
Abstract
Insulin secretion from pancreatic islet beta cells is acutely regulated by a complex interplay of metabolic and electrogenic events. The electrogenic mechanism regulating insulin secretion from beta cells is commonly referred to as the ATP-sensitive K+ (KATP) channel dependent pathway. Briefly, an increase in ATP and, perhaps more importantly, a decrease in ADP stimulated by glucose metabolism depolarises the beta cell by closing KATP channels. Membrane depolarisation results in the opening of voltage-dependent Ca2+ channels, and influx of Ca2+ is the main trigger for insulin secretion. Repolarisation of pancreatic beta cell action potential is mediated by the activation of voltage-dependent K+ (Kv) channels. Various Kv channel homologues have been detected in insulin secreting cells, and recent studies have shown a role for specific Kv channels as modulators of insulin secretion. Here we review the evidence supporting a role for Kv channels in the regulation of insulin secretion and discuss the potential and the limitations for beta-cell Kv channels as therapeutic targets. Furthermore, we review recent investigations of mechanisms regulating Kv channels in beta cells, which suggest that Kv channels are active participants in the regulation of beta-cell electrical activity and insulin secretion.
Similar content being viewed by others

Glucose-stimulated insulin secretion from pancreatic beta cells is regulated by a series of electrogenic events leading to exocytosis of insulin containing granules (Fig. 1). These events, including depolarisation resulting from closure of ATP-sensitive K+ (KATP) channels, opening of voltage-dependent Ca2+ channels (VDCCs), increased intracellular Ca2+ ([Ca2+]i), and subsequent repolarisation of the membrane by voltage-dependent K+ (Kv) and Ca2+-sensitive voltage-dependent K+ (KCa) channel activation, are collectively referred to as KATP channel dependent stimulus-secretion coupling. Although KATP channel independent signals from glucose metabolism have been established as an important component of stimulus-secretion coupling within the last 10 years [1, 2], the ionic mechanisms are of primary importance in triggering and maintaining insulin secretion. The importance of the KATP channel dependent pathway is shown by the reliance of the KATP channel independent pathway on increased [Ca2+]i, and by the fact that second phase insulin secretion, the component thought most closely linked with the KATP channel independent pathway, is strongly affected by agents that perturb beta-cell membrane potential responses [3, 4, 5]. There is evidence for a KATP channel independent and Ca2+-independent stimulation of insulin secretion by glucose [6], however this effect requires activation of the cAMP/PKA and PKC signalling pathways. As suggested previously [7] the KATP channel dependent and independent signalling pathways could be more appropriately referred to as the triggering and amplifying pathways, respectively.

The KATP channel dependent mechanism for glucose-stimulated insulin secretion. Rises in circulating glucose concentrations increase intracellular ATP and decrease intracellular ADP, closing ATP-sensitive K+ (KATP) channels. This results in membrane depolarisation, opening voltage-dependent Ca2+ channels (VDCCs) and allowing a rise in the intracellular Ca2+ concentration ([Ca2+]i) that is the main trigger for insulin secretion. Also upon membrane depolarisation, voltage-dependent K+ (Kv) channels open to repolarise the action potential, limit Ca2+ entry through VDCCs, and limit insulin secretion. The sulphonylurea drugs (SU's) stimulate insulin secretion by blocking KATP channels, and tetraethylammonium (TEA) enhances insulin secretion in a glucose-dependent manner by blocking Kv channels
The ability of glucose to cause depolarisation of pancreatic beta cells was first recognised in 1968 [8], and in the 1970's this was attributed to a reduction in whole cell K+ permeability [9, 10]. In the 1980's, K+ channels that are closed by glucose [11] and ATP [12] were identified in rat beta cells. These were subsequently shown in mouse beta cells to be the same channel [13], closure of which precedes depolarisation-induced Ca2+ influx [14]. The KATP channel responsible for transducing the metabolic signal to an electrical response (Kir6.2) and its regulatory sulphonylurea receptor (SUR1) subunit were cloned in 1995 (Fig. 2) [15, 16, 17]. SUR binding and antagonism of KATP channels [18] is the primary mechanism of the anti-diabetic sulphonylurea drugs [19]. Conversely, the sulphonamide drug diazoxide opens KATP channels [20], preventing insulin secretion [21], and is used to treat hyperinsulinaemia, particularly hyperinsulinaemia in polycystic ovary syndrome, pancreatic insulinomas, and cases of persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) that are not attributable to KATP channel defects [22, 23, 24].

Some important ion channels in pancreatic beta cells. Examples are shown for the membrane topology of some of the ion channels expressed in insulin-secreting cells. All of these channels contain one or more pore-forming loop (P-loop) and all of the voltage-sensitive channels contain a transmembrane 'voltage-sensor' (+++). Also shown are sites important for sensitivity to hanatoxin (HaTX), external tetraethylammonium (TEA), sulphonylurea drugs (glibenclamide and tolbutamide), diazoxide (diaz.) and dihydropyridines (DHP- such as nifedipine). Truncating the Kv2.1 α-subunit at the site shown (scissors) resulted in our dominant-negative Kv2 construct. Functional Kv, BK (large-conductance and Ca2+ sensitive K+ channels) and KATP channels are formed by the tetrameric assembly of subunits while the VDCC pore forming subunit contains four repeated domains with similar architecture to Kv α-subunits. Although not shown, multiple accessory (non-pore forming) subunits are required for VDCC function
Action potentials in rodent beta cells were shown to result largely from activation of a Ca2+ rather than a Na+ current [25, 26]. The activation of single VDCCs in mouse beta cells was first inferred from voltage noise analysis by Atwater et al. [27]. Subsequently, VDCC currents were measured from isolated mouse [28] and human [29] beta cells. Transcripts encoding L-type Ca2+ channels (Fig. 2) thought to be largely responsible for Ca2+ influx in beta cells were identified in the early 1990's [30, 31, 32], however there is still some controversy regarding the role of other Ca2+ channels as N-type, P/Q-type and T-type channels have also been detected in insulin-secreting cells [33]. Clinically, some cases of PHHI, particularly those resulting from KATP channel defects which are therefore not responsive to diazoxide, could be treated with VDCC antagonists [34, 35, 36].
In the early 1980's the Drosophila melanogaster locus Shaker was determined to contain a gene encoding a Kv channel (Fig. 2) [37]. Subsequently, the shaker gene was cloned [38, 39, 40], followed by the first mammalian homologues in 1988 (mouse) and 1989 (rat) [41, 42]. There are 11 mammalian Kv channel families and various related families (EAG related, KCNQ or KvLQT and KCa) currently known (Table 1) [43, 44, 45, 46, 47, 48, 49, 50, 51]. Due to this diversity, coupled with the existence of heteromultimeric channels and lack of selective antagonists, the specific Kv channels mediating beta-cell repolarisation and their role in insulin secretion are only now becoming known. Recent work identifies Kv2.1 as a major contributor to voltage-dependent outward K+ currents in insulinoma cells and rodent pancreatic beta cells [3, 52]. Kv2.1 has been shown, with both a dominant-negative strategy and selective antagonists, to regulate excitability, [Ca2+]i dynamics and insulin secretion in insulinoma cells and rodent models [3, 52, 53]. Furthermore, regulation of Kv channels could contribute to the modulation of insulin secretion as evidenced by the studies showing regulation of beta-cell Kv channels by exocytotic SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins, insulin-stimulating incretin hormones, and products of glucose metabolism [54, 55, 56].
Here we discuss the identification, role and regulation of Kv channels in pancreatic beta cells as well as their potential and their limitations as therapeutic targets for the treatment of Type 2 diabetes. The evidence supporting a role for these channels in the regulation of insulin secretion is reviewed. We discuss the different voltage-dependent K+ current components observed in insulin secreting cells, their likely molecular correlates, and recent work identifying a role for specific Kv channels as regulators of secretion. The potential for tissue-specific differences in Kv channel function is also examined, as this could provide a key to the appropriate targeting of therapeutic agents. Finally, we examine evidence that hormonal stimuli and intracellular mechanisms can acutely regulate beta-cell Kv channels as a potential means for modulating excitability and insulin secretion.
Evidence supporting a role for voltage-dependent K+ currents as regulators of insulin secretion
Voltage-dependent K+ currents and beta cell stimulus-secretion coupling
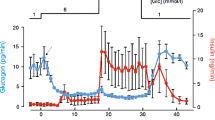
Early evidence for the role of depolarisation as a key feature of stimulus-secretion coupling in the beta cell was provided by studies carried out using high resistance microelectrode measurement of electrical activity in mouse islet cells [8], which was subsequently linked to insulin secretion [25, 57, 58]. It was clear from these early studies that glucose stimulates action potentials in pancreatic beta cells. As early as 1952, the work of Hodgkin and Huxley showed that action potential repolarisation in giant squid axon was an active process mediated by activation of a K+ permeability [59, 60]. It could therefore be reasonably hypothesised, even in the late 1960's and early 1970's that a repolarising K+ current is involved in regulating beta-cell electrical activity and insulin secretion. The first direct evidence that repolarising outward K+ currents are involved in insulin secretion is from studies showing that the general Kv and KCa channel antagonist tetraethylammonium (TEA) prolongs mouse beta-cell action potentials [61] and enhances insulin secretion from rat islets (Fig. 3) [4, 5, 62]. Since these initial experiments, numerous studies have examined the effect of TEA on beta-cell insulin secretion, electrical activity and [Ca2+]i signalling [3, 52, 63, 64, 65, 66, 67]. Although TEA also antagonises KATP channels at higher concentrations, the general conclusion from these studies is that blocking Kv (and possibly KCa) channels potently enhances insulin secretion in a glucose-dependent manner.

