
triciribine
References
Hering BJ, Kandaswamy R, Ansite JD et al (2005) Single-donor, marginal-dose islet transplantation in patients with type 1 diabetes. JAMA 293:830–835
Paraskevas S, Maysinger D, Wang R, Duguid TP, Rosenberg L (2000) Cell loss in isolated human islets occurs by apoptosis. Pancreas 20:270–276
Aikin R, Rosenberg L, Paraskevas S, Maysinger D (2004) Inhibition of caspase-mediated PARP-1 cleavage results in increased necrosis in isolated islets of Langerhans. J Mol Med 82:389–397
Datta SR, Brunet A, Greenberg ME (1999) Cellular survival: a play in three Akts. Genes Dev 13:2905–2927
Toker A, Newton AC (2000) Cellular signaling: pivoting around PDK-1. Cell 103:185–188
Hresko RC, Murata H, Mueckler M (2003) Phosphoinositide-dependent kinase-2 is a distinct protein kinase enriched in a novel cytoskeletal fraction associated with adipocyte plasma membranes. J Biol Chem 278:21615–21622
Downward J (2004) PI 3-kinase, Akt and cell survival. Semin Cell Dev Biol 15:177–182
Tuttle RL, Gill NS, Pugh W et al (2001) Regulation of pancreatic beta-cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 7:1133–1137
Bernal-Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA (2001) Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 108:1631–1638
Rao P, Roccisana J, Takane KK et al (2005) Gene transfer of constitutively active akt markedly improves human islet transplant outcomes in diabetic severe combined immunodeficient mice. Diabetes 54:1664–1675
Aikin R, Maysinger D, Rosenberg L (2004) Cross-talk between PI3K/AKT and JNK mediates survival of isolated human islets. Endocrinology 145:4522–4531
Contreras JL, Smyth CA, Bilbao G, Young CJ, Thompson JA, Eckhoff DE (2002) Simvastatin induces activation of the serine-threonine protein kinase AKT and increases survival of isolated human pancreatic islets. Transplantation 74:1063–1069
Aikin R, Rosenberg L, Maysinger D (2000) Phosphatidylinositol 3-kinase signaling to Akt mediates survival in isolated canine islets of Langerhans. Biochem Biophys Res Commun 277:455–461
Liu W, Chin-Chance C, Lee EJ, Lowe WL Jr (2002) Activation of phosphatidylinositol 3-kinase contributes to insulin-like growth factor I-mediated inhibition of pancreatic beta-cell death. Endocrinology 143:3802–3812
Dickson LM, Lingohr MK, McCuaig J et al (2001) Differential activation of protein kinase B and p70(S6)K by glucose and insulin-like growth factor 1 in pancreatic beta-cells (INS-1). J Biol Chem 276:21110–21120
Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ (2002) Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J Biol Chem 277:49676–49684
Srinivasan S, Bernal-Mizrachi E, Ohsugi M, Permutt MA (2002) Glucose promotes pancreatic islet beta-cell survival through a PI 3-kinase/Akt-signaling pathway. Am J Physiol Endocrinol Metab 283:E784–E793
Froud T, Ricordi C, Baidal DA et al (2005) Islet transplantation in type 1 diabetes mellitus using cultured islets and steroid-free immunosuppression: Miami experience. Am J Transplant 5:2037–2046
Rijkelijkhuizen JK, Van Der Burg MP, Tons A, Terpstra OT, Bouwman E (2006) Pretransplant culture selects for high-quality porcine islets. Pancreas 32:403–407
Johansson U, Olsson A, Gabrielsson S, Nilsson B, Korsgren O (2003) Inflammatory mediators expressed in human islets of Langerhans: implications for islet transplantation. Biochem Biophys Res Commun 308:474–479
Ricordi C, Lacy PE, Finke EH, Olack BJ, Scharp DW (1988) Automated method for isolation of human pancreatic islets. Diabetes 37:413–420
Janjic D, Wollheim CB (1992) Islet cell metabolism is reflected by the MTT (tetrazolium) colorimetric assay. Diabetologia 35:482–485
Yang L, Dan HC, Sun M et al (2004) Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res 64:4394–4399
Barnett SF, Defeo-Jones D, Fu S et al (2005) Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J 385:399–408
Kau TR, Schroeder F, Ramaswamy S et al (2003) A chemical genetic screen identifies inhibitors of regulated nuclear export of a Forkhead transcription factor in PTEN-deficient tumor cells. Cancer Cell 4:463–476
Burgering BM, Coffer PJ (1995) Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature 376:599–602
Brange J, Langkjoer L (1993) Insulin structure and stability. Pharm Biotechnol 5:315–350
De Meyts P, Whittaker J (2002) Structural biology of insulin and IGF1 receptors: implications for drug design. Nat Rev Drug Discov 1:769–783
Blum G, Gazit A, Levitzki A (2003) Development of new insulin-like growth factor-1 receptor kinase inhibitors using catechol mimics. J Biol Chem 278:40442–40454
Parrizas M, Gazit A, Levitzki A, Wertheimer E, LeRoith D (1997) Specific inhibition of insulin-like growth factor-1 and insulin receptor tyrosine kinase activity and biological function by tyrphostins. Endocrinology 138:1427–1433