The effects of a Kv channel antagonist on pancreatic beta cells. The effects of the general Kv channel antagonist tetraethylammonium (TEA) on rat beta cells, rat insulinoma cells and isolated rat islets are shown. (A) Blocking Kv currents with 15 mmol/l TEA prevents the after-hyperpolarisation and prolongs rat beta-cell action potentials generated by current injection. (B) Blocking Kv currents with 20 mmol/l TEA enhances the membrane potential (grey line) and [Ca2+]i (black line) responses of rat beta cells (INS-1) to glucose as measured by DiBAC and Fura-2-AM fluorescence respectively (courtesy S.R. Smukler and A.M.F. Salapatek). (C) Blocking Kv currents in isolated rat islets did not affect insulin secretion under low (2.5 mmol/l) glucose conditions, but dose-dependently enhanced insulin secretion stimulated with 15 mmol/l glucose
Inherent glucose (depolarisation)-dependence of secretion stimulated by Kv channel antagonists
The realisation that a Kv channel antagonist acts as a glucose-dependent insulinotropic agent (Fig. 3) raises the possibility of developing Kv channel based therapeutics. The enormous interest in potential glucose-dependent therapeutics is evidenced by current research into glucose-dependent secretagogues such as the incretin hormones glucagon-like peptide-1 (GLP-1) and gastric inhibitory polypeptide (GIP) and their analogues [68], the imidazoline compounds [69, 70], and agonists of the protein kinase A (PKA) signalling pathway [71]. The theory behind the glucose-dependence of Kv channel antagonists is quite simple. Beta-cell Kv channels are closed under basal (i.e. non-stimulatory) conditions. Evidence supporting this includes the inability of TEA to alter resting membrane potential in mouse beta cells [61] and the fact that voltage-dependent K+ currents in rodent beta cells and insulinoma cells activate only at membrane potentials well above the resting values [28, 52, 66, 72, 73, 74, 75]. Therefore, inhibiting these channels in the absence of stimulatory glucose will have no effect on insulin secretion (Fig. 3). Only after glucose-dependent closure of KATP channels and subsequent membrane depolarisation, will Kv channels open to restore the outward flux of K+, and inhibition under these conditions prevents or delays action potential repolarisation (Fig. 3) [61]. Therefore, the insulinotropic effect of Kv channel inhibition is not strictly glucose-dependent, but depolarisation-dependent. This is highlighted by the ability of TEA and 4-aminopyridine (4-AP, another general Kv antagonist) to enhance insulin secretion from rat islets and insulinoma cells stimulated by sulphonylureas, even without glucose [52, 75]. Additionally, TEA has also been shown to enhance the membrane electrical activity of rat beta cells depolarised with the sulphonylurea tolbutamide [76]. Glucose-dependent stimulation of insulin secretion from rodent islets and insulinoma cells has also been reported for 4-AP [77], the peptide Kv2.1 antagonist hanatoxin [53], and a small molecule Kv2.1 antagonist termed compound 1 (C-1, a bispidine derivative related to class III anti-arrhythmic agents) [3]. Recent research has been aimed at identifying the molecular mediators of beta-cell voltage-dependent outward K+ currents to gain a better understanding of beta-cell stimulus-secretion coupling and with the hope that this might lead to the development of beta cell-specific therapies.
Voltage-dependent K+ channel expression in insulin-secreting cells
Electrophysiological evidence for different repolarising currents
Generally speaking, Kv currents are classified based on their biophysical and pharmacological properties (i.e. A-current/delayed-rectifier, sensitivity to block by 4-AP or TEA, and Ca2+ sensitivity). A-currents activate and inactivate quickly upon a step membrane potential depolarisation, giving rise to the characteristic waveform for which reason they are named (Fig. 4A). Delayed-rectifier currents activate more slowly and do not inactivate (or inactivate slowly over seconds; Fig. 4A). One should be cautious, however, when classifying currents based on these broad definitions since it is now clear that current kinetics can be heavily influenced by the experimental conditions, such as the presence or absence of regulatory subunits, phosphorylation state, temperature, redox state and/or concentration of O2 [56, 78, 79, 80]. The same channel therefore could be classified as a delayed-rectifier or A-current, or have a different pharmacology, under different experimental conditions. Certain voltage-dependent K+ currents are also dependent on or enhanced by [Ca2+]i. These are denoted as KCa and are sensitive to external TEA.

A-currents and delayed-rectifier currents and their presence in insulin-secreting cells. (A) Examples of A-currents (cloned Kv1.4) and delayed-rectifier currents (cloned Kv2.1) measured at 32–35°C using the voltage-clamp protocols shown. (B) Experiments done at 32–35°C, A-current components and delayed-rectifier components can be separated in rat beta cells (left panels) by expressing a dominant-negative Kv2 construct (average of six cells), or in MIN6 insulinoma cells (mouse- right panels) using the voltage-clamp protocol shown (average of eight cells). In each case, subtraction of the resulting currents yields the A-current component shown
The voltage-dependent outward K+ currents responsible for repolarising pancreatic beta cells were first described in mouse beta cells in 1986 [28] and subsequently in human beta cells in 1991 [29]. The slow activation and inactivation kinetics of these currents, usually observed in insulin-secreting cells in experiments at room temperature, place them in the broad category of delayed-rectifier K+ currents. More recent studies have shown that one can indeed also detect A-currents in insulin-secreting cells (Fig. 4B). A TEA-insensitive and 4-AP-sensitive A-current component was first described in mouse beta cells in 1989 [81]. Our group has subsequently identified a TEA-insensitive A-current component in rat beta cells [52], and recently demonstrated a large A-current component in MIN6 (mouse) insulinoma cells patch-clamped at near-physiological temperatures [3]. Interestingly, this large rapidly-inactivating component, unlike classical A-currents, was readily blocked by external application of TEA, similar to the rapidly-inactivating component described by Gopel et al. [82] in mouse beta cells of intact islets. As well, our group has recently shown that fast inactivation of TEA-sensitive rat beta-cell Kv currents can be regulated by temperature and the intracellular NADPH/NADP+ ratio [56]. These TEA-sensitive and -insensitive transient components likely reflect activation of separate channels and apparently differ between species.
It is recognised that voltage-dependent outward K+ currents in insulin secreting cells are comprised of both Ca2+-dependent (KCa) and a Ca2+-independent (Kv) current components. Studies by us and others suggest that the Kv and KCa components contribute 80 to 85% and 15 to 20% of total voltage-dependent outward currents respectively [3, 74, 83]. Single Kv and KCa channels have also been resolved in insulinoma (HIT) cells [84] and mouse beta cells [72, 74]. Based on pharmacological properties the KCa component seems to be composed mostly of large-conductance charybdotoxin/iberiotoxin-sensitive channels (called BKCa channels as opposed to small conductance or SKCa channels) [85, 86], although a number of studies have detected atypical components that are not blocked by selective antagonists [67, 87, 88]. The role of the Ca2+-sensitive current components is unclear as inhibitors of both large- and small-conductance KCa channels fail to affect insulin secretion from rodent islets [52, 89, 90].
Molecular correlates of beta-cell voltage-dependent K+ channels
Kv channels are formed by the tetrameric assembly of 6-transmembrane (TM) domain α-subunits (Fig. 2) [44, 91]. This is in contrast to KATP channels that are composed of four 2-TM domain pore forming subunits and four regulatory sulphonylurea receptor subunits (Fig. 2). Kv α-subunits can co-assemble as hetero-tetramers in a family specific manner (see Table 1 for the known Kv and related channel family members), and some of these associate with cytosolic or transmembrane regulatory subunits. Due to this, the combinations of naturally occurring, functionally distinct, channels is enormous, and makes identification of channels solely based on kinetic and pharmacological properties unreliable. Numerous Kv channel α-subunits have been detected in insulin-secreting cells (Table 2). The majority of studies have been carried out using reverse transcriptase-PCR (RT-PCR) identification of mRNA transcripts, and in general studies examining immortalised cell lines detect a larger number of Kv channel mRNA transcripts than in primary cells. Less sensitive, and therefore more likely to detect relevant subunits expressed abundantly at the protein level, are more recent western blot and immunohistochemical studies (Table 2).
Although many pore-forming Kv channel subunits have been detected in insulin-secreting cells, until recently little was known about which channels contribute to beta-cell repolarising currents and therefore regulate insulin secretion. This is, in part, due to the lack of appropriate pharmacological and/or molecular agents. Studies with the limited number of Kv1 antagonists suggest little contribution from this family [52, 83], although Kv1.1 might contribute somewhat in the HIT-T15 insulinoma cell line [83]. The ability of these antagonists to block heterotetrameric channels is not known, however. Using a dominant-negative approach, our group has shown that Kv1 and Kv2 channels mediate 20 and 60% of the delayed rectifier currents, respectively in rat beta cells, and knock out of these channels in rat islets enhances glucose-dependent insulin secretion [52]. Similar results were obtained in the HIT-T15 insulinoma cell line where the Kv1 and Kv2 dominant-negative constructs inhibited currents by 30 and 70%, respectively [52]. The Kv1 family channel that, in our experience, is detected with the greatest abundance is Kv1.4, for which there are no selective antagonists. It is unlikely that this channel contributes to the insulinotropic effect of TEA however, since it is insensitive to the commonly used concentrations of this compound when applied extracellularly. Kv1.4, and possibly Kv4.2 [54], therefore could contribute to the TEA-insensitive inactivating current in rat beta cells, although the small amplitude of the inactivating currents observed [52] does not seem to reflect the abundance of these channels at the protein level [52, 54]. As for the TEA-sensitive A-current detected in mouse beta cells [81] and mouse insulinoma cells [3], it seems likely that this current is mediated by Kv3.4, detected in mouse beta cells by immunohistochemistry [82], since this fast-inactivating Kv channel is sensitive to TEA.
We now know that Kv2.1 mediates the majority of the voltage-dependent outward K+ current in rat and mouse beta cells. The first report of Kv2.1 protein expression in an insulin-secreting cell (βTC-neo) was in 1996 [66]. However, these investigators were unable at the time to discern the absolute contribution of this channel in these cells due to the lack of an appropriate pharmacological inhibitor. Using a dominant-negative strategy, we were able to show that Kv2.1 contributes 60 to 70% of the voltage-dependent outward K+ current in rat beta cells and HIT-T15 insulinoma cells and does indeed regulate glucose-stimulated insulin secretion from rat islets [52]. Although the dominant-negative construct we used is expected to 'knockout' all Kv2 family channels, Kv2.2 is the only other Kv2 channel known to form functional channels, and this could not be detected by RT-PCR [52]. Further studies have investigated the contribution of Kv2.1 in beta cells using recently-identified selective antagonists. Using the novel Kv2.1 antagonist C-1 (described above) we showed that Kv2.1 contributes up to 85% of the steady-state outward K+ current in mouse beta cells and MIN6 insulinoma cells. In this study, antagonism of Kv2.1 with C-1 enhanced the membrane potential and [Ca2+]i responses of insulin-secreting cells to glucose, and stimulated insulin secretion from MIN6 insulinoma cells and perfused mouse pancreas in a glucose-dependent manner. These results are supported by a recent report that the Kv2.1 peptide antagonist hanatoxin blocks voltage-dependent K+ current in mouse beta cells, enhances Ca2+ oscillations in mouse islets, and augments insulin secretion from rat islets and βTC3 insulinoma cells [53].
Kv channels and human beta cells
To our knowledge, there has only been one study to investigate human beta-cell voltage-dependent K+ currents, which were reported to be similar to those observed in rodent beta cells and insulinoma cells [29]. There is also little known regarding Kv channel expression in human beta cells. Our group has recently reported expression of Kv2.1 protein in human islets [55], and we have observed Kv1.5, 1.6 and 2.1 mRNA transcripts in human islets (MacDonald and Wheeler, unpublished observations). Additionally, mRNA transcripts for Kv1.5 have been detected in human insulinoma cells [92]. A number of Kv channel regulatory β-subunits (described below) have been detected in human islet cDNA by PCR [93]. In this study Kvβ1.1, 2.1 and 2.2 were detected with high abundance, while Kvβ1.2, 1.3 and 3 were detected at lower levels. Additionally, one report showed the presence of the Kv-related human-ERG (hERG) K+ channel (Table 1) in human islets [94]. Although these channels are thought to facilitate action potential repolarisation and perhaps modulate glucose-dependent bursting, the hERG antagonist WAY123 398 did not enhance glucose-stimulated insulin secretion in that study making the true role of these channels unclear.
Potential contribution of Kv channels in diabetes
Few studies have examined the possible role of Kv channels in the development of beta-cell defects and diabetes. Genes encoding both Kv1.7 and Kv3.3 have been mapped to a region of chromosome 19 (19q13.3–13.4) containing a diabetes susceptibility locus [95], although the incidence of Kv channel polymorphism in a diabetic cohort has not been investigated. Mutations in Kv and related channels are known to play important roles in disorders such as familial long QT syndrome, episodic ataxia type 1, benign familial neonatal convulsion, familial and thyrotoxic hypokalemic periodic paralysis, and autosomal dominant deafness [96]. Polymorphism of Kv channels or their numerous regulatory proteins could lead to loss or gain of function. Either situation might contribute to the pathogenesis of the beta-cell defect. Increased Kv channel function would compromise glucose-stimulated insulin secretion by causing premature repolarisation of the action potential. Decreased Kv channel function could lead to beta-cell over-excitability and increased cytosolic Ca2+, possibly leading to apoptosis through activation of mitochondrial permeability transition pores and/or the caspase cascade [97].
Alterations in Kv channel function could also contribute indirectly to the development of diabetes. One recent study reports that Kv1.3 -/- mice had an increased basal metabolic rate and were resistant to the development of obesity, a well known risk factor for Type 2 diabetes, in response to a high fat diet [98]. In addition, it should be noted that channel function might be altered by external factors, rather than through genetic mutation. For example, high glucose or exogenous superoxide both decreased Kv current density and 4-AP induced constriction in rat small coronary arteries, an effect which is implicated in the pathogenesis of vascular complications [99]. It remains to be determined whether a similar glucotoxic effect on Kv currents occurs in pancreatic beta cells. To our knowledge no study has investigated Kv channel function in beta cells from diabetic humans or animal models of diabetes. The role, if any, of Kv channels in the development of diabetes is unknown.
The potential basis for tissue-specific differences in Kv2.1 currents
One clear difficulty for those wishing to develop Kv channel antagonists as potential therapeutics is the wide range of tissues that express these channels. In particular, Kv2.1 mRNA transcripts were recently identified in placenta, lung, liver, skeletal muscle, kidney, pancreas, spleen, thymus, prostate, testes, ovary, small intestine, colon and blood leukocytes [47]. Thus, there is the likelihood for undesirable side effects of any Kv2.1 antagonist that is not appropriately targeted. Although the problem of specificity must be dealt with regarding the potential use of Kv channel antagonists to treat Type 2 diabetes, similar problems have been (and are continually being) dealt with in other fields. For example, Kv1.3 antagonists are being investigated for their immune-suppressive effects, and have even been studied in animal models [100], and Kv channel antagonists are also being studied for their ability to limit adhesion and proliferation of some cancer cells [101, 102, 103]. It is possible that although the Kv2.1 α-subunit is widely expressed, the functional properties of native Kv2.1 channels differ between tissues. These differences could be sufficient to allow tissue-specific targeting of an antagonist. As discussed in the following sections, there are a number of ways in which tissue-specific differences in Kv channels can result, including tissue specificity of modulatory α-subunit or regulatory β-subunit expression, post-translational processing and/or channel localisation.
Modulatory α-subunits
It was mentioned above that there are at least 11 Kv channel families currently known (Table 2 only addresses the expression of Kv1–4). To date, no studies on insulin-secreting cells have investigated the expression of subunits from the Kv5–11 families. Subunits of these families have not been shown to form functional channels alone, but rather associate with members of the Kv1–4 families as modulator α-subunits (Table 1) [46, 47, 48, 49, 104]. Kv2.1 in particular, is often a target for these modulatory subunits, which have been shown to alter this channel's biophysical properties including kinetics of activation and inactivation, voltage-dependencies of activation and inactivation, and recovery times [47, 48, 105, 106, 107, 108]. One recent study shows mRNA expression in human pancreas of Kv10.1 and 11.1, which can form heterotetrameric channels with, and modulate the properties of, Kv2.1 [47]. In this study, the modulator α-subunits Kv6.3, 10.1 and 11.1 showed a more selective tissue distribution than Kv2.1. It seems then, that although there are only two known members of the Kv2 channel family, the tissue-specific biophysical characteristics of Kv2.1 can be determined by the expression of modulatory α-subunits. Heterogeneity of Kv2.1 currents resulting from modulatory α-subunit expression has been speculated to exist in the heart [106], and could result in fundamentally different Kv2.1 containing channels in different tissues.
Regulatory β-subunits
Kv channel heterogeneity can also result from tissue-specific differences in the expression of cytoplasmic regulatory proteins such as Kvβ-subunits. These subunits interact with the channel N-terminus and modulate channel function and expression [109]. The Kvβ subunits are related to the NADPH-dependent oxidoreductase family of enzymes and are postulated to confer intracellular redox sensitivity to Kv α-subunits (this will be discussed further below). As mentioned above, human islets express Kvβ1–3 to varying degrees and the same Kvβ-subunit expression profile was detected by PCR of rat islet and insulinoma cell (INS-1) cDNA, while Kvβ2 was detected by western blot of rat islet protein [93]. Currently no known Kvβ subunits associate directly with Kv2.1 [110, 111]. However, a novel Kv2.1-associated 38-kD neuronal protein similar to Kvβ subunits has been described [112], and Kv2.1 interacts with cytosolic KChAP (K + Channel Accessory Protein) which can in turn bind Kvβ1.2 and Kvβ2 [113]. Additionally, one group reports that co-expression of Kv2.1 with Kvβ2.1 in HEK293 cells confers sensitivity of the Kv2.1 current to hypoxia [114]. It remains to be determined however, whether there is a Kv2.1 specific β-subunit that can be detected in vivo. It is possible that a so far unknown β-subunit, or other cytoplasmic regulatory subunit, could confer tissue-specific differences to Kv2.1 channels.
Channel localisation
Another potential mechanism contributing to tissue-specific differences in Kv channels is the functional localisation of particular channels. In beta cells it is known that L-type VDCCs preferentially support insulin secretion compared with other Ca2+ channel types [33]. The L-type channels are localised to sites of exocytosis by their interaction with the exocytotic SNARE proteins where, when opened by depolarisation, they allow local [Ca2+] increases in the vicinity of the secretory granule. Sub-cellular spatial localisation of Kv2.1 has been shown in mouse retina cells [115] and in rat hippocampal neurones [116], where a C-terminal targeting sequence allows spatial separation from other Kv channels, even its closely related family member Kv2.2 [117, 118]. As discussed below, we have recently described an interaction between Kv2.1 and the SNARE proteins SNAP-25 and syntaxin 1A which might serve to localise the channel to sites of exocytosis [55]. In that study, we detected a binding interaction between SNAP-25 and the C-terminus of Kv2.1, which could not be accounted for in terms of functional channel regulation, but could contribute to channel localisation. Another potential mechanism regulating sub-cellular localisation of Kv channels is their association with detergent insoluble lipid rafts. Kv2.1 preferentially targets to lipid rafts in a heterologous expression system and in rat brain, disruption of which by cholesterol depletion alters channel function [119]. Furthermore, Kv2.1 targets to a different lipid raft population than Kv1.5 when expressed in mouse Ltk- cells, as Kv1.5 but not Kv2.1 was found to co-localise with caveolin and follow caveolin re-distribution after microtubule disruption [120]. Therefore, the spatial localisation of Kv2.1 in various tissues (and the physiological processes with which the channel is associated with) will determine the effects of any potential antagonist.