Paraskevas S, Aikin R, Maysinger D et al (1999) Activation and expression of ERK, JNK, and p38 MAP-kinases in isolated islets of Langerhans: implications for cultured islet survival. FEBS Lett 455:203–208
Aguirre V, Uchida T, Yenush L, Davis R, White MF (2000) The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307). J Biol Chem 275:9047–9054
Paraskevas S, Aikin R, Maysinger D et al (2001) Modulation of JNK and p38 stress activated protein kinases in isolated islets of Langerhans: insulin as an autocrine survival signal. Ann Surg 233:124–133
Rothenberg PL, Willison LD, Simon J, Wolf BA (1995) Glucose-induced insulin receptor tyrosine phosphorylation in insulin-secreting beta-cells. Diabetes 44:802–809
Velloso LA, Carneiro EM, Crepaldi SC, Boschero AC, Saad MJ (1995) Glucose- and insulin-induced phosphorylation of the insulin receptor and its primary substrates IRS-1 and IRS-2 in rat pancreatic islets. FEBS Lett 377:353–357
Leibiger B, Leibiger IB, Moede T et al (2001) Selective insulin signaling through A and B insulin receptors regulates transcription of insulin and glucokinase genes in pancreatic beta cells. Mol Cell 7:559–570
Doyle ME, Egan JM (2003) Pharmacological agents that directly modulate insulin secretion. Pharmacol Rev 55:105–131
Wicksteed B, Alarcon C, Briaud I, Lingohr MK, Rhodes CJ (2003) Glucose-induced translational control of proinsulin biosynthesis is proportional to preproinsulin mRNA levels in islet beta-cells but not regulated via a positive feedback of secreted insulin. J Biol Chem 278:42080–42090
Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ (2003) Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes 52:974–983
Navarro-Tableros V, Sanchez-Soto MC, Garcia S, Hiriart M (2004) Autocrine regulation of single pancreatic beta-cell survival. Diabetes 53:2018–2023
Polastri L, Galbiati F, Bertuzzi F et al (2002) Secretory defects induced by immunosuppressive agents on human pancreatic beta-cells. Acta Diabetol 39:229–233
Lingohr MK, Dickson LM, McCuaig JF, Hugl SR, Twardzik DR, Rhodes CJ (2002) Activation of IRS-2-mediated signal transduction by IGF-1, but not TGF-alpha or EGF, augments pancreatic beta-cell proliferation. Diabetes 51:966–976
Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M (2001) Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes 50:2237–2243
Ris F, Hammar E, Bosco D et al (2002) Impact of integrin–matrix matching and inhibition of apoptosis on the survival of purified human beta-cells in vitro. Diabetologia 45:841–850
Khwaja A, Rodriguez-Viciana P, Wennstrom S, Warne PH, Downward J (1997) Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. EMBO J 16:2783–2793
Hammar E, Parnaud G, Bosco D et al (2004) Extracellular matrix protects pancreatic beta-cells against apoptosis: role of short- and long-term signaling pathways. Diabetes 53:2034–2041
Maes E, Pipeleers D (1984) Effects of glucose and 3′,5′-cyclic adenosine monophosphate upon reaggregation of single pancreatic B-cells. Endocrinology 114:2205–2209
Plas DR, Talapatra S, Edinger AL, Rathmell JC, Thompson CB (2001) Akt and Bcl-xL promote growth factor-independent survival through distinct effects on mitochondrial physiology. J Biol Chem 276:12041–12048
McClintock DS, Santore MT, Lee VY et al (2002) Bcl-2 family members and functional electron transport chain regulate oxygen deprivation-induced cell death. Mol Cell Biol 22:94–104
Menger MD, Yamauchi J, Vollmar B (2001) Revascularization and microcirculation of freely grafted islets of Langerhans. World J Surg 25:509–515
Acknowledgements
This work was supported by the Juvenile Diabetes Research Foundation, the Canadian Institutes for Health Research (CIHR), and the Canadian Diabetes Association (CDA). Organs were obtained in collaboration with Quebec-Transplant. L. Rosenberg is a National Scientist supported by the Fonds de la Recherches en Santé du Quebec. S. Hanley is supported by a fellowship from the CDA and the CIHR. M. Lipsett is supported by a fellowship from the Diabetic Children’s Foundation and the CIHR. The authors would also like to thank J. Ding for technical assistance and J. Tam for critically reviewing the manuscript.
Duality of interest
The authors declare no conflicts of interest with regards to the present manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Aikin, R., Hanley, S., Maysinger, D. et al. Autocrine insulin action activates Akt and increases survival of isolated human islets. Diabetologia 49, 2900–2909 (2006). https://doi.org/10.1007/s00125-006-0476-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0476-0