Post-translational phosphorylation
Kv2.1 is subject to a high level of constitutive post-translational phosphorylation early in biosynthesis when expressed in COS-1 cells [121]. The result is a channel subunit that, when subjected to sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), appears much larger (Mr 108 000) than the Mr 95 000 predicted by the amino acid sequence. This increase in size does not appear to involve N-linked glycosylation [122]. A range of apparent molecular weights have been detected for Kv2.1 α-subunits in different tissues. Kv2.1 α-subunits have been described either as Mr 130 000 in the rat brain [123], as Mr 132 000 in PC12 cells [124], as Mr 125 000 in rat aortic myocytes [125], and as Mr 130 000 in rat atrial and ventricular myocytes [126, 127], while rat mesenteric artery smooth muscle cells express what seems to be an unphosphorylated Mr 95 000 Kv2.1 subunit [128]. Insulin secreting cells express a Kv2.1 α-subunit that is approximately Mr 108 000 [3, 55, 66]. It was subsequently shown that phosphorylation occurs mainly on serine residues, and that the extent of channel phosphorylation, particularly at the channels' C-terminus, can alter its voltage-dependent activation [129]. It seems possible then, that the tissue specific differences in post-translational phosphorylation of Kv2.1 can result in channels that are distinct, both biophysically and in their interactions with other proteins. This may be exploited to aid in the tissue targeting of any potential therapeutic agent.
Regulation of Kv channel activity in beta cells
Kv channels associate with and are regulated by the SNARE complex
SNARE proteins constitute the molecular machinery regulating vesicle docking and fusion. Vesicle-associated SNARE proteins (or v-SNAREs) include the vesicle-associated membrane proteins (VAMPs or synaptobrevins). SNARE proteins associated with the target membrane (or t-SNAREs) include SNAP-25 and syntaxin. The molecular mechanism of exocytosis in pancreatic beta cells has been extensively reviewed elsewhere [130]. It is now becoming clear that, while the SNARE complex mediates the molecular events of exocytosis upon elevation of [Ca2+]i, it is also functionally coupled to the excitatory machinery in a unit termed the 'excitosome'. This hypothesis results largely from studies showing that SNARE proteins can associate with and regulate VDCCs [83, 131, 132, 133].
Recent work has shown that SNARE proteins can also associate with and regulate Kv channels. Particularly, syntaxin 1A was found to associate with Kv1.1 and augment channel inactivation by enhancing the efficacy of the Kvβ1.1 subunit which was co-expressed with Kv1.1 in this study [134]. Additionally, it was recently shown that SNAP-25 can regulate the activity of Kv1.1 in the HIT-T15 insulinoma cell line [83], in part by slowing channel activation while enhancing slow inactivation. This interaction is likely of little relevance to insulin secretion as Kv1.1 is not expressed in rat or human islets (Table 2) and the Kv1.1 inhibitor dendrotoxin has little effect on insulin secretion and outward currents, even in the same HIT-T15 cell line [52]. In oesophageal smooth muscle cells, SNAP-25 was found to inhibit both Kv and KCa currents, causing a leftward shift of voltage-dependent activation of KCa but not Kv channels [135]. The molecular correlates of these currents were not identified in this study. We recently reported that SNAP-25 and syntaxin 1A can bind Kv2.1 and that SNAP-25 can inhibit Kv2.1 in beta cells by approximately 40% through an interaction with the Kv2.1 α-subunit N-terminus [55]. In this study the effect of SNAP-25 on the biophysical properties of the current was not investigated. Interestingly, it was determined that the inhibitory effect of SNAP-25 was specific to Kv2.1 in primary rat beta cells as compared to other Kv channels. In addition to the interaction with the channel N-terminus, we also detected a binding interaction between SNAP-25 and a C-terminal fragment of the channel to which no functional role was ascribed. As mentioned above, this interaction could be important in terms of channel localisation, as C-terminal Kv2.1 sequences are known to be involved in channel targeting [118]. The potential importance of the C-terminus in channel localisation is supported by a recent study showing overexpression of syntaxin 1A disrupts membrane targeting of cloned Kv2.1 through an interaction with the C-terminus [136]. In this study syntaxin 1A also reduced Kv currents in rat beta cells and modulated cloned Kv2.1 activation kinetics and voltage-dependence of steady-state inactivation.
Although the benefits of a close association between the SNARE complex and VDCCs seem clear (local delivery of the Ca2+ trigger), it is more difficult to imagine the benefit of a close association with Kv2.1. Since little is known about how ion channels could mediate local changes in membrane potential, it is possible that Kv2.1 could locally modulate VDCC activity. Alternatively, the SNARE proteins might have an important role in direct regulation of excitability through their interaction with VDCCs and Kv channels. Another possibility is that these associations allow for the multi-protein excitosome complex to be regulated as a single functional unit. In fact, as discussed below, all of the currently identified excitosome components (SNARE complex, VDCCs and Kv channels) are subject to hormonal (GLP-1) regulation. Together, these studies support the existence of an excitosome composed of both secretory and excitatory machinery.
Hormonal regulation of beta-cell Kv channels
Numerous studies have described hormone-mediated alterations in voltage-dependent K+ currents, both excitatory and inhibitory. The best characterised of these effects is β-adrenergic-mediated voltage-dependent K+ current down-regulation in lymphocytes [137] and up-regulation in cardiac myocytes [138]. In both of these tissues, the cAMP/PKA-signalling pathway has been implicated in the regulation of these channels [139, 140]. Reports suggest that cAMP can reduce voltage-dependent K+ currents in murine lymphocytes [139], a pituitary cell line [141], and a human melanoma cell line [101]. In contrast, cAMP enhances voltage-dependent K+ currents in cardiac myocytes [140], a study that has been confirmed at the single channel level in frog atrial myocytes [142] and the giant squid axon [143]. Phosphorylation can occur directly on the channel, as PKA phosphorylation of an atrial Kv channel near the amino terminus enhances channel activity [144] and phosphorylation of Kv1.1 channel α-subunits regulates the extent of inhibition by a regulatory β-subunit [145]. Phosphorylation of β-subunits themselves can also modulate the regulatory interaction with pore forming α-subunits [146]. It has recently been shown that regulation of a cardiac voltage-dependent K+ channel (KCNQ, also called KvLQT) by cAMP requires the expression of an A kinase-anchoring protein (AKAP15/18 or AKAP79) [147]. Additionally, an increase in voltage-dependent K+ current is implicated in epinephrine-induced inhibition of the glucose-dependent increase in [Ca2+]i in ob/ob and wild-type mouse beta cells [148] as the effect was reversed by TEA. Interestingly, the inhibitory effect of epinephrine on [Ca2+]i was also reversed by the adenylyl cyclase activator forskolin [148]. Therefore, we believe that there is mounting evidence to suggest that hormonal modulation of Kv currents is physiologically important.
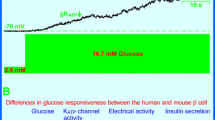
The Kv2.1 α-subunit contains two PKA phosphorylation sites on the C-terminus and a conserved PKC phosphorylation site on the cytoplasmic loop between the 4th and 5th transmembrane domains (Fig. 2). Inhibition of PKA leads to a reduction in cloned Kv2.1 current [149], while phosphorylation at the conserved PKC phosphorylation site suppresses Kv1 channels [150, 151, 152]. GLP-1 is proposed to enhance glucose-stimulated insulin secretion by regulating the activity of several ion channels (KATP, VDCC, NSCC) involved in KATP channel-dependent insulin secretion as well as the secretory machinery itself [153]. Since beta-cell Kv currents are potent glucose-dependent regulators of insulin secretion (Fig. 4), we hypothesised that the physiological secretagogue GLP-1 could regulate Kv channel function. Indeed, we have found that GLP-1 and the GLP-1 receptor agonist exendin 4 inhibit voltage-dependent outward K+ currents in rat beta cells voltage-clamped in the whole-cell configuration by approximately 40% in a cAMP/PKA dependent manner and prolongs the time-course of beta-cell repolarisation following transient depolarisation by current injection [54].
The ability of GLP-1 to reduce beta-cell Kv currents seems contradictory to the known effects of PKA phosphorylation on the cloned Kv2.1 channel (above). One recent study suggests that cAMP signalling was not sufficient in itself to antagonise voltage-dependent K+ currents in INS-1 insulinoma cells [75]. Although this discrepancy may result from differences in the models studied (primary beta cell vs insulinoma cell vs heterologuous expression), it is possible that additional GLP-1 signalling pathways [153] are involved. We are currently undertaking studies to investigate the latter possibility. Additionally, we have now determined that the mechanism of Kv current block by GLP-1 involves an approximately 20 mV leftward shift in the voltage-dependence of steady-state inactivation (MacDonald and Wheeler, unpublished), effectively reducing the number of available channels. Although GLP-1 receptor activation inhibits voltage-dependent outward K+ currents in rat beta cells in the absence of glucose, this effect could still contribute to the glucose-dependence of GLP-1's insulinotropic effect, as Kv channels are not normally expected to be active until after glucose-induced depolarisation of the cell membrane (Fig. 1) [154]. The absolute contribution of Kv channel inhibition to the insulinotropic effect of GLP-1 is unknown and is currently under investigation.
Kv channels as intracellular redox Sensors: Potential role in glucose-stimulated electrical activity
Certain Kv channels, particularly the regulatory β-subunits associated with the pore forming α-subunits, have the ability to act as sensors of intracellular redox potential [155, 156, 157] and to regulate channels dependant on their NADPH-dependent oxidoreductase activity [158, 159]. At least one study has shown that mutation of the Kvβ (Kvβ1.1) oxidoreductase active site attenuated the ability of this subunit to confer fast inactivation to a Kv1 channel (Kv1.5) [159]. Others have shown that mutation of regions putatively involved in NADPH co-factor binding alter the ability of β-subunits to promote channel surface expression [158, 160]. Regulatory β-subunits have also been implicated in the regulation of Kv channels in response to changes in [O2] [114]. Although we have stressed the role of β-subunits as potential redox sensors, direct redox modulation of a Kv α-subunit (particularly cysteine residues) could also modify channel properties [78].
It was proposed in 1986 that, analogous to the mechanism mediating hypoxia-induced pulmonary vasoconstriction, an increased intracellular redox potential links beta-cell metabolism to K+ channel function, contributing to membrane excitability and glucose-stimulated insulin secretion [161]. Recent studies have strongly suggested that NADPH production via the malate-aspartate shuttle [162, 163, 164], also called pyruvate cycling [165], might be an important metabolic signal. Indeed, metabolisable insulin secretagogues increase the NADPH/NADP+ ratio in rodent islets [166, 167] and inhibition of NADPH formation reduces glucose-stimulated insulin secretion from rat islets [168, 169]. Recent evidence suggests the existence of membrane associated aldehyde oxidoreductase-like enzyme activity in rat islets [170] and, as mentioned above, a number of oxidoreductase-like Kvβ subunits (Kvβ1, 2 and 3) are expressed in human and rat islets and INS-1 insulinoma cells [93]. The ability of the aldehyde reductase antagonist diphenylhydantoin to prevent glucose-stimulated insulin secretion from rat islets supports a role for an NADPH-oxidoreductase activity in stimulus-secretion coupling [170, 171, 172, 173], although it is suggested that this effect could be related to the ability of diphenylhydantoin to block Na+ channels. However, the role for Na+ channels in stimulus-secretion coupling is unclear; in fact, rodent beta-cell voltage-dependent Na+ channels are thought to be completely or nearly completely inactivated within the operating membrane potential range of rodent beta cells [73, 82], but could be active in human beta cells [174].
Recently, we have described a potent regulation of native Kv2.1 channels in primary rat beta cells by the cytoplasmic NADPH/NADP+ ratio [56]. In that study, increasing the intracellular redox potential by raising the intracellular NADPH/NADP+ ratio caused beta-cell Kv2.1 currents to inactivate quickly and more completely, and caused a leftward shift in the voltage-dependence of steady-state inactivation (meaning that more channels were already inactivated, and therefore unavailable). This has important implications since the metabolic generation of NADPH could reduce the efficacy of Kv channels in repolarising the beta cell (Fig. 5). We propose that the metabolic generation of NADPH contributes to beta-cell electrical excitability in response to glucose by reducing the effective ability of Kv currents to hyperpolarise the cell membrane (Fig. 5). This model represents a modification of the 1986 proposal [161] and is similar to the effect of [O2] on pulmonary vascular smooth muscle cells, where Kv2.1 channel function is modulated by O2 dependent changes in intracellular redox potential [175]. This cannot, however, account for the KATP channel independent pathway as defined by the ability of glucose to stimulate insulin secretion when cells are held depolarised with diazoxide and high K+ [1, 2]. This model is most applicable to glucose-stimulated electrical activity (which is of course directly related to secretion), and could in part account for the ability of glucose to modify beta-cell electrical responses even when KATP channels are already closed by sulphonylureas [76, 176]. It should be noted that other mechanisms have been proposed for this observation including the glucose-dependent regulation of VDCCs [177], volume-sensitive anion currents [76] and Na+/K+ ATPase activity [178]. It is likely that other, unknown, targets of NADPH or other metabolic signals such as long chain CoA's mediate the true membrane potential independent (though largely Ca2+ dependent) stimulation of insulin secretion. Future studies should investigate the possibility that regulation of beta-cell Kv2.1 channel inactivation by NADPH contributes to the electrical response of beta cells.

An electrogenic model for glucose-stimulated insulin secretion. In this model, glucose metabolism causes a rise in both intracellular ATP and NADPH and decreases intracellular ADP and NADP+. KATP channels are closed by the reciprocal changes in ATP and ADP, leading to membrane depolarisation, activation of VDCCs and entry of Ca2+. Kv channels also activate upon membrane depolarisation (although slower than VDCCs) to repolarise beta-cell action potentials. An increased NADPH/NADP+ ratio can augment membrane excitability and Ca2+ entry by causing greater inactivation of Kv channels (both in terms of steady-state voltage dependence and faster inactivation kinetics), meaning that fewer Kv channels are available to repolarise the membrane and limit the activity of VDCCs (shown as a positive effect on VDCC activity). Activation of the G-protein coupled glucagon-like peptide-1 (GLP-1) receptor also antagonises beta-cell Kv channels, through a cAMP and PKA dependent pathway. This effect is expected to increase insulin secretion by prolonging glucose-stimulated action potentials, thereby enhancing the activity of VDCCs and the entry of Ca2+
Conclusion
There is clear evidence supporting a role for voltage-dependent K+ channels, in particular Kv2.1, in the regulation of insulin secretion. These currents repolarise beta-cell action potentials when triggered by a glucose-induced KATP channel closure. Blocking Kv currents prolong the action potential and therefore increases the activity of VDCC's and entry of Ca2+. Recent evidence supports the localisation of Kv channels to the excitosome complex, where ion channels and secretory SNARE proteins interact to control the complex events underlying secretion. Beta-cell Kv channels are also targets of the G-protein coupled GLP-1 receptor and signals from glucose metabolism, pathways which could be physiologically relevant to the control of insulin secretion. The glucose-dependence of the insulinotropic effect of Kv inhibitors make these channels promising targets for the development of hypoglycaemic therapeutics. Further studies characterising tissue specific differences in Kv2.1 currents and the roles and regulation of the various Kv channels expressed in insulin-secreting cells may lead to the development of agents with sufficient beta-cell specificity to be considered for therapeutic use.
Abbreviations
- [Ca2+]i :
-
intracellular Ca2+
- 4-AP:
-
4-aminopyridine
- GLP-1:
-
glucagon-like peptide-1
- IHC:
-
immunohistochemistry
- ISH:
-
in situ hybridisation
- KATP :
-
ATP-sensitive K+
- KCa :
-
Ca2+-sensitive voltage-dependent K+
- Kv:
-
voltage-dependent K+
- NSCC:
-
non-selective cation channel
- PHHI:
-
persistent hyperinsulinemic hypoglycaemia of infancy
- PKA:
-
protein kinase A
- PKC:
-
protein kinase C
- RT-PCR:
-
reverse transcriptase-PCR
- SNAP-25:
-
synaptosome-associated protein of 25-kilodaltons
- SNARE:
-
soluble N-ethylmaleimide-sensitive factor attachment protein receptor
- SUR:
-
sulphonylurea receptor
- TEA:
-
tetraethylammonium
- VDCC:
-
voltage-dependent Ca2+ channel
- WB:
-
western blot
References
Aizawa T, Sato Y, Komatsu M (2002) Importance of nonionic signals for glucose-induced biphasic insulin secretion. Diabetes 51 [Suppl 1]:S96–S98
Prentki M, Tornheim K, Corkey BE (1997) Signal transduction mechanisms in nutrient-induced insulin secretion. Diabetologia 40 [Suppl 2]:S32–S41
MacDonald PE, Sewing S, Wang J et al. (2002) Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic β-cells enhances glucose-dependent insulin secretion. J Biol Chem 277:44938–44945
Henquin JC, Meissner HP, Preissler M (1979) 9-Aminoacridine- and tetraethylammonium-induced reduction of the potassium permeability in pancreatic β-cells. Effects on insulin release and electrical properties. Biochim Biophys Acta 587:579–592
Herchuelz A, Thonnart N, Carpinelli A, Sener A, Malaisse WJ (1980) Regulation of calcium fluxes in rat pancreatic islets: the role of K+ conductance. J Pharmacol Exp Ther 215:213–220
Komatsu M, Schermerhorn T, Aizawa T, Sharp GW (1995) Glucose stimulation of insulin release in the absence of extracellular Ca2+ and in the absence of any increase in intracellular Ca2+ in rat pancreatic islets. Proc Natl Acad Sci USA 92:10728–10732
Henquin JC (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Dean PM, Matthews EK (1968) Electrical activity in pancreatic islet cells. Nature 219:389–390
Henquin JC (1978) D-glucose inhibits potassium efflux from pancreatic islet cells. Nature 271:271–273
Sehlin J, Taljedal IB (1975) Glucose-induced decrease in Rb+ permeability in pancreatic β-cells. Nature 253:635–636
Ashcroft FM, Harrison DE, Ashcroft SJ (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature 312:446–448
Cook DL, Hales CN (1984) Intracellular ATP directly blocks K+ channels in pancreatic β-cells. Nature 311:271–273
Rorsman P, Trube G (1985) Glucose dependent K+-channels in pancreatic β-cells are regulated by intracellular ATP. Pflugers Arch 405:305–309
Arkhammar P, Nilsson T, Rorsman P, Berggren PO (1987) Inhibition of ATP-regulated K+ channels precedes depolarization-induced increase in cytoplasmic free Ca2+ concentration in pancreatic β-cells. J Biol Chem 262:5448–5454
Inagaki N, Gonoi T, Clement JP et al. (1995) Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science 270:1166–1170
Sakura H, Ammala C, Smith PA, Gribble FM, Ashcroft FM (1995) Cloning and functional expression of the cDNA encoding a novel ATP- sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Lett 377:338–344
Aguilar-Bryan L, Nichols CG, Wechsler SW et al. (1995) Cloning of the β-cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science 268:423–426
Sturgess NC, Ashford ML, Cook DL, Hales CN (1985) The sulphonylurea receptor may be an ATP-sensitive potassium channel. Lancet 2:474–475
Ashcroft SJ (2000) The β-cell KATP channel. J Membr Biol 176:187–206
Trube G, Rorsman P, Ohno-Shosaku T (1986) Opposite effects of tolbutamide and diazoxide on the ATP-dependent K+ channel in mouse pancreatic β-cells. Pflugers Arch 407:493–499
Yabo R, Viktora J, Staquet M, Wolff F (1965) Studies concerning the hyperglycemic effects of diazoxide and its mode of action. Diabetes 14:591–594
Stabile BE (1997) Islet cell tumors. Gastroenterologist 5:213–232
Shirland L (2001) When it is more than transient neonatal hypoglycemia: hyperinsulinemia—a case study challenge. Neonatal Netw 20:5–11
Taylor AE (2000) Insulin-lowering medications in polycystic ovary syndrome. Obstet Gynecol Clin North Am 27:583–595
Meissner HP, Schmelz H (1974) Membrane potential of β-cells in pancreatic islets. Pflugers Arch 351:195–206
Dean PM, Matthews EK (1970) Electrical activity in pancreatic islet cells: effect of ions. J Physiol 210:265–275
Atwater I, Dawson CM, Eddlestone GT, Rojas E (1981) Voltage noise measurements across the pancreatic β-cell membrane: calcium channel characteristics. J Physiol 314:195–212
Rorsman P, Trube G (1986) Calcium and delayed potassium currents in mouse pancreatic β-cells under voltage-clamp conditions. J Physiol (Lond) 374:531–550
Kelly RP, Sutton R, Ashcroft FM (1991) Voltage-activated calcium and potassium currents in human pancreatic β-cells. J Physiol (Lond) 443:175–192
Perez-Reyes E, Wei XY, Castellano A, Birnbaumer L (1990) Molecular diversity of L-type calcium channels. Evidence for alternative splicing of the transcripts of three non-allelic genes. J Biol Chem 265:20430–20436
Yaney GC, Wheeler MB, Wei X et al. (1992) Cloning of a novel α1-subunit of the voltage-dependent calcium channel from the β-cell. Mol Endocrinol 6:2143–2152
Seino S, Chen L, Seino M (1992) Cloning of the α1 subunit of a voltage-dependent calcium channel expressed in pancreatic β-cells. Proc Natl Acad Sci USA 89:584–588
Satin LS (2000) Localized calcium influx in pancreatic β-cells: its significance for Ca2+-dependent insulin secretion from the islets of Langerhans. Endocrine 13:251–262
Bas F, Darendeliler F, Demirkol D, Bundak R, Saka N, Gunoz H (1999) Successful therapy with calcium channel blocker (nifedipine) in persistent neonatal hyperinsulinemic hypoglycemia of infancy. J Pediatr Endocrinol Metab 12:873–878
Eichmann D, Hufnagel M, Quick P, Santer R (1999) Treatment of hyperinsulinaemic hypoglycaemia with nifedipine. Eur J Pediatr 158:204–206
Sanke T, Nanjo K, Kondo M, Nishi M, Moriyama Y, Miyamura K (1986) Effect of calcium antagonists on reactive hypoglycemia associated with hyperinsulinemia. Metabolism 35:924–927
Salkoff L (1983) Genetic and voltage-clamp analysis of a Drosophila potassium channel. Cold Spring Harb Symp Quant Biol 48 Pt 1:221–231
Kamb A, Tseng-Crank J, Tanouye MA (1988) Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron 1:421–430
Pongs O, Kecskemethy N, Muller R, Krah-Jentgens I, Baumann A, Kiltz HH, Canal I, Llamazares S, Ferrus A (1988) Shaker encodes a family of putative potassium channel proteins in the nervous system of Drosophila. EMBO J 7:1087–1096
Tempel BL, Papazian DM, Schwarz TL, Jan YN, Jan LY (1987) Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science 237:770–775
Christie MJ, Adelman JP, Douglass J, North RA (1989) Expression of a cloned rat brain potassium channel in Xenopus oocytes. Science 244:221–224
Tempel BL, Jan YN, Jan LY (1988) Cloning of a probable potassium channel gene from mouse brain. Nature 332:837–839
Sano Y, Mochizuki S, Miyake A et al. (2002) Molecular cloning and characterization of Kv6.3, a novel modulatory subunit for voltage-gated K+ channel Kv2.1. FEBS Lett 512:230–234
Christie MJ (1995) Molecular and functional diversity of K+ channels. Clin Exp Pharmacol Physiol 22:944–951
Dilks D, Ling HP, Cockett M, Sokol P, Numann R (1999) Cloning and expression of the human Kv4.3 potassium channel. J Neurophysiol 81:1974–1977
Hugnot JP, Salinas M, Lesage F et al. (1996) Kv8.1, a new neuronal potassium channel subunit with specific inhibitory properties towards Shab and Shaw channels. EMBO J 15:3322–3331
Ottschytsch N, Raes A, Van Hoorick D, Snyders DJ (2002) Obligatory heterotetramerization of three previously uncharacterized Kv channel α-subunits identified in the human genome. Proc Natl Acad Sci USA 99:7986–7991
Salinas M, Duprat F, Heurteaux C, Hugnot JP, Lazdunski M (1997) New modulatory α-subunits for mammalian Shab K+ channels. J Biol Chem 272:24371–24379
Stocker M, Kerschensteiner D (1998) Cloning and tissue distribution of two new potassium channel α-subunits from rat brain. Biochem Biophys Res Commun 248:927–934
Bauer CK, Schwarz JR (2001) Physiology of EAG K+ channels. J Membr Biol 182:1–15
Robbins J (2001) KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther 90:1–19
MacDonald PE, Ha XF, Wang J et al. (2001) Members of the Kv1 and Kv2 voltage-dependent K+ channel families regulate insulin secretion. Mol Endocrinol 15:1423–1435
Tamarina NA, Kuznetsov A, Dukes ID, Philipson LH (2002) Delayed rectifier potassium channel Kv2.1 role in β-cell physiology and insulin secretion. Diabetes 51:P1525 (abstract)
MacDonald PE, Salapatek AMF, Wheeler MB (2002) Glucagon-like peptide-1 receptor activation antagonizes voltage-dependent repolarizing K+ currents in β-cells: A possible glucose-dependent insulinotropic mechanism. Diabetes 51:S443–S447
MacDonald PE, Wang G, Tsuk S et al. (2002) Synaptosome-associated protein of 25 kilodaltons modulates Kv2.1 voltage-dependent K+ channels in neuroendocrine islet β-cells through an interaction with the channel N-terminus. Mol Endocrinol 16:2452–2461
MacDonald PE, Salapatek AMF, Wheeler MB (2003) Temperature and redox state dependence of native Kv2.1 currents in rat pancreatic β-cells. J Physiol 546:647–653
Dean PM, Matthews EK (1970) Glucose-induced electrical activity in pancreatic islet cells. J Physiol 210:255–264
Pace CS, Price S (1972) Electrical responses of pancreatic islet cells to secretory stimuli. Biochem Biophys Res Commun 46:1557–1563
Hodgkin AL, Huxley AF (1952) Currents carried by sodium and potassium ions through the membrane of the gian axon of Loligo. J Physiol (Lond) 116:449–472
Hodgkin AL, Huxley AF (1952) The components of membrane conductance in the giant axon of Loligo. J Physiol (Lond) 116:473–496
Atwater I, Ribalet B, Rojas E (1979) Mouse pancreatic β-cells: tetraethylammonium blockage of the potassium permeability increase induced by depolarization. J Physiol (Lond) 288:561–574
Henquin JC (1977) Tetraethylammonium potentiation of insulin release and inhibition of rubidium efflux in pancreatic islets. Biochem Biophys Res Commun 77:551–556
Henquin JC (1990) Role of voltage- and Ca2+-dependent K+ channels in the control of glucose-induced electrical activity in pancreatic β-cells. Pflugers Arch 416:568–572
Philipson LH, Rosenberg MP, Kuznetsov A, Lancaster ME, Worley JF III, Roe MW, Dukes ID (1994) Delayed rectifier K+ channel overexpression in transgenic islets and β-cells associated with impaired glucose responsiveness. J Biol Chem 269:27787–27790
Eberhardson M, Tengholm A, Grapengiesser E (1996) The role of plasma membrane K+ and Ca2+ permeabilities for glucose induction of slow Ca2+ oscillations in pancreatic β-cells. Biochim Biophys Acta 1283:67–72
Roe MW, Worley JF III, Mittal AA et al. (1996) Expression and function of pancreatic β-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J Biol Chem 271:32241–32246
Gopel SO, Kanno T, Barg S, Eliasson L, Galvanovskis J, Renstrom E, Rorsman P (1999) Activation of Ca2+-dependent K+ channels contributes to rhythmic firing of action potentials in mouse pancreatic β cells. J Gen Physiol 114:759–770
Creutzfeldt W (2001) The entero-insular axis in type 2 diabetes-incretins as therapeutic agents. Exp Clin Endocrinol Diabetes 109 [Suppl 2]:S288–S303
Efanov AM, Zaitsev SV, Mest HJ et al. (2001) The novel imidazoline compound BL11282 potentiates glucose-induced insulin secretion in pancreatic β-cells in the absence of modulation of KATP channel activity. Diabetes 50:797–802
Morgan NG, Chan SL, Mourtada M, Monks LK, Ramsden CA (1999) Imidazolines and pancreatic hormone secretion. Ann NY Acad Sci 881:217–228
Hashiguchi S, Yada T, Arima T (2001) A new hypoglycemic agent, JTT-608, evokes protein kinase A-mediated Ca2+ signaling in rat islet β-cells: strict regulation by glucose, link to insulin release, and cooperation with glucagon-like peptide-1 (7–36) amide and pituitary adenylate cyclase-activating polypeptide. J Pharmacol Exp Ther 296:22–30
Bokvist K, Rorsman P, Smith PA (1990) Effects of external tetraethylammonium ions and quinine on delayed rectifying K+ channels in mouse pancreatic β-cells. J Physiol (Lond) 423:311–325
Gopel S, Kanno T, Barg S, Galvanovskis J, Rorsman P (1999) Voltage-gated and resting membrane currents recorded from β-cells in intact mouse pancreatic islets. J Physiol 521 Pt 3:717–728
Smith PA, Bokvist K, Arkhammar P, Berggren PO, Rorsman P (1990) Delayed rectifying and calcium-activated K+ channels and their significance for action potential repolarization in mouse pancreatic β-cells. J Gen Physiol 95:1041–1059
Su J, Yu H, Lenka N, Hescheler J, Ullrich S (2001) The expression and regulation of depolarization-activated K+ channels in the insulin-secreting cell line INS-1. Pflugers Arch 442:49–56
Best L (2002) Evidence that glucose-induced electrical activity in rat pancreatic β-cells does not require KATP channel inhibition. J Membr Biol 185:193–200
Ahren B, Leander S, Lundquist I (1981) Effects of 4-aminopyridine on insulin secretion and plasma glucose levels in intact and adrenalectomized-chemically sympathectomized mice. Eur J Pharmacol 74:221–226
Ruppersberg JP, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M (1991) Regulation of fast inactivation of cloned mammalian IKA channels by cysteine oxidation. Nature 352:711–714
Walaas SI, Greengard P (1991) Protein phosphorylation and neuronal function. Pharmacol Rev 43:299–349
Archer SL, Souil E, Dinh-Xuan AT et al. (1998) Molecular identification of the role of voltage-gated K+ channels, Kv1.5 and Kv2.1, in hypoxic pulomnary vasoconstriction and control of resting membrane potential in rat pulmonary artery myocytes. J Clin Invest 101:2319–2330
Smith PA, Bokvist K, Rorsman P (1989) Demonstration of A-currents in pancreatic islet cells. Pflugers Arch 413:441–443
Gopel SO, Kanno T, Barg S, Rorsman P (2000) Patch-clamp characterisation of somatostatin-secreting δ-cells in intact mouse pancreatic islets. J Physiol 528:497–507
Ji J, Tsuk S, Salapatek AMF, Huang X et al. (2002) The 25-kDa synaptosome associated protein (SNAP-25) binds and inhibits delayed rectifier potassium channels in secretory cells. J Biol Chem 277:20195–20204
Fatherazi S, Cook DL (1991) Specificity of tetraethylammonium and quinine for three K channels in insulin-secreting cells. J Membr Biol 120:105–114
Li ZW, Ding JP, Kalyanaraman V, Lingle CJ (1999) RINm5f cells express inactivating BK channels whereas HIT cells express noninactivating BK channels. J Neurophysiol 81:611–624
Oosawa Y, Ashcroft SJ, Ashcroft FM (1992) Ca2+-activated K+ channels from an insulin-secreting cell line incorporated into planar lipid bilayers. Diabetologia 35:619–623
Ammala C, Bokvist K, Larsson O, Berggren PO, Rorsman P (1993) Demonstration of a novel apamin-insensitive calcium-activated K+ channel in mouse pancreatic β-cells. Pflugers Arch 422:443–448
Kozak JA, Misler S, Logothetis DE (1998) Characterization of a Ca2+-activated K+ current in insulin-secreting murine βTC-3 cells. J Physiol (Lond) 509 (Pt 2):355–370
Kukuljan M, Goncalves AA, Atwater I (1991) Charybdotoxin-sensitive KCa channel is not involved in glucose-induced electrical activity in pancreatic β-cells. J Membr Biol 119:187–195
Lebrun P, Malaisse WJ, Herchuelz A (1983) Activation, but not inhibition, by glucose of Ca2+-dependent K+ permeability in the rat pancreatic β-cell. Biochim Biophys Acta 731:145–150
Yellen G (2002) The voltage-gated potassium channels and their relatives. Nature 419:35–42
Philipson LH, Hice RE, Schaefer K, LaMendola J, Bell GI, Nelson DJ, Steiner DF (1991) Sequence and functional expression in Xenopus oocytes of a human insulinoma and islet potassium channel. Proc Natl Acad Sci USA 88:53–57
Chouinard SW, Lu F, Ganetzky B, MacDonald MJ (2000) Evidence for voltage-gated potassium channel β-subunits with oxidoreductase motifs in human and rodent pancreatic β cells. Recept Channels 7:237–243
Rosati B, Marchetti P, Crociani O, Lecchi M, Lupi R, Arcangeli A, Olivotto M, Wanke E (2000) Glucose- and arginine-induced insulin secretion by human pancreatic β-cells: the role of HERG K+ channels in firing and release. FASEB J 14:2601–2610
Kalman K, Nguyen A, Tseng-Crank J, Dukes ID, Chandy G, Hustad CM, Copeland NG, Jenkins NA, Mohrenweiser H, Brandriff B, Cahalan M, Gutman GA, Chandy KG (1998) Genomic organization, chromosomal localization, tissue distribution, and biophysical characterization of a novel mammalian Shaker-related voltage-gated potassium channel, Kv1.7. J Biol Chem 273:5851–5857
Hatta S, Sakamoto J, Horio Y (2002) Ion channels and diseases. Med Electron Microsc 35:117–126
Chandra J, Zhivotovsky B, Zaitsev S, Juntti-Berggren L, Berggren PO, Orrenius S (2001) Role of apoptosis in pancreatic β-cell death in diabetes. Diabetes 50 [Suppl 1]:S44–S47
Xu J, Koni PA, Wang P et al. (2003) The voltage-gated potassium channel Kv1.3 regulates energy homeostasis and body weight. Hum Mol Genet 12:551–559
Liu Y, Gutterman DD (2002) The coronary circulation in diabetes: influence of reactive oxygen species on K+ channel-mediated vasodilation. Vascul Pharmacol 38:43–49
Cahalan MD, Chandy KG (1997) Ion channels in the immune system as targets for immunosuppression. Curr Opin Biotechnol 8:749–756
Nilius B, Wohlrab W (1992) Potassium channels and regulation of proliferation of human melanoma cells. J Physiol 445:537–548
Zhou Q, Kwan HY, Chan HC, Jiang JL, Tam SC, Yao X (2003) Blockage of voltage-gated K+ channels inhibits adhesion and proliferation of hepatocarcinoma cells. Int J Mol Med 11:261–266
Rouzaire-Dubois B, DuBois JM (1998) K+ channel block-induced mammalian neuroblastoma cell swelling: a possible mechanism to influence proliferation. J Physiol 510 (Pt 1):93–102
Salinas M, Weille J de, Guillemare E, Lazdunski M, Hugnot JP (1997) Modes of regulation of shab K+ channel activity by the Kv8.1 subunit. J Biol Chem 272:8774–8780
Post MA, Kirsch GE, Brown AM (1996) Kv2.1 and electrically silent Kv6.1 potassium channel subunits combine and express a novel current. FEBS Lett 399:177–182
Kerschensteiner D, Stocker M (1999) Heteromeric assembly of Kv2.1 with Kv9.3: effect on the state dependence of inactivation. Biophys J 77:248–257
Kramer JW, Post MA, Brown AM, Kirsch GE (1998) Modulation of potassium channel gating by coexpression of Kv2.1 with regulatory Kv5.1 or Kv6.1 alpha-subunits. Am J Physiol 274:C1501–C1510
Patel AJ, Lazdunski M, Honore E (1997) Kv2.1/Kv9.3, a novel ATP-dependent delayed-rectifier K+ channel in oxygen-sensitive pulmonary artery myocytes. EMBO J 16:6615–6625
Pongs O, Leicher T, Berger M, Roeper J, Bahring R, Wray D, Giese KP, Silva AJ, Storm JF (1999) Functional and molecular aspects of voltage-gated K+ channel β-subunits. Ann NY Acad Sci 868:344–355
Heinemann SH, Rettig J, Graack HR, Pongs O (1996) Functional characterization of Kv channel β-subunits from rat brain. J Physiol (Lond) 493:625–633
Rhodes KJ, Keilbaugh SA, Barrezueta NX, Lopez KL, Trimmer JS (1995) Association and colocalization of K+ channel α- and β-subunit polypeptides in rat brain. J Neurosci 15:5360–5371
Trimmer JS (1991) Immunological idendification and characteriazation of a delayed rectifier K+ channel polypeptide in rat brain. Proc Natl Acad Sci USA 88:10764–10768
Wible BA, Yang Q, Kuryshev YA, Accili EA, Brown AM (1998) Cloning and expression of a novel K+ channel regulatory protein, KChAP. J Biol Chem 273:11745–11751
Coppock EA, Martens JR, Tamkun MM (2001) Molecular basis of hypoxia-induced pulmonary vasoconstriction: role of voltage-gated K+ channels. Am J Physiol Lung Cell Mol Physiol 281:L1–L12
Klumpp DJ, Song EJ, Pinto LH (1995) Identification and localization of K+ channels in the mouse retina. Vis Neurosci 12:1177–1190
Maletic-Savatic M, Lenn NJ, Trimmer JS (1995) Differential spatiotemporal expression of K+ channel polypeptides in rat hippocampal neurons developing in situ and in vitro. J Neurosci 15:3840–3851
Scannevin RH, Murakoshi H, Rhodes KJ, Trimmer JS (1996) Identification of a cytoplasmic domain important in the polarized expression and clustering of the Kv2.1 K+ channel. J Cell Biol 135:1619–1632
Lim ST, Antonucci DE, Scannevin RH, Trimmer JS (2000) A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 25:385–397
Martens JR, Navarro-Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM (2000) Differential targeting of Shaker-like potassium channels to lipid rafts. J Biol Chem 275:7443–7446
Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM (2001) Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J Biol Chem 276:8409–8414
Shi G, Kleinklaus AK, Marrion NV, Trimmer JS (1994) Properties of Kv2.1 K+ channels expressed in transfected mammalian cells. J Biol Chem 269:23204–23211
Shi G, Trimmer JS (1999) Differential asparagine-linked glycosylation of voltage-gated K+ channels in mammalian brain and in transfected cells. J Membr Biol 168:265–273
Frech GC, VanDongen AM, Schuster G, Brown AM, Joho RH (1989) A novel potassium channel with delayed rectifier properties isolated from rat brain by expression cloning. Nature 340:642–645
Sharma N, D'Arcangelo G, Kleinlaus A, Halegoua S, Trimmer JS (1993) Nerve growth factor regulates the abundance and distribution of K+ channels in PC12 cells. J Cell Biol 123:1835–1843
Belevych AE, Beck R, Tammaro P, Poston L, Smirnov SV (2002) Developmental changes in the functional characteristics and expression of voltage-gated K+ channel currents in rat aortic myocytes. Cardiovasc Res 54:152–161
Barry DM, Trimmer JS, Merlie JP, Nerbonne JM (1995) Differential expression of voltage-gated K+ channel subunits in adult rat heart. Relation to functional K+ channels? Circ Res 77:361–369
Huang B, Qin D, El Sherif N (2001) Spatial alterations of Kv channels expression and K+ currents in post-MI remodeled rat heart. Cardiovasc Res 52:246–254
Xu C, Lu Y, Tang G, Wang R (1999) Expression of voltage-dependent K+ channel genes in mesenteric artery smooth muscle cells. Am J Physiol 277:G1055–G1063
Murakoshi H, Shi G, Scannevin RH, Trimmer JS (1997) Phosphorylation of the Kv2.1 K+ channel alters voltage-dependent activation. Mol Pharmacol 52:821–828
Gerber SH Sudhof TC (2002) Molecular determinants of regulated exocytosis. Diabetes 51:S3–S11
Wiser O, Bennet MK, Atlas D (1996) Functional interaction of syntaxin and SNAP-25 with voltage-sensitive L- and N-type Ca2+ channels. EMBO J 15:4100–4110
Wiser O, Trus M, Hernandez A, Renstrom E, Barg S, Rorsman P, Atlas D (1999) The voltage sensitive Lc-type Ca2+ channel is functionally coupled to the exocytotic machinery. Proc Natl Acad Sci USA 96:248–253
Barg S, Ma X, Eliasson L et al. (2001) Fast exocytosis with few Ca2+ channels in insulin-secreting mouse pancreatic β-cells. Biophys J 81:3308–3323
Fili O, Michaelevski I, Bledi Y et al. (2001) Direct interaction of a brain voltage-gated K+ channel with syntaxin 1A: functional impact on channel gating. J Neurosci 21:1964–1974
Ji J, Salapatek AM, Lau H, Wang G, Gaisano HY, Diamant NE (2002) SNAP-25, a SNARE protein, inhibits two types of K channels in esophageal smooth muscle. Gastroenterology 122:994–1006
Leung YM, Kang Y, Gao X et al. (2003) Syntaxin 1A binds to the cytoplasmic C-terminus of Kv2.1 to regulate channel gating and trafficking. J Biol Chem 278:17532–17538
Soliven B, Nelson DJ (1990) β-adrenergic modulation of K+ current in human T lymphocytes. J Membr Biol 117:263–274
Walsh KB, Begenisich TB, Kass RS (1989) β-adrenergic modulation of cardiac ion channels. Differential temperature sensitivity of potassium and calcium currents. J Gen Physiol 93:841–854
Choquet D, Sarthou P, Primi D, Cazenave PA, Korn H (1987) Cyclic AMP-modulated potassium channels in murine B cells and their precursors. Science 235:1211–1214
Walsh KB, Kass RS (1988) Regulation of a heart potassium channel by protein kinase A and C. Science 242:67–69
Chung S, Kaczmarek LK (1995) Modulation of the inactivation of voltage-dependent potassium channels by cAMP. J Neurosci 15:3927–3935
Duchatelle-Gourdon I, Hartzell HC (1990) Single delayed rectifier channels in frog atrial cells. Effects of β-adrenergic stimulation. Biophys J 57:903–909
Perozo E, Vandenberg CA, Jong DS, Bezanilla F (1991) Single channel studies of the phosphorylation of K+ channels in the squid giant axon. I. Steady-state conditions. J Gen Physiol 98:1–17
Huang XY, Morielli AD, Peralta EG (1994) Molecular basis of cardiac potassium channel stimulation by protein kinase A. Proc Natl Acad Sci USA 91:624–628
Levin G, Chikvashvili D, Singer-Lahat D, Peretz T, Thornhill WB, Lotan I (1996) Phosphorylation of a K+ channel α-subunit modulates the inactivation conferred by a β-subunit. Involvement of cytoskeleton. J Biol Chem 271:29321–29328
Kwak YG, Hu NN, Wei J et al. (1999) Protein kinase A phosphorylation alters Kvβ1.3 subunit-mediated inactivation of the Kv1.5 potassium channel. J Biol Chem 274:13928–13932
Potet F, Scott JD, Mohammad-Panah R, Escande D, Baro I (2001) AKAP proteins anchor cAMP-dependent protein kinase to KvLQT1/IsK channel complex. Am J Physiol Heart Circ Physiol 280:H2038–H2045
Fournier L, Whitfield JF, Xiang H, Schwartz JL, Begin-Heick N (1993) K+ channel and α2-adrenergic effects on glucose-induced Ca2+ i surges: aberrant behavior in ob/ob mice. Am J Physiol 264:C1458–C1465
Wilson GG, O'Neill CA, Sivaprasadarao A, Findlay JB, Wray D (1994) Modulation by protein kinase A of a cloned rat brain potassium channel expressed in Xenopus oocytes. Pflugers Arch 428:186–193
Boland LM, Jackson KA (1999) Protein kinase C inhibits Kv1.1 potassium channel function. Am J Physiol 277:C100–C110
Murray KT, Fahrig SA, Deal KK et al. (1994) Modulation of an inactivating human cardiac K+ channel by protein kinase C. Circ Res 75:999–1005
Conley EC (1999) VLG K Kv1-Shak. In: Conley EC, Brammar WJ (eds) The ion channel facts book IV: voltage-gated channels. Academic Press, London, pp 347–523
MacDonald PE, El-kholy W, Riedel MJ, Salapatek AMF, Light PE, Wheeler MB (2002) The multiple actions of GLP-1 on the process of glucose-stimulated insulin secretion. Diabetes 51:S434–S442
Dukes ID, Philipson LH (1996) K+ channels: generating excitement in pancreatic β-cells. Diabetes 45:845–853
Gulbis JM, Mann S, MacKinnon R (1999) Structure of a voltage-dependent K+ channel β-subunit. Cell 97:943–952
Chouinard SW, Wilson GF, Schlimgen AK, Ganetzky B (1995) A potassium channel β-subunit related to the aldo-keto reductase superfamily is encoded by the Drosophila hyperkinetic locus. Proc Natl Acad Sci USA 92:6763–6767
McCormack T, McCormack K (1994) Shaker K+ channel β-subunits belong to an NAD(P)H-dependent oxidoreductase superfamily. Cell 79:1133–1135
Peri R, Wible BA, Brown AM (2001) Mutations in the Kvβ2 binding site for NADPH and their effects on Kv1.4. J Biol Chem 276:738–741
Bahring R, Milligan CJ, Vardanyan V et al. (2001) Coupling of voltage-dependent potassium channel inactivation and oxidoreductase active site of Kvβ subunits. J Biol Chem 276:22923–22929
Campomanes CR, Carroll KI, Manganas LN et al. (2002) Kv β-subunit oxidoreductase activity and Kv1 potassium channel trafficking. J Biol Chem 277:8298–8305
Archer SL, Will JA, Weir EK (1986) Redox status in the control of pulmonary vascular tone. Herz 11:127–141
MacDonald MJ (1995) Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem 270:20051–20058
Fahien LA, MacDonald MJ (2002) The succinate mechanism of insulin release. Diabetes 51:2669–2676
MacDonald MJ (2003) The export of metabolites from mitochondria and anaplerosis in insulin secretion. Biochim Biophys Acta 1619:77–88
Lu D, Mulder H, Zhao P et al. (2002) 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc Natl Acad Sci USA 99:2708–2713
Ashcroft SJ, Christie MR (1979) Effects of glucose on the cytosolic ration of reduced/oxidized nicotinamide-adenine dinucleotide phosphate in rat islets of Langerhans. Biochem J 184:697–700
Hedeskov CJ, Capito K, Thams P (1987) Cytosolic ratios of free [NADPH]/[NADP+] and [NADH]/[NAD+] in mouse pancreatic islets, and nutrient-induced insulin secretion. Biochem J 241:161–167
Ammon HP, Steinke J (1972) 6-Amnionicotinamide (6-AN) as a diabetogenic agent. In vitro and in vivo studies in the rat. Diabetes 21:143–148
MacDonald MJ, Ammon HP, Patel T, Steinke J (1974) Failure of 6-aminonicotinamide to inhibit the potentiating effect of leucine and arginine on glucose-induced insulin release in vitrol. Diabetologia 10:761–765
Laclau M, Lu F, MacDonald MJ (2001) Enzymes in pancreatic islets that use NADP(H) as a cofactor including evidence for a plasma membrane aldehyde reductase. Mol Cell Biochem 225:151–160
Kizer JS, Vargas-Gordon M, Brendel K, Bressler R (1970) The in vitro inhibition of insulin secretion by diphenylhydantoin. J Clin Invest 49:1942–1948
Levin SR, Grodsky GM, Hagura R, Smith D (1972) Comparison of the inhibitory effects of diphenylhydantoin and diazoxide upon insulin secretion from the isolated perfused pancreas. Diabetes 21:856–862
Matthews EK, Sakamoto Y (1975) Pancreatic islet cells: electrogenic and electrodiffusional control of membrane potential. J Physiol 246:439–457
Misler S, Barnett DW, Gillis KD, Pressel DM (1992) Electrophysiology of stimulus-secretion coupling in human β-cells. Diabetes 41:1221–1228
Michelakis ED, Rebeyka I, Wu X et al. (2002) O2 sensing in the human ductus arteriosus: regulation of voltage-gated K+ channels in smooth muscle cells by a mitochondrial redox sensor. Circ Res 91:478–486
Henquin JC (1998) A minimum of fuel is necessary for tolbutamide to mimic the effects of glucose on electrical activity in pancreatic β-cells. Endocrinology 139:993–998
Smith PA, Rorsman P, Ashcroft FM (1989) Modulation of dihydropyridine-sensitive Ca2+ channels by glucose metabolism in mouse pancreatic β-cells. Nature 342:550–553
Owada S, Larsson O, Arkhammar P et al. (1999) Glucose decreases Na+/K+-ATPase activity in pancreatic β-cells. An effect mediated via Ca2+-independent phospholipase A2 and protein kinase C-dependent phosphorylation of the α-subunit. J Biol Chem 274:2000–2008
Betsholtz C, Baumann A, Kenna S et al. (1990) Expression of voltage-gated K+ channels in insulin-producing cells. Analysis by polymerase chain reaction. FEBS Lett 263:121–126
Acknowledgements
The authors wish to thank Dr. P. Brubaker, Dr. D. Drucker and J. Joseph for critical review of the revised manuscript. The present work was supported by a research grant from the Canadian Institutes of Health Research (CIHR: MOP-49521). PEM is a CIHR Fellow and MBW is a CIHR Investigator.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
MacDonald, P.E., Wheeler, M.B. Voltage-dependent K+ channels in pancreatic beta cells: Role, regulation and potential as therapeutic targets. Diabetologia 46, 1046–1062 (2003). https://doi.org/10.1007/s00125-003-1159-8
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1159-